
Highly Repetitive Genome of Coniella granati (syn. Pilidiella granati), the Causal Agent of Pomegranate Fruit Rot, Encodes a Minimalistic Proteome with a Streamlined Arsenal of Effector Proteins


 , , ,
, , ,  ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Genome Assembly
2.2. Analysis of Repetitive DNA
2.3. Annotation of Protein-Coding Genes and Their Putative Functions
2.4. Coniella spp. Comparative Genomics
2.5. Prediction of Pathogenicity Genes
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Culture
4.2. Genome Sequencing and Assembly
4.3. Transcriptome Sequencing for Gene Annotation Supporting Evidence
4.4. Prediction of DNA Repeats, Protein-Coding Genes and Gene Functions
4.5. Comparative Genomics between Coniella spp.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alvarez, L.V.; Groenewald, J.Z.; Crous, P.W. Revising the Schizoparmaceae: Coniella and its synonyms Pilidiella and Schizoparme. Stud. Mycol. 2016, 85, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Mincuzzi, A.; Ippolito, A. Pomegranate: Postharvest Fungal Diseases and Control. In New Advances in Postharvest Technology; Kahramanoğlu, I., Ed.; IntechOpen: Rijeka, Croatia, 2023. [Google Scholar]
- Thomidis, T. Pathogenicity and characterization of Pilidiella granati causing pomegranate diseases in Greece. Eur. J. Plant Pathol. 2015, 141, 45–50. [Google Scholar] [CrossRef]
- EFSA Panel on Plant Health; Bragard, C.; Baptista, P.; Chatzivassiliou, E.; Di Serio, F.; Gonthier, P.; Jaques Miret, J.A.; Justesen, A.F.; MacLeod, A.; Magnusson, C.S.; et al. Pest categorisation of Coniella granati. EFSA J. 2023, 21, e07848. [Google Scholar] [PubMed]
- Palou, L.; Guardado, A.; Montesinos-Herrero, C. First report of Penicillium spp. and Pilidiella granati causing postharvest fruit rot of pomegranate in Spain. New Dis. Rep. 2010, 22, 21. [Google Scholar] [CrossRef]
- Gerin, D.; Moncini, L.; Faretra, F.; Pollastro, S.; Chimienti, N.; Simone, G.; De Miccolis, A. Characterization of Coniella granati isolates causing pomegranate decline in Italy. Plant Dis. 2024, 108, 451–460. [Google Scholar] [CrossRef]
- Linaldeddu, B.T.; Bregant, C.; Ruzzon, B.; Montecchio, L. Coniella granati and Phytophthora palmivora the main pathogens involved in pomegranate dieback and mortality in north-eastern Italy. Ital. J. Mycol. 2020, 49, 92–100. [Google Scholar]
- Tziros, G.; Tzavella-Klonari, K. Pomegranate fruit rot caused by Coniella granati confirmed in Greece. Plant Pathol. 2008, 57, 783. [Google Scholar] [CrossRef]
- Mincuzzi, A.; Garganese, F.; Ippolito, A.; Sanzani, S. First report of Pilidiella granati causing postharvest fruit rot on pomegranate in southern Italy. J. Plant Pathol. 2016, 98, 377. [Google Scholar]
- Pollastro, S.; Dongiovanni, C.; Gerin, D.; Pollastro, P.; Fumarola, G.; De Miccolis Angelini, R.; Faretra, F. First report of Coniella granati as a causal agent of pomegranate crown rot in southern Italy. Plant Dis. 2016, 100, 1498. [Google Scholar] [CrossRef]
- Szendrei, L.; Toth, A.; Palkovics, L.; Salamon, P.; Petroczy, M. First report of Coniella granati causing leaf spot of pomegranate (Punica granatum L.) in Hungary. Plant Dis. 2022, 106, 2995. [Google Scholar] [CrossRef]
- Çeliker, N.M.; Uysal, A.; Çetinel, B.; Poyraz, D. Crown rot on pomegranate caused by Coniella granati in Turkey. Australas. Plant Dis. Notes 2012, 7, 161–162. [Google Scholar] [CrossRef]
- Levy, E.; Elkind, G.; Ben-Arie, R.; Ben-Ze’ev, I. First report of Coniella granati causing pomegranate fruit rot in Israel. Phytoparasitica 2011, 39, 403–405. [Google Scholar] [CrossRef]
- Mahadevakumar, S.; Shreenidhi, M.; Janardhana, G.R. First report of Coniella granati associated with dieback and fruit rot of pomegranate (Punica granatum L.) in India. J. Plant Pathol. 2019, 101, 787. [Google Scholar] [CrossRef]
- Kwon, J.-H.; Park, C.-S. Fruit rot of pomegranate (Punica granatum) caused by Coniella granati in Korea. Res. Plant Dis. 2002, 8, 215–219. [Google Scholar] [CrossRef]
- Chen, Y.; Shao, D.D.; Zhang, A.F.; Yang, X.; Zhou, M.G.; Xu, Y.L. First Report of a Fruit Rot and Twig Blight on Pomegranate (Punica granatum) Caused by Pilidiella granati in Anhui Province of China. Plant Dis. 2014, 98, 695. [Google Scholar] [CrossRef] [PubMed]
- Kc, A.; Vallad, G. First report of Pilidiella granati causing fruit rot and leaf spots on pomegranate in Florida. Plant Dis. 2016, 100, 1238. [Google Scholar] [CrossRef]
- Cintora-Martínez, E.A.; Leyva-Mir, S.G.; Ayala-Escobar, V.; Ávila-Quezada, G.D.; Camacho-Tapia, M.; Tovar-Pedraza, J.M. Pomegranate fruit rot caused by Pilidiella granati in Mexico. Australas. Plant Dis. Notes 2017, 12, 1–3. [Google Scholar] [CrossRef]
- Jabnoun-Khiareddine, H.; Ibrahim, N.; Aydi Ben Abdallah, R.; Mars, M.; Daami-Remadi, M. Response of Tunisian pomegranate (Punica granatum L.) cultivars and several plant hosts to Coniella granati (Saccardo). J. Hortic. 2018, 5, 1000245. [Google Scholar] [CrossRef]
- Lennox, C.; Mostert, L.; Venter, E.; Laubscher, W.; Meitz-Hopkins, J. First report of Coniella granati fruit rot and dieback on pomegranate in the western cape of South Africa. Plant Dis. 2018, 102, 821. [Google Scholar] [CrossRef]
- Mincuzzi, A.; Ippolito, A.; Brighenti, V.; Marchetti, L.; Benvenuti, S.; Ligorio, A.; Pellati, F.; Sanzani, S.M. The effect of polyphenols on pomegranate fruit susceptibility to Pilidiella granati provides insights into disease tolerance mechanisms. Molecules 2020, 25, 515. [Google Scholar] [CrossRef]
- Michailides, T.; Puckett, R.; Morgan, D. Pomegranate Decay Caused by Pilidiella granati in California. In Phytopathology; American Phytopathological Society: St. Paul, MN, USA, 2010; p. S83. [Google Scholar]
- Sharma, R.; Tegta, R. Incidence of dry rot of pomegranate in Himachal Pradesh and its management. Acta Hortic. 2011, 890, 491–499. [Google Scholar] [CrossRef]
- Jabnoun-Khiareddine, H.; Ibrahim, N.; Aydi Ben Abdallah, R.; Mars, M.; Kthiri, Z.; Daami-Remadi, M. Coniella granati (Saccardo) a new potential threat to pomegranate (Punica granatum L.) in Tunisia causing twig dieback and Fruit Rot. J. Plant Pathol. Microbiol. 2018, 9, 450. [Google Scholar] [CrossRef]
- Tsafouros, A.; Tsalgatidou, P.C.; Boutsika, A.; Delis, C.; Mincuzzi, A.; Ippolito, A.; Zambounis, A. Deciphering the Interaction between Coniella granati and Pomegranate Fruit Employing Transcriptomics. Life 2024, 14, 752. [Google Scholar] [CrossRef] [PubMed]
- Mahadevakumar, S.; Deepika, Y.S.; Amruthesh, K.N.; Lakshmidevi, N. First Report of Coniella granati Associated with Dieback of Rose (Rosa sp.) in India. Plant Dis. 2022, 106, 1304. [Google Scholar] [CrossRef] [PubMed]
- Sutton, B. The Coelomycetes. Fungi Imperfecti with Pycnidia, Acervuli and Stromata; Commonwealth Mycological Institute: Kew, UK, 1980. [Google Scholar]
- Van Niekerk, J.M.; Groenewald, J.Z.; Verkley, G.J.; Fourie, P.H.; Wingfield, M.J.; Crous, P.W. Systematic reappraisal of Coniella and Pilidiella, with specific reference to species occurring on Eucalyptus and Vitis in South Africa. Mycol. Res. 2004, 108 Pt 3, 283–303. [Google Scholar] [CrossRef] [PubMed]
- Maung, M.T. Biodiversity survey of coelomycetes in Burma. Australas. Mycol. 2008, 27, 74–110. [Google Scholar]
- Alaka, P.; Rao, V. A Compendium of Fungi on Legumes from India; Scientific Publishers: Jodhpur, India, 1998. [Google Scholar]
- Mahla, R.; Ashok, M. Evaluation of fungicides against leaf spot (Pseuducercosporella granati) of pomegranate. Pestology 1989, 13, 22–24. [Google Scholar]
- Gaikwad, A. Synergy between carbendazim and mancozeb in controlling leaf and fruit spots of pomegranate. J. Maharashtra Agric. Univ. 2000, 25, 165–167. [Google Scholar]
- Xavier, K.; Kc, A.; Vallad, G. Fungicide application timing essential for the management of leaf spot and fruit rot on pomegranate (Punica granatum L.) in Florida. Plant Dis. 2020, 104, 1629–1637. [Google Scholar] [CrossRef]
- Yang, X.; Gu, C.Y.; Sun, J.Z.; Bai, Y.; Zang, H.Y.; Chen, Y. Biological Activity of Pyraclostrobin against Coniella granati Causing Pomegranate Crown Rot. Plant Dis. 2021, 105, 3538–3544. [Google Scholar] [CrossRef]
- Thomidis, T.; Filotheou, A. Evaluation of five essential oils as bio-fungicides on the control of Pilidiella granati rot in pomegranate. Crop Prot. 2016, 89, 66–71. [Google Scholar] [CrossRef]
- Munhuweyi, K.; Lennox, C.L.; Meitz-Hopkins, J.C.; Caleb, O.J.; Opara, U.L. Major diseases of pomegranate (Punica granatum L.), their causes and management—A review. Sci. Hortic 2016, 211, 126–139. [Google Scholar] [CrossRef]
- Munhuweyi, K.; Caleb, O.J.; Lennox, C.L.; van Reenen, A.J.; Opara, U.L. In vitro and in vivo antifungal activity of chitosan-essential oils against pomegranate fruit pathogens. Postharvest Biol. Technol. 2017, 129, 9–22. [Google Scholar] [CrossRef]
- Brighenti, V.; Iseppi, R.; Pinzi, L.; Mincuzzi, A.; Ippolito, A.; Messi, P.; Sanzani, S.M.; Rastelli, G.; Pellati, F. Antifungal Activity and DNA Topoisomerase Inhibition of Hydrolysable Tannins from Punica granatum L. Int. J. Mol. Sci. 2021, 22, 4175. [Google Scholar] [CrossRef] [PubMed]
- Mincuzzi, A.; Picciotti, U.; Sanzani, S.M.; Garganese, F.; Palou, L.; Addante, R.; Ragni, M.; Ippolito, A. Postharvest Diseases of Pomegranate: Alternative Control Means and a Spiderweb Effect. J. Fungi 2023, 9, 808. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Tan, X.; Liu, J.; Luo, W.; Huang, S. Identification of an endophytic biocontrol strain NS03 and its efficacy in controlling pomegranate dry fruit rot disease. Acta Hortic 2015, 1089, 145–151. [Google Scholar]
- Ma, Y.; Tan, X.; Huang, S.; Zhang, X.; Zang, L.; Niu, X. Identification of a biocontrol strain Z2 against pomegranate dry rot and optimisation of its cultural conditions. Acta Phytopathol. Sin. 2015, 45, 425–437. [Google Scholar]
- Tekiner, N.; Kotan, R.; Tozlu, E.; Dadaşoğlu, F. Biological control of Coniella granati Saccardo in pomegranate. Univ. J. Agric. 2020, 8, 18–24. [Google Scholar] [CrossRef]
- Zhou, S.; Li, B. Genome Sequence Resource of Coniella vitis, a Fungal Pathogen Causing Grape White Rot Disease. Mol. Plant-Microbe Interact. 2020, 33, 787–789. [Google Scholar] [CrossRef]
- Liu, R.; Wang, Y.; Li, P.; Sun, L.; Jiang, J.; Fan, X.; Liu, C.; Zhang, Y. Genome Assembly and Transcriptome Analysis of the Fungus Coniella diplodiella during Infection on Grapevine (Vitis vinifera L.). Front. Microbiol. 2020, 11, 599150. [Google Scholar] [CrossRef]
- Raudabaugh, D.B.; Iturriaga, T.; Carver, A.; Mondo, S.; Pangilinan, J.; Lipzen, A.; He, G.; Amirebrahimi, M.; Grigoriev, I.V.; Miller, A.N. Coniella lustricola, a new species from submerged detritus. Mycol. Prog. 2018, 17, 191–203. [Google Scholar] [CrossRef]
- Valero-Jimenez, C.A.; Veloso, J.; Staats, M.; van Kan, J.A.L. Comparative genomics of plant pathogenic Botrytis species with distinct host specificity. BMC Genom. 2019, 20, 203. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, W.T.; Lu, R.S.; Chen, M.; Liu, J.; Sun, X.Q.; Zhang, Y.M. Genome Sequence Resource for Colletotrichum gloeosporioides, an Important Pathogenic Fungus Causing Anthracnose of Dioscorea alata. Plant Dis. 2023, 107, 893–895. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Darwish, O.; Alkharouf, N.W.; Musungu, B.; Matthews, B.F. Analysis of the genome sequence of Phomopsis longicolla: A fungal pathogen causing Phomopsis seed decay in soybean. BMC Genom. 2017, 18, 688. [Google Scholar] [CrossRef] [PubMed]
- Derbyshire, M.; Denton-Giles, M.; Hegedus, D.; Seifbarghy, S.; Rollins, J.; Van Kan, J.; Seidl, M.F.; Faino, L.; Mbengue, M.; Navaud, O.; et al. The complete genome sequence of the phytopathogenic fungus Sclerotinia sclerotiorum reveals insights into the genome architecture of broad host range pathogens. Genome Biol. Evol. 2017, 9, 593–618. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Hane, J.K.; Paxman, J.; Jones, D.A.; Oliver, R.P.; De Wit, P. “CATAStrophy”, a genome-informed trophic classification of filamentous plant pathogens—How many different types of filamentous plant pathogens are there? Front. Microbiol. 2020, 10, 3088. [Google Scholar] [CrossRef]
- Testa, A.C.; Oliver, R.P.; Hane, J.K. OcculterCut: A Comprehensive Survey of AT-Rich Regions in Fungal Genomes. Genome Biol. Evol. 2016, 8, 2044–2064. [Google Scholar] [CrossRef]
- Mincuzzi, A.; Sanzani, S.M.; Palou, L.; Ragni, M.; Ippolito, A. Postharvest rot of pomegranate fruit in southern Italy: Characterization of the main pathogens. J. Fungi 2022, 8, 475. [Google Scholar] [CrossRef]
- Belay, Z.A.; Caleb, O.J.; Vorster, A.; Van Heerden, C.; Opara, U.L. Transcriptomic changes associated with husk scald incidence on pomegranate fruit peel during cold storage. Food Res. Int. 2020, 135, 109285. [Google Scholar] [CrossRef]
- Palou, L.; Taberner, V.; Guardado, A.; Del Río, M.Á.; Montesinos-Herrero, C. Incidence and etiology of postharvest fungal diseases of pomegranate (Punica granatum cv. Mollar de Elche) in Spain. Phytopathol. Mediterr. 2013, 478–489. [Google Scholar]
- Wang, Y.; Wu, J.; Yan, J.; Guo, M.; Xu, L.; Hou, L.; Zou, Q. Comparative genome analysis of plant ascomycete fungal pathogens with different lifestyles reveals distinctive virulence strategies. BMC Genom. 2022, 23, 34. [Google Scholar] [CrossRef]
- Van de Wouw, A.P.; Cozijnsen, A.J.; Hane, J.K.; Brunner, P.C.; McDonald, B.A.; Oliver, R.P.; Howlett, B.J. Evolution of linked avirulence effectors in Leptosphaeria maculans is affected by genomic environment and exposure to resistance genes in host plants. PLoS Pathog. 2010, 6, e1001180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xie, S.; Zhao, Y.; Meng, X.; Song, L.; Feng, H.; Huang, L. Hce2 domain-containing effectors contribute to the full virulence of Valsa mali in a redundant manner. Mol. Plant Pathol. 2019, 20, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Ben M’Barek, S.; Cordewener, J.H.; Tabib Ghaffary, S.M.; van der Lee, T.A.; Liu, Z.; Mirzadi Gohari, A.; Mehrabi, R.; America, A.H.; Robert, O.; Friesen, T.L.; et al. FPLC and liquid-chromatography mass spectrometry identify candidate necrosis-inducing proteins from culture filtrates of the fungal wheat pathogen Zymoseptoria tritici. Fungal Genet. Biol. 2015, 79, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Ashwin, N.M.R.; Barnabas, L.; Ramesh Sundar, A.; Malathi, P.; Viswanathan, R.; Masi, A.; Agrawal, G.K.; Rakwal, R. CfPDIP1, a novel secreted protein of Colletotrichum falcatum, elicits defense responses in sugarcane and triggers hypersensitive response in tobacco. Appl. Microbiol. Biotechnol. 2018, 102, 6001–6021. [Google Scholar] [CrossRef]
- Chen, S.; Songkumarn, P.; Venu, R.; Gowda, M.; Bellizzi, M.; Hu, J.; Liu, W.; Ebbole, D.; Meyers, B.; Mitchell, T.; et al. Identification and characterization of in planta–expressed secreted effector proteins from Magnaporthe oryzae that induce cell death in rice. Mol. Plant Microbe Interact. 2013, 26, 191–202. [Google Scholar] [CrossRef]
- Crowhurst, R.N.; Binnie, S.J.; Bowen, J.K.; Hawthorne, B.T.; Plummer, K.M.; Rees-George, J.; Rikkerink, E.H.; Templeton, M.D. Effect of disruption of a cutinase gene (cutA) on virulence and tissue specificity of Fusarium solani f. sp. cucurbitae race 2 toward Cucurbita maxima and C. moschata. Mol. Plant Microbe Interact. 1997, 10, 355–368. [Google Scholar] [CrossRef]
- Snelders, N.C.; Rovenich, H.; Petti, G.C.; Rocafort, M.; van den Berg, G.C.M.; Vorholt, J.A.; Mesters, J.R.; Seidl, M.F.; Nijland, R.; Thomma, B. Microbiome manipulation by a soil-borne fungal plant pathogen using effector proteins. Nat. Plants 2020, 6, 1365–1374. [Google Scholar] [CrossRef]
- Hong, Y.; Yang, Y.; Zhang, H.; Huang, L.; Li, D.; Song, F. Overexpression of MoSM1, encoding for an immunity-inducing protein from Magnaporthe oryzae, in rice confers broad-spectrum resistance against fungal and bacterial diseases. Sci. Rep. 2017, 7, 41037. [Google Scholar] [CrossRef]
- Yang, G.; Tang, L.; Gong, Y.; Xie, J.; Fu, Y.; Jiang, D.; Li, G.; Collinge, D.B.; Chen, W.; Cheng, J. A cerato-platanin protein SsCP1 targets plant PR1 and contributes to virulence of Sclerotinia sclerotiorum. New Phytol. 2018, 217, 739–755. [Google Scholar] [CrossRef] [PubMed]
- Thon, M.; Al-Abdallah, Q.; Hortschansky, P.; Brakhage, A.A. The thioredoxin system of the filamentous fungus Aspergillus nidulans: Impact on development and oxidative stress response. J. Biol. Chem. 2007, 282, 27259–27269. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.J.; Gardiner, R.B.; Day, A.W. Melanin synthesis by Sclerotinia sclerotiorum. Mycologia 2009, 101, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Starratt, A.N.; Ross, L.M.; Lazarovits, G. 1,8-Dihydroxynaphthalene monoglucoside, a new metabolite of Sclerotinia sclerotiorum, and the effect of tricyclazole on its production. Can. J. Microbiol. 2002, 48, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Ishii, Y.; Honda, A.; Masunaka, A.; Tsuge, T.; Yamamoto, M.; Ohtani, K.; Fukumoto, T.; Gomi, K.; Peever, T.; et al. Function of genes encoding acyl-CoA synthetase and enoyl-CoA hydratase for host-selective ACT-toxin biosynthesis in the tangerine pathotype of Alternaria alternata. Phytopathology 2009, 99, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, S.; Niu, Q.; Xu, G.; Lu, C.; Zhang, J. Genomic Sequence Resource of Talaromyces albobiverticillius, the Causative Pathogen of Pomegranate Pulp Rot Disease. J. Fungi 2023, 9, 909. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Palmer, J.; Stajich, J. Funannotate (Version 1.8.15) [Computer software]. 2023. Available online: https://github.com/nextgenusfs/funannotate (accessed on 12 April 2023).
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Jones, D.A.; Rozano, L.; Debler, J.W.; Mancera, R.L.; Moolhuijzen, P.M.; Hane, J.K. An automated and combinative method for the predictive ranking of candidate effector proteins of fungal plant pathogens. Sci. Rep. 2021, 11, 19731. [Google Scholar]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, C.L.; Booth, T.J.; van Wersch, B.; van Grieken, L.; Medema, M.H.; Chooi, Y.-H. Cblaster: A remote search tool for rapid identification and visualization of homologous gene clusters. Bioinform. Adv. 2021, 1, vbab016. [Google Scholar] [CrossRef]
- Gilchrist, C.L.; Chooi, Y.-H. Clinker & clustermap. js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [PubMed]
- van den Belt, M.; Gilchrist, C.; Booth, T.J.; Chooi, Y.-H.; Medema, M.H.; Alanjary, M. CAGECAT: The CompArative GEne Cluster Analysis Toolbox for rapid search and visualisation of homologous gene clusters. BMC Bioinform. 2023, 24, 181. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Marcais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Findeiss, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Goodwin, S.B.; M’Barek, S.B.; Dhillon, B.; Wittenberg, A.H.; Crane, C.F.; Hane, J.K.; Foster, A.J.; Van der Lee, T.A.; Grimwood, J.; Aerts, A.; et al. Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. 2011, 7, e1002070. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| (A) Genome assembly | |
| Total | 46.8 Mb/46,832,344 bp |
| Sequence number | 2009 |
| N50 length | 23,311 bp |
| N50 number | 399 |
| Max length | 220,902 bp |
| Mean length | 35,815 bp |
| (B) Repetitive DNA | |
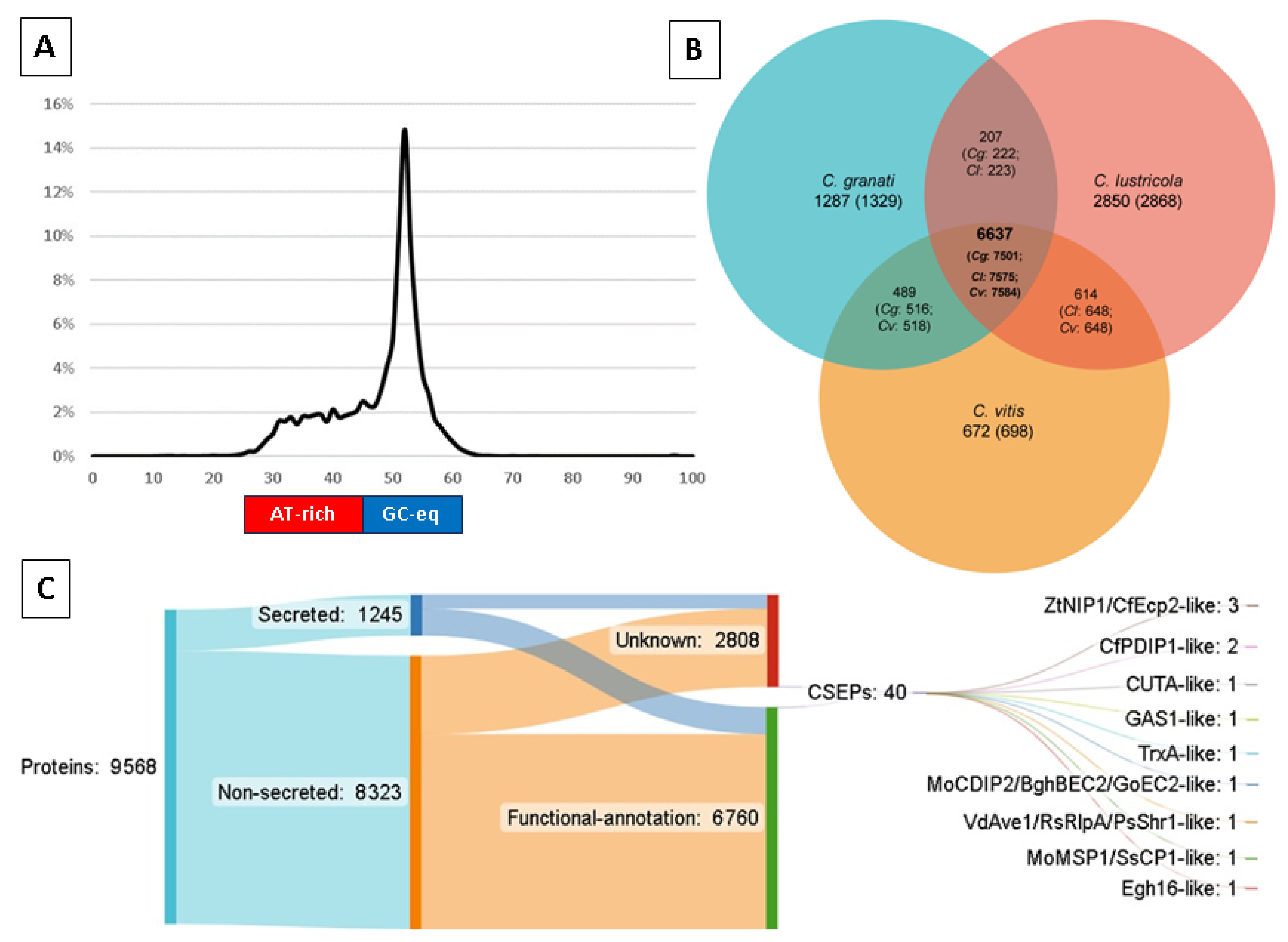
| Proportion of genome in AT-rich regions | 26.9% |
| Repetitive DNA | 11.69 Mb (24.95%) |
| Retroelements | 15% |
| LINEs | 0.58% |
| LTR elements | 14.42% |
| Copia-like | 3.34% |
| Gypsy-like | 11.08% |
| DNA transposons | 1.07% |
| Unclassified | 6.94% |
| Low complexity/small RNA/Simple repeats | 1.93% |
| (C) Predicted Pathogenicity Features | |
| CATAStrophy-predicted infection type | Hemibiotrophic: extracellular mesotroph (0.94); monomertroph (1); saprotroph (1); |
| Protein-coding genes | 9568 |
| Functionally-annotated proteins | 9086 (95%) |
| Secreted proteins | 1245 (13%) |
| Candidate pathogenicity effectors (Predector ≥ 1.5) | 40 |
| Feature | C. granati | C. lustricola | C. vitis | C. diplodiella * |
|---|---|---|---|---|
| Host range | Pomegranate | Saprophyte | Grapevine | Grapevine |
| Isolate | Ph1 | B22-T-1 | QNYT13637 | WR01 |
| Assembly size (Mb) | 46.83 | 36.56 | 41.54 | 40.93 * |
| Sequence number | 2009 | 634 | 22 | 13 * |
| N50 number | 399 | 76 | 5 | * |
| N50 length (Mb) | 0.02 | 0.14 | 3.20 | 3.99 |
| Repetitive (%) | 26.9 | * | 5.76 | 12.74 |
| Gene annotations | 9568 | 11,317 | 7985 (published) * 9448 (this study) | 9403 * |
| BUSCO % completeness | 84.8% | 96.0% | 99.3% | 97.6% |
| (A) % sequences aligned (x vs. y) | C. granati | C. lustricola | C. vitis |
| C. granati | - | 64.3% | 78.3% |
| C. lustricola | 84.54 | - | 85.8% |
| C. vitis | 100% | 95.45% | - |
| (B) % bases aligned (x vs. y) | C. granati | C. lustricola | C. vitis |
| C. granati | - | 12% | 33.48% |
| C. lustricola | 15.4% | - | 15.4% |
| C. vitis | 40% | 14.5% | - |
| (C) SNPs (number and %total) | Cg-vs.-Cl | Cg-vs.-Cv | Cl-vs.-Cv |
| A-C | 35,949 (4.9%) | 81,114 (4.9%) | 33,220 (4.5%) |
| A-G * | 122,165 (16.5%) | 279,243 (16.8%) | 111,223 (15%) |
| A-T | 23,446 (3.2%) | 51,250 (3.1%) | 23,029 (3.11%) |
| C-A * | 32,134 (4.3%) | 72,791 (4.4%) | 34,545 (4.7%) |
| C-G | 47,737 (6.4%) | 92,555 (5.6%) | 50,542 (6.8%) |
| C-T * | 109,902 (14.9%) | 254,516 (15.3%) | 117,764 (15.9%) |
| G-A * | 109,974 (14.9%) | 254,462 (15.3%) | 115,929 (15.7%) |
| G-C | 46,769 (6.3%) | 91,853 (5.5%) | 50,407 (6.8%) |
| G-T | 31,558 (4.3%) | 73,048 (4.4%) | 34,621 (4.7%) |
| T-A | 23,339 (3.1%) | 51,022 (3.1%) | 22,848 (3.1%) |
| T-C * | 121,043 (16.4%) | 277,912 (16.7%) | 111,806 (15.1%) |
| T-G | 35,746 (4.8%) | 80,624 (4.9%) | 33,452 (4.5%) |
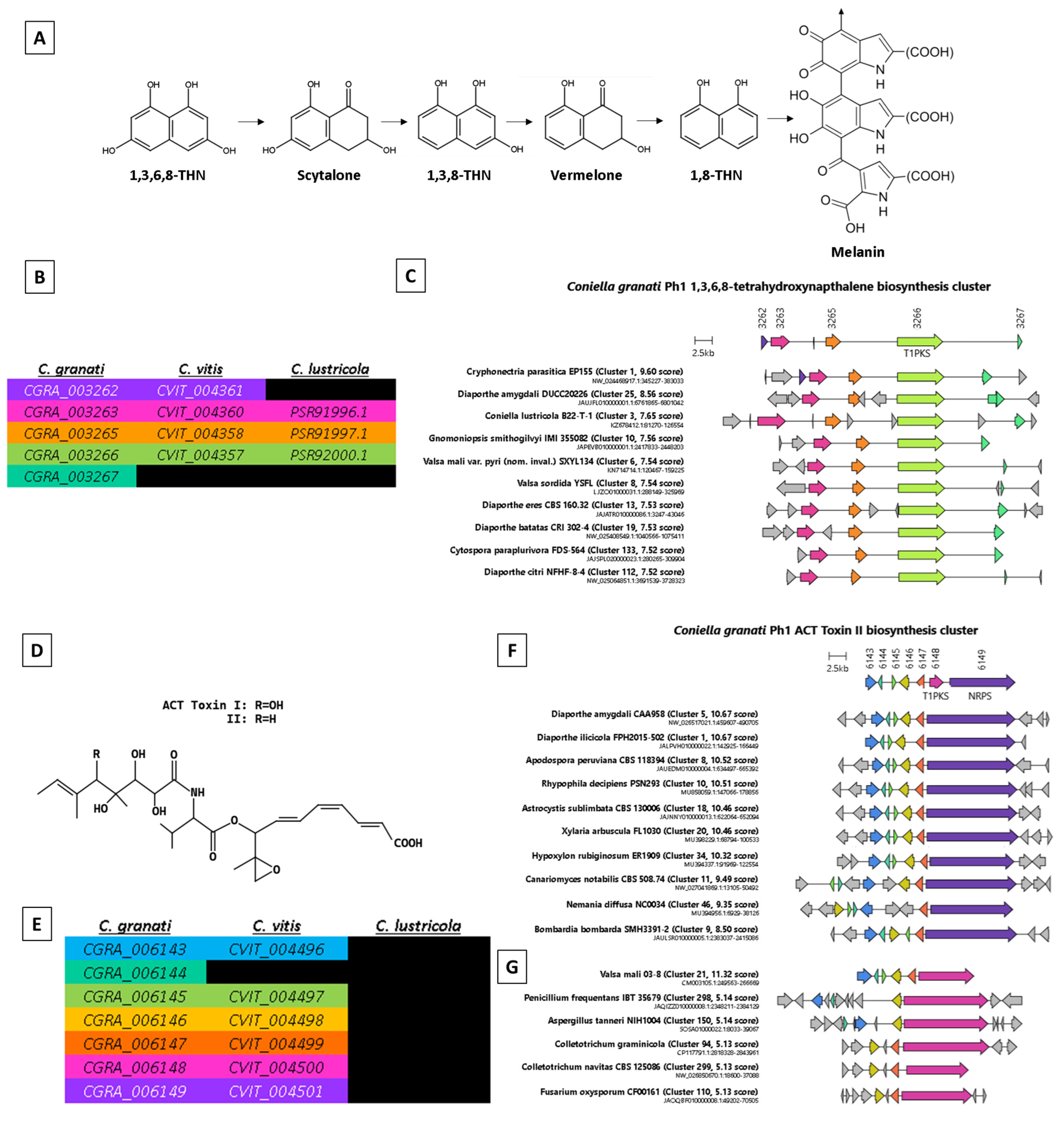
| Locus | PAV * | Score | Effector Homology and Functional Annotations | #Cys | Len (aa) | Distance (bp) | Region Type |
|---|---|---|---|---|---|---|---|
| PGRA_006204 | Core | 3.086 | Homology:CfEcp2,ZtNIP1; Pfam:PF14856(Hce2); | 4 | 227 | 9970 | End |
| PGRA_007290 | Cg-Cl | 2.646 | [No match] | 7 | 141 | 3056 | AT |
| PGRA_008218 | Core | 2.505 | Homology: ZtNIP1,CfEcp2; Pfam:PF14856(Hce2); Localiser:chloroplast; | 5 | 197 | 782 | End |
| PGRA_002694 | Cg-Cv | 2.474 | [No match] | 0 | 228 | 23,880 | End |
| PGRA_001911 | Core | 2.443 | [No match] | 4 | 170 | 18,286 | End |
| PGRA_008765 | Core | 2.427 | [No match] | 12 | 137 | 3446 | End |
| PGRA_009449 | Core | 2.398 | PHIbase:CUTA(KO-unaffected pathogenicity); Pfam:PF01083(Cutinase) | 6 | 195 | 22 | End |
| PGRA_006664 | Core | 2.339 | PHIbase: GAS1(KO-reduced virulence); Pfam:PF11327(Egh16-like); Localiser:nucleus; | 4 | 257 | 1446 | AT |
| PGRA_002414 | Core | 2.299 | [No match] | 4 | 224 | 13,631 | AT |
| PGRA_004539 | Cg | 2.162 | Pfam:PF00488(MutS_V); | 2 | 249 | 2890 | AT |
| PGRA_008027 | Cg-Cv | 2.138 | [No match] | 12 | 184 | 26 | AT |
| PGRA_003492 | Cg-Cl | 2.134 | Localiser:nucleus; | 8 | 181 | 5414 | End |
| PGRA_008885 | Core | 2.095 | Pfam:PF14273(DUF4360); | 4 | 217 | 4169 | End |
| PGRA_008860 | Cg-Cv | 2.092 | [No match] | 6 | 120 | 2506 | AT |
| PGRA_009224 | Core | 2.084 | Homology: CfPDIP1; | 6 | 113 | 2644 | End |
| PGRA_007015 | Core | 2.068 | Homology: CfPDIP1; | 8 | 130 | 7150 | End |
| PGRA_008976 | Cg | 2.045 | PHIbase:TrxA,Thioredoxin_1(KO-reduced virulence); Pfam:PF00085(Thioredoxin); | 5 | 137 | 74 | End |
| PGRA_001981 | Core | 2.044 | Pfam:PF11327(Egh16-like); | 4 | 239 | 7609 | End |
| PGRA_004164 | Cg-Cl | 2.016 | Homology: GoEC2,MoCDIP2,BghBEC2; Pfam:PF05730(CFEM); Localiser:nucleus; | 7 | 154 | 255 | End |
| PGRA_005485 | Core | 1.994 | Homology:ZtNIP1,CfEcp2; Pfam:PF14856(Hce2); | 4 | 167 | 95 | End |
| PGRA_001358 | Core | 1.982 | Pfam:PF06766(Hydrophobin_2); | 8 | 97 | 23,442 | AT |
| PGRA_006064 | Core | 1.953 | Localiser:nucleus; | 8 | 143 | 6677 | End |
| PGRA_007124 | Cg-Cv | 1.936 | Pfam:PF01822(WSC); | 7 | 125 | 7176 | AT |
| PGRA_009571 | Cg | 1.911 | [No match] | 6 | 75 | 35 | AT |
| PGRA_005443 | Core | 1.908 | [No match] | 9 | 194 | 5754 | End |
| PGRA_001444 | Core | 1.898 | [No match] | 8 | 127 | 17,504 | End |
| PGRA_005889 | Core | 1.892 | [No match] | 8 | 196 | 14,164 | End |
| PGRA_008276 | Core | 1.869 | [No match] | 0 | 101 | 708 | AT |
| PGRA_009699 | Cg | 1.864 | Pfam:PF00135(COesterase); | 2 | 171 | 245 | End |
| PGRA_002373 | Core | 1.79 | Pfam:PF00085(Thioredoxin); | 2 | 175 | 17,357 | End |
| PGRA_001309 | Core | 1.762 | Pfam:PF01161(PBP); | 10 | 156 | 22,013 | AT |
| PGRA_008695 | Cg | 1.695 | [No match] | 1 | 204 | 2994 | End |
| PGRA_008501 | Core | 1.647 | [No match] | 9 | 212 | 2852 | End |
| PGRA_000088 | Core | 1.644 | Pfam:PF01083(Cutinase); | 5 | 253 | 6346 | AT |
| PGRA_000913 | Core | 1.628 | Pfam:PF01105(EMP24_GP25L); | 3 | 222 | 0 | AT |
| PGRA_008930 | Core | 1.623 | Pfam:PF10270(MMgT); | 1 | 139 | 1778 | End |
| PGRA_002139 | Core | 1.59 | [No match] | 7 | 207 | 440 | AT |
| PGRA_004189 | Core | 1.577 | Pfam:PF11327(Egh16-like); | 4 | 244 | 14,924 | End |
| PGRA_009219 | Core | 1.534 | Homology: RsRlpA,VdAve1,PsShr1; | 5 | 132 | 68 | AT |
| PGRA_006644 | Core | 1.521 | Homology: SsCP1,MoMSP1 | 10 | 221 | 189 | AT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zambounis, A.; Maniatis, E.I.; Mincuzzi, A.; Gray, N.; Hossain, M.; Tsitsigiannis, D.I.; Paplomatas, E.; Ippolito, A.; Schena, L.; Hane, J.K. Highly Repetitive Genome of Coniella granati (syn. Pilidiella granati), the Causal Agent of Pomegranate Fruit Rot, Encodes a Minimalistic Proteome with a Streamlined Arsenal of Effector Proteins. Int. J. Mol. Sci. 2024, 25, 9997. https://doi.org/10.3390/ijms25189997
Zambounis A, Maniatis EI, Mincuzzi A, Gray N, Hossain M, Tsitsigiannis DI, Paplomatas E, Ippolito A, Schena L, Hane JK. Highly Repetitive Genome of Coniella granati (syn. Pilidiella granati), the Causal Agent of Pomegranate Fruit Rot, Encodes a Minimalistic Proteome with a Streamlined Arsenal of Effector Proteins. International Journal of Molecular Sciences. 2024; 25(18):9997. https://doi.org/10.3390/ijms25189997
Chicago/Turabian StyleZambounis, Antonios, Elisseos I. Maniatis, Annamaria Mincuzzi, Naomi Gray, Mohitul Hossain, Dimitrios I. Tsitsigiannis, Epaminondas Paplomatas, Antonio Ippolito, Leonardo Schena, and James K. Hane. 2024. "Highly Repetitive Genome of Coniella granati (syn. Pilidiella granati), the Causal Agent of Pomegranate Fruit Rot, Encodes a Minimalistic Proteome with a Streamlined Arsenal of Effector Proteins" International Journal of Molecular Sciences 25, no. 18: 9997. https://doi.org/10.3390/ijms25189997