
Potential of NRF2 Inhibitors—Retinoic Acid, K67, and ML-385—In Overcoming Doxorubicin Resistance in Promyelocytic Leukemia Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Comparison of HL-60 and HL-60/DR Cells
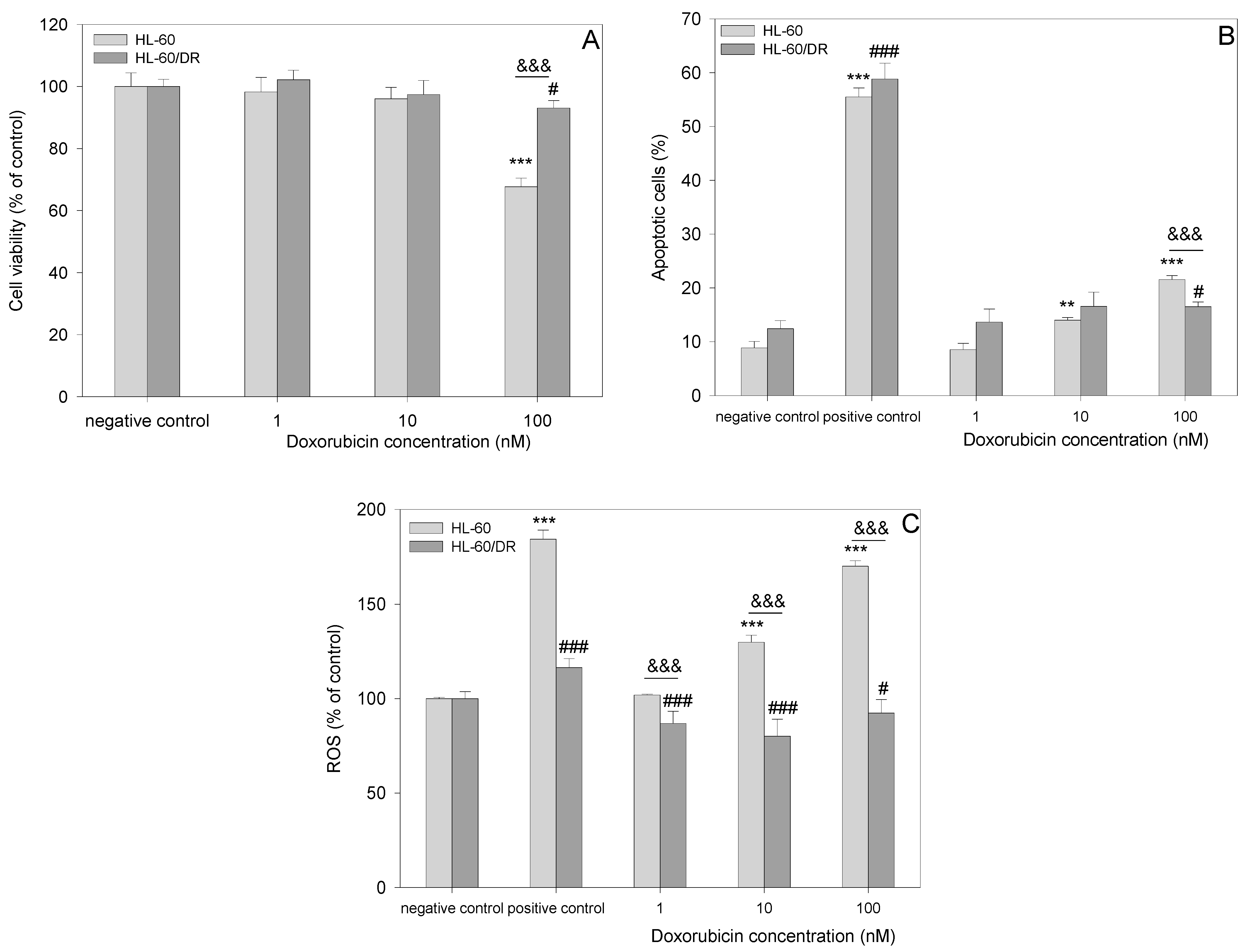
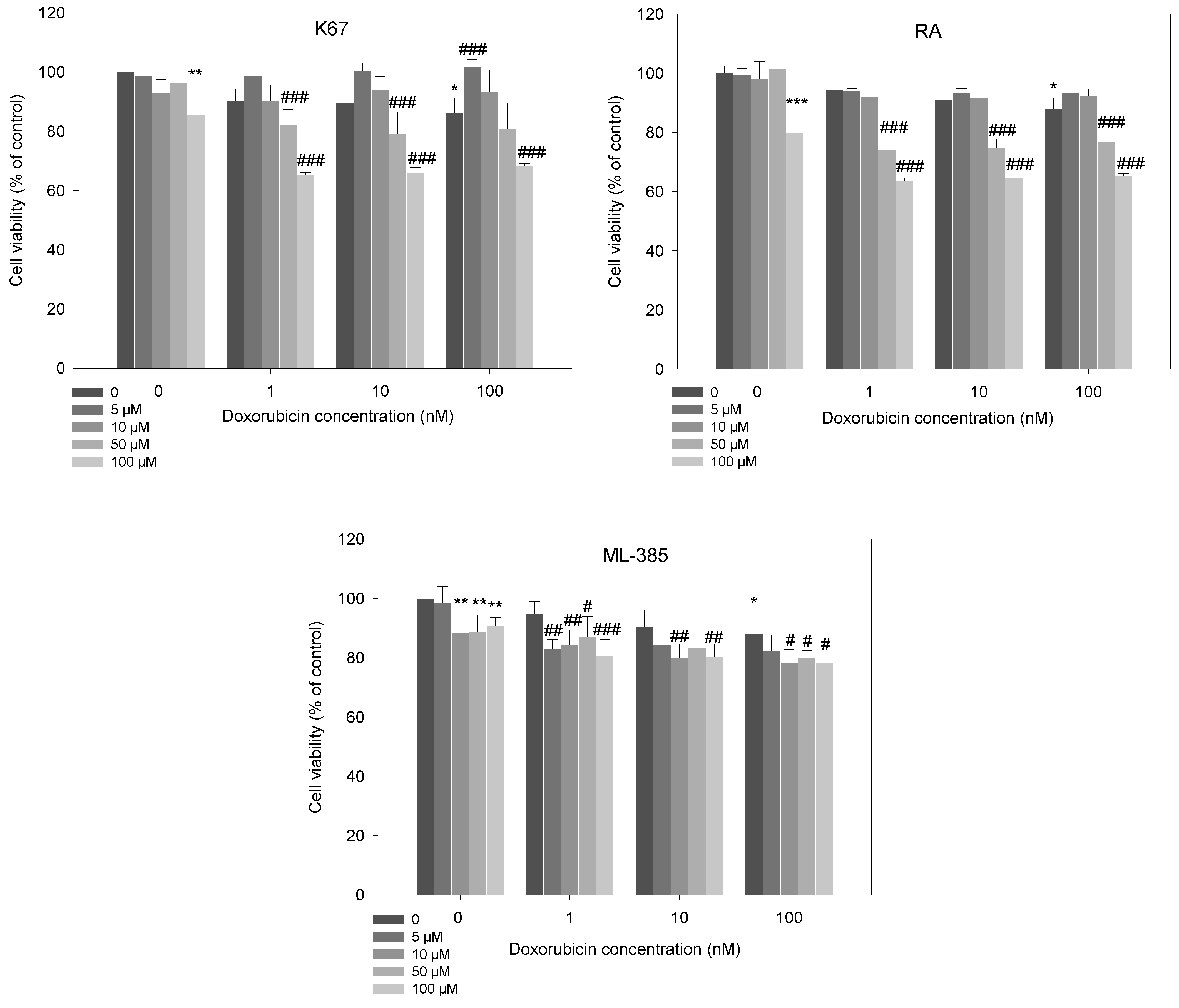
2.2. Cell Viability Studies
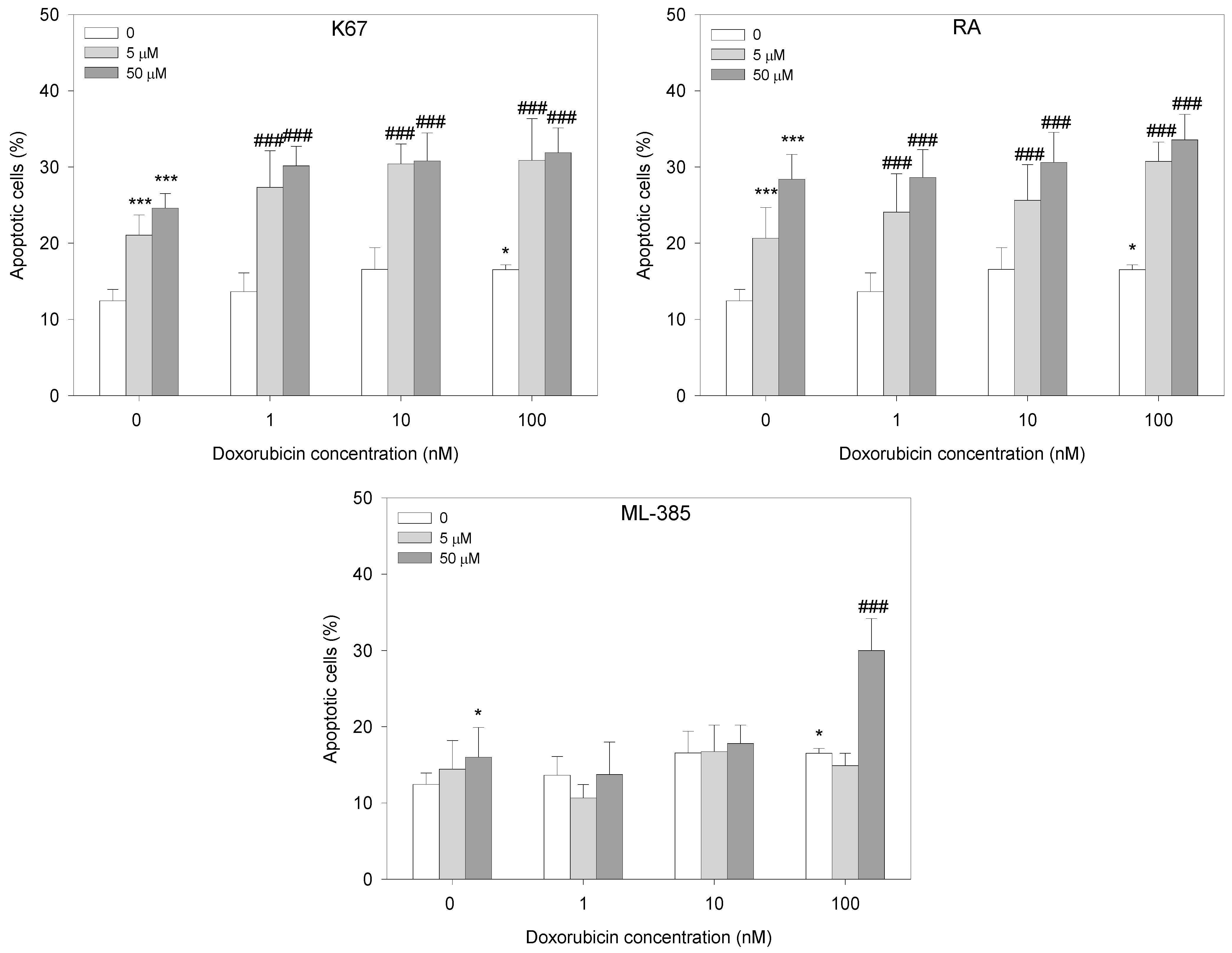
2.3. Apoptosis
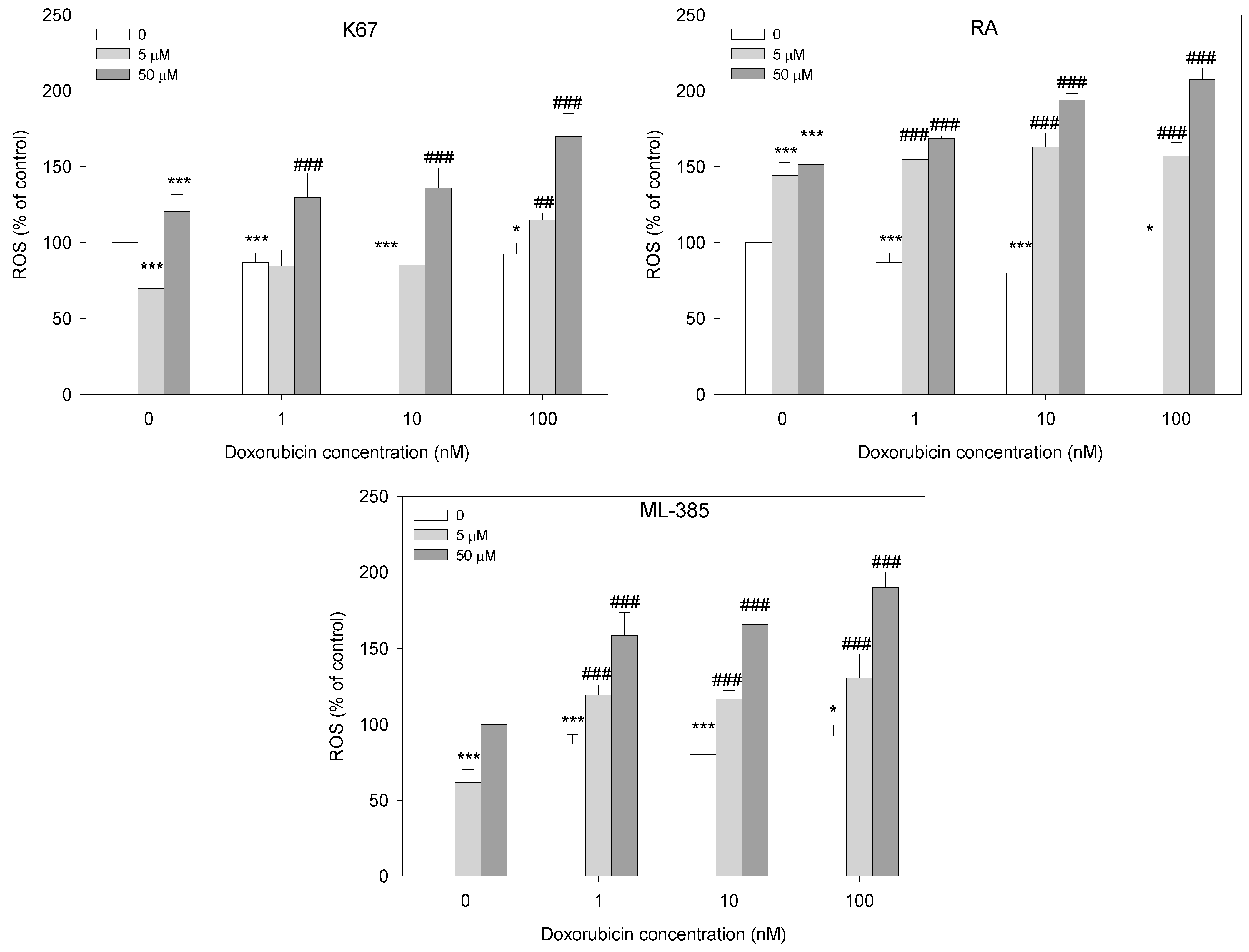
2.4. Reactive Oxygen Species Generation
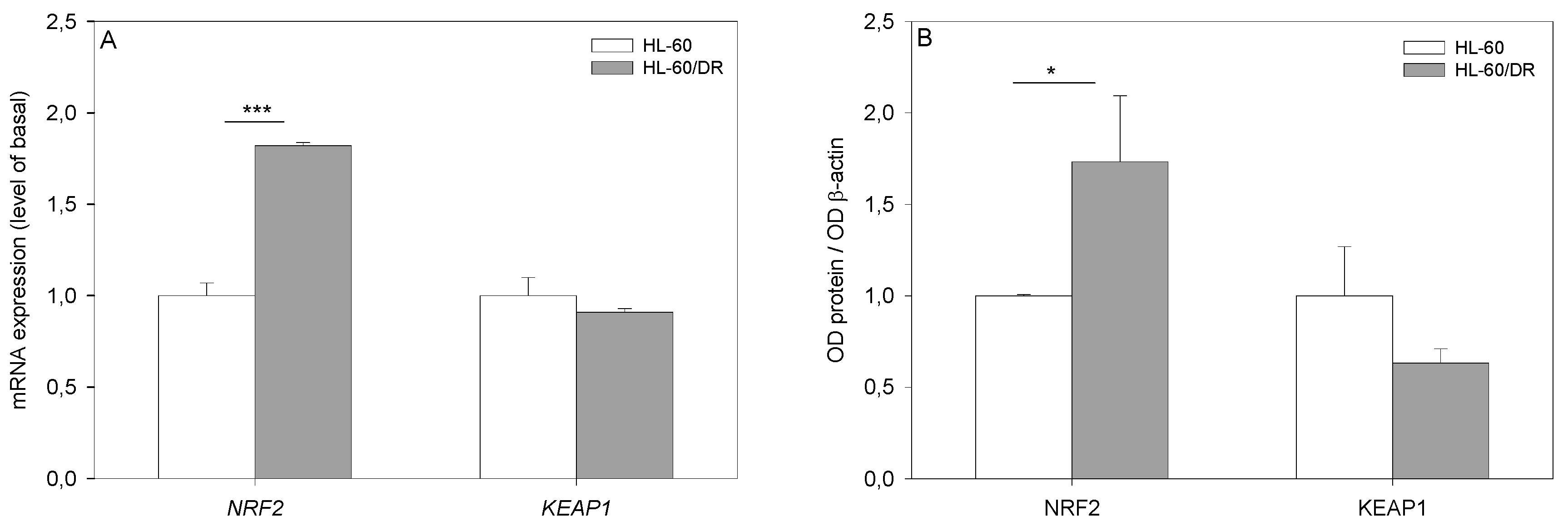
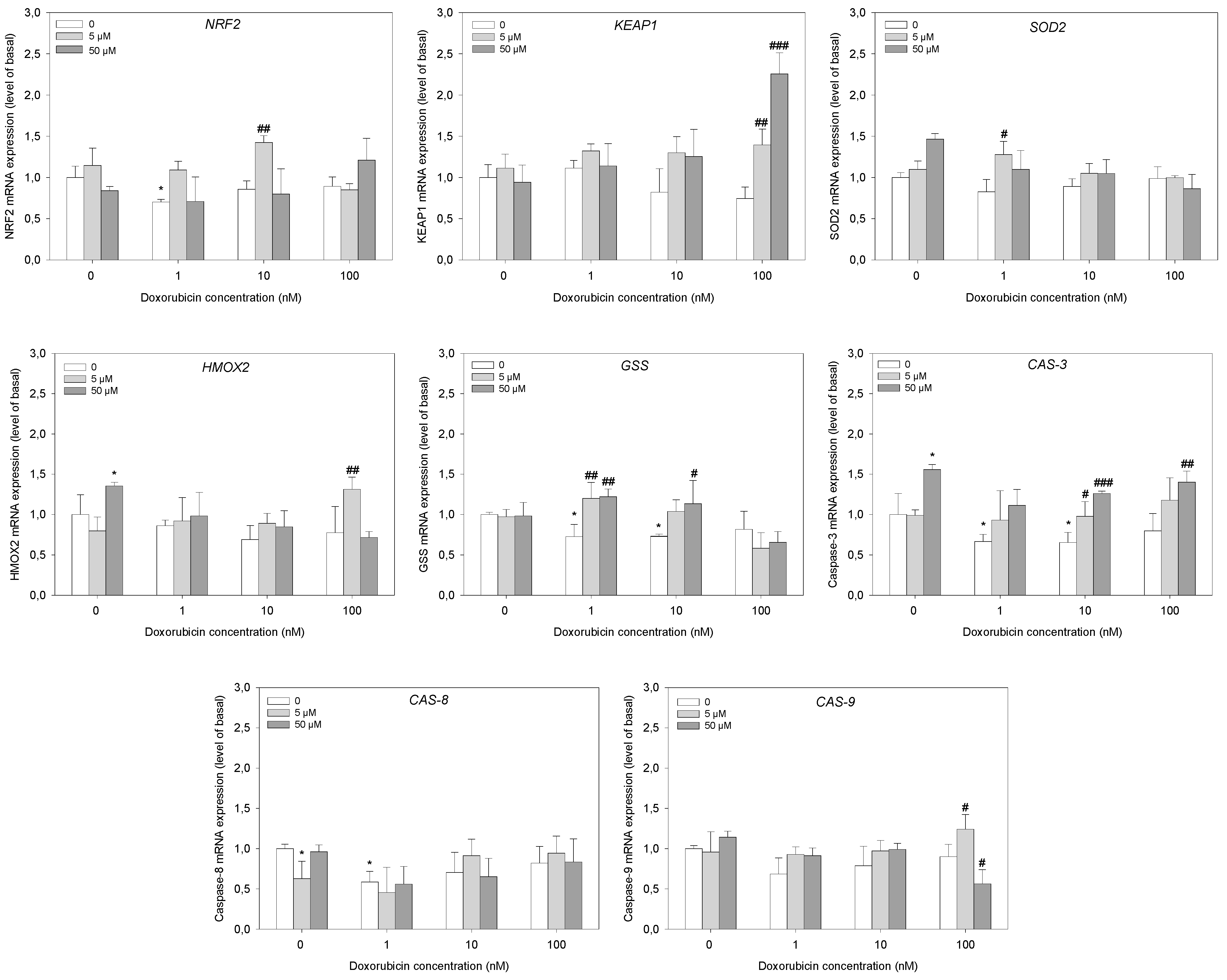
2.5. Gene Expression Analysis
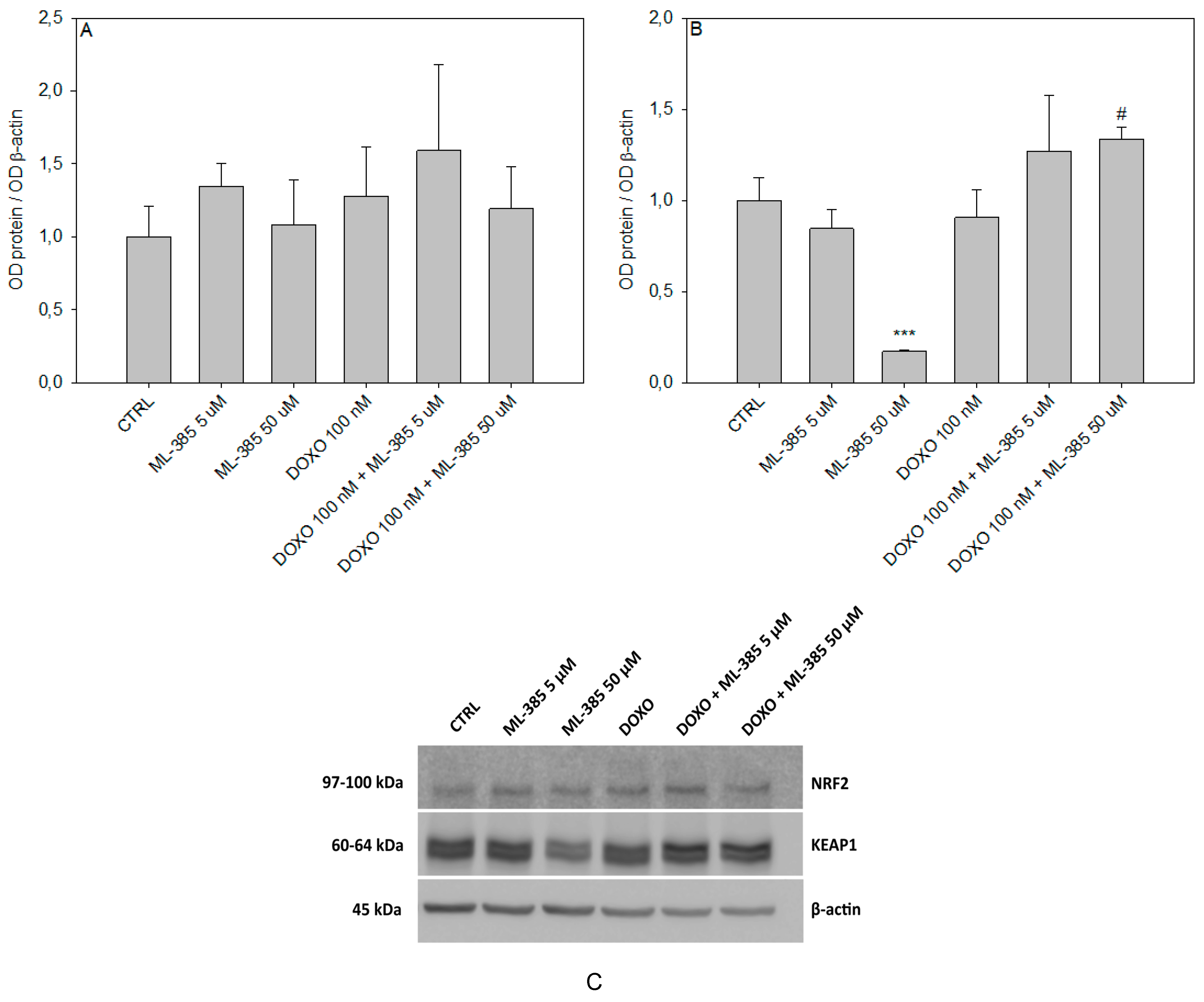
2.6. Western Blot Analysis
3. Discussion
4. Materials and Methods
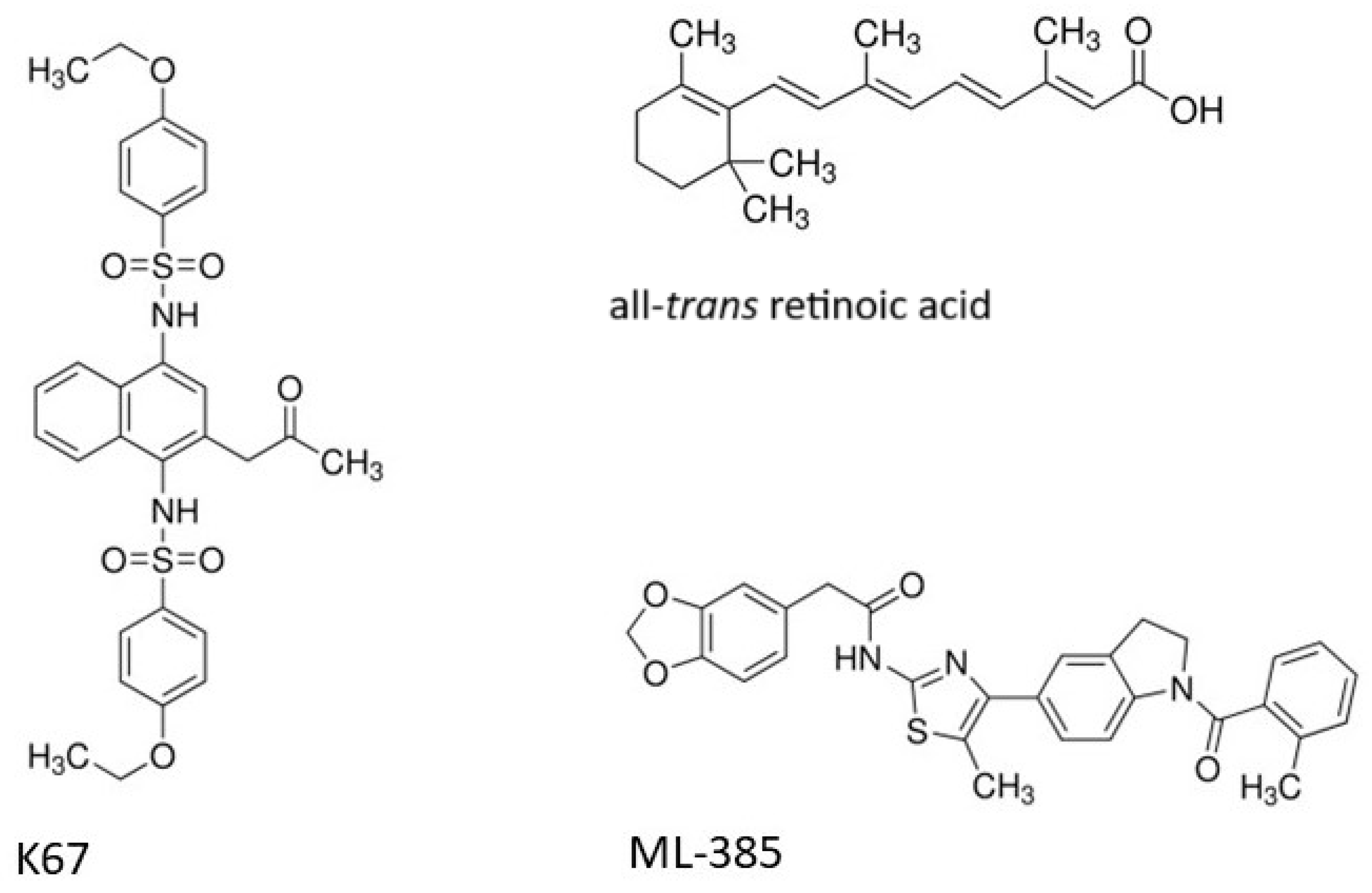
4.1. Chemicals
4.2. Cell Culture
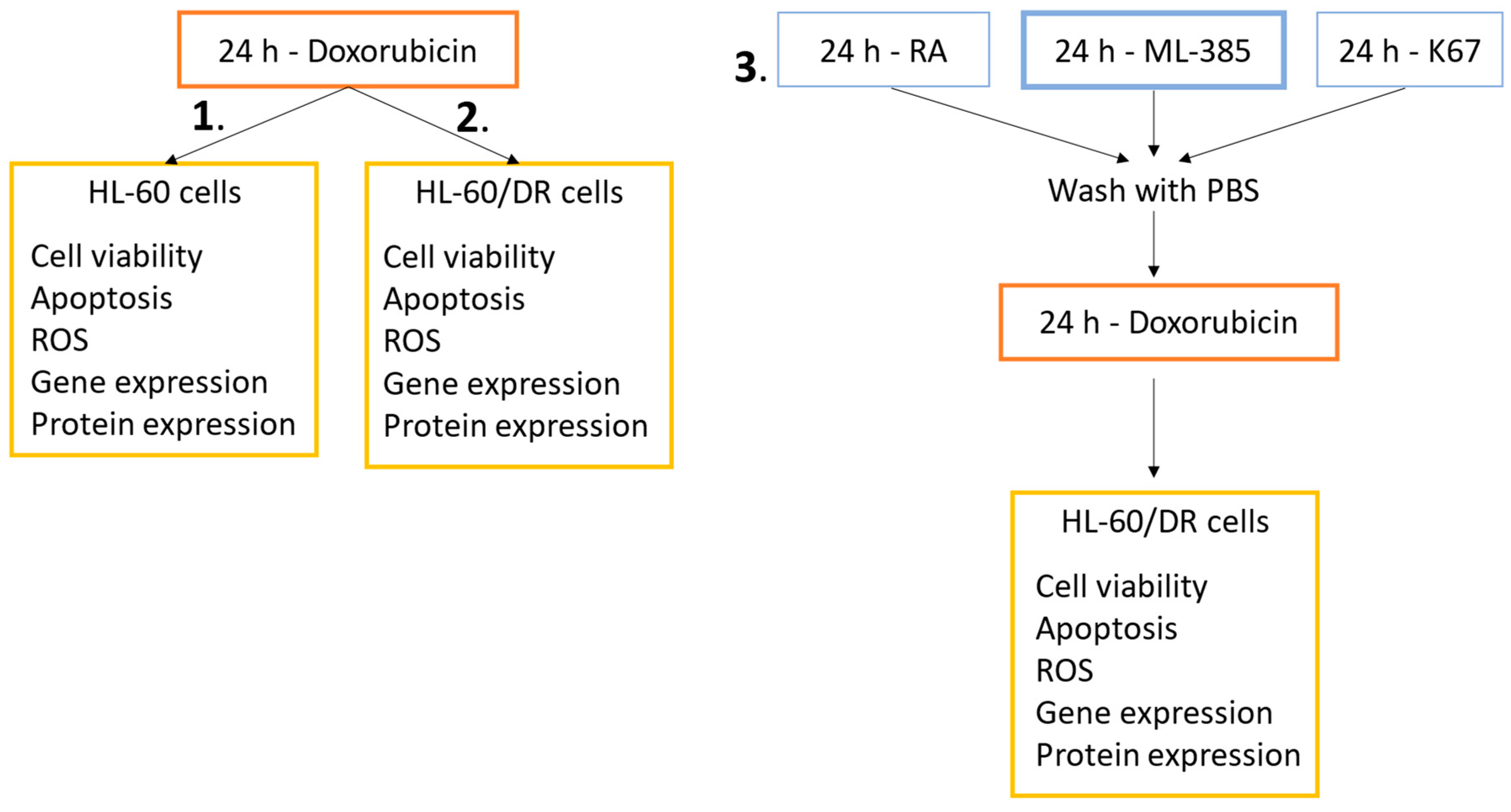
4.3. Experimental Schemes
4.4. Cell Viability Resazurin Assay
4.5. Evaluation of Oxidative Stress
4.6. Apoptosis
4.7. Gene Expression Analysis
4.8. Western Blot
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 Regulatory Network Provides an Interface between Redox and Intermediary Metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Ryoo, I.; Kwak, M.-K. Regulatory Crosstalk between the Oxidative Stress-Related Transcription Factor Nfe2l2/Nrf2 and Mitochondria. Toxicol. Appl. Pharmacol. 2018, 359, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. The Nrf2/HO-1 Axis in Cancer Cell Growth and Chemoresistance. Oxidative Med. Cell. Longev. 2016, 2016, 1958174. [Google Scholar] [CrossRef]
- Osburn, W.; Kensler, T. Nrf2 Signaling: An Adaptive Response Pathway for Protection against Environmental Toxic Insults. Mutat. Res./Rev. Mutat. Res. 2008, 659, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in Cancers: A Double-edged Sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Shim, G.; Manandhar, S.; Shin, D.; Kim, T.-H.; Kwak, M.-K. Acquisition of Doxorubicin Resistance in Ovarian Carcinoma Cells Accompanies Activation of the NRF2 Pathway. Free Radic. Biol. Med. 2009, 47, 1619–1631. [Google Scholar] [CrossRef]
- Hu, T.; Pan, C.; Zhang, T.; Ni, M.; Wang, W.; Zhang, S.; Chen, Y.; Wang, J.; Fang, Q. Nrf2 Overexpression Increases the Resistance of Acute Myeloid Leukemia to Cytarabine by Inhibiting Replication Factor C4. Cancer Gene Ther. 2022, 29, 1773–1790. [Google Scholar] [CrossRef]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1–NRF2 Interaction in Non-Small-Cell Lung Cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef]
- Wang, X.-J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 Enhances Resistance of Cancer Cells to Chemotherapeutic Drugs, the Dark Side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef]
- Lau, A.; Villeneuve, N.; Sun, Z.; Wong, P.; Zhang, D. Dual Roles of Nrf2 in Cancer. Pharmacol. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lu, L.; Yan, S.; Yi, H.; Yao, H.; Wu, D.; He, G.; Tao, X.; Deng, X. Autophagy and Doxorubicin Resistance in Cancer. Anti-Cancer Drugs 2018, 29, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Al-malky, H.S.; Al Harthi, S.E.; Osman, A.-M.M. Major Obstacles to Doxorubicin Therapy: Cardiotoxicity and Drug Resistance. J. Oncol. Pharm. Pract. 2020, 26, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Ling, G.; Wang, X.; Tan, N.; Cao, J.; Li, W.; Zhang, Y.; Jiang, J.; Sun, Q.; Jiang, Y.; Wang, W.; et al. Mechanisms and Drug Intervention for Doxorubicin-Induced Cardiotoxicity Based on Mitochondrial Bioenergetics. Oxidative Med. Cell. Longev. 2022, 2022, 7176282. [Google Scholar] [CrossRef]
- Rivankar, S. An Overview of Doxorubicin Formulations in Cancer Therapy. J. Can. Res. Ther. 2014, 10, 853. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-Induced Cardiotoxicity: An Update on the Molecular Mechanism and Novel Therapeutic Strategies for Effective Management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, S.; Zarrabi, A.; Hashemi, F.; Zabolian, A.; Saleki, H.; Azami, N.; Hamzehlou, S.; Farahani, M.V.; Hushmandi, K.; Ashrafizadeh, M.; et al. Nrf2 Signaling Pathway in Chemoprotection and Doxorubicin Resistance: Potential Application in Drug Discovery. Antioxidants 2021, 10, 349. [Google Scholar] [CrossRef]
- Valenzuela, M.; Glorieux, C.; Stockis, J.; Sid, B.; Sandoval, J.M.; Felipe, K.B.; Kviecinski, M.R.; Verrax, J.; Calderon, P.B. Retinoic Acid Synergizes ATO-Mediated Cytotoxicity by Precluding Nrf2 Activity in AML Cells. Br. J. Cancer 2014, 111, 874–882. [Google Scholar] [CrossRef]
- Butsri, S.; Kukongviriyapan, V.; Senggunprai, L.; Kongpetch, S.; Prawan, A. All-trans-retinoic Acid Induces RARB-dependent Apoptosis via ROS Induction and Enhances Cisplatin Sensitivity by NRF2 Downregulation in Cholangiocarcinoma Cells. Oncol. Lett. 2022, 23, 179. [Google Scholar] [CrossRef]
- Kim, D.; Choi, B.; Ryoo, I.; Kwak, M.-K. High NRF2 Level Mediates Cancer Stem Cell-like Properties of Aldehyde Dehydrogenase (ALDH)-High Ovarian Cancer Cells: Inhibitory Role of All-Trans Retinoic Acid in ALDH/NRF2 Signaling. Cell Death Dis. 2018, 9, 896. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Q.; Ye, L.; Ye, X.; Yang, W.; Zhang, H.; Zhou, X.; Ren, Y.; Ma, L.; Zhang, X.; et al. All-Trans Retinoic Acid Enhances the Cytotoxic Effect of Decitabine on Myelodysplastic Syndromes and Acute Myeloid Leukaemia by Activating the RARα-Nrf2 Complex. Br. J. Cancer 2023, 128, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. P62/Sqstm1 Promotes Malignancy of HCV-Positive Hepatocellular Carcinoma through Nrf2-Dependent Metabolic Reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, D.; Ohe, T.; Takahashi, K.; Imamura, R.; Kojima, H.; Okabe, T.; Ichimura, Y.; Komatsu, M.; Yamamoto, M.; Nagano, T.; et al. Inhibitors of the Protein–Protein Interaction between Phosphorylated P62 and Keap1 Attenuate Chemoresistance in a Human Hepatocellular Carcinoma Cell Line. Free Radic. Res. 2020, 54, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamamoto, M. The KEAP1–NRF2 System as a Molecular Target of Cancer Treatment. Cancers 2020, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.-Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef]
- Bartman, A.E.; Raeisi, M.; Peiris, C.D.; Jacobsen, I.E.; Martin, D.B.C.; Doorn, J.A. A Novel Analog of the Natural Product Fraxinellone Protects against Endogenous and Exogenous Neurotoxicants. ACS Chem. Neurosci. 2024, 15, 2612–2622. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 Pathway: Mechanisms of Activation and Dysregulation in Cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef]
- Ji, L.; Moghal, N.; Zou, X.; Fang, Y.; Hu, S.; Wang, Y.; Tsao, M.S. The NRF2 Antagonist ML385 Inhibits PI3K-mTOR Signaling and Growth of Lung Squamous Cell Carcinoma Cells. Cancer Med. 2023, 12, 5688–5702. [Google Scholar] [CrossRef]
- Wu, X.; Chen, S.; Huang, K.; Lin, G. Triptolide Promotes Ferroptosis by Suppressing Nrf2 to Overcome Leukemia Cell Resistance to Doxorubicin. Mol. Med. Rep. 2022, 27, 17. [Google Scholar] [CrossRef]
- Ganapathi, R.; Hoeltge, G.; Casey, G.; Grabowski, D.; Neelon, R.; Ford, J. Acquisition of Doxorubicin Resistance in Human Leukemia HL-60 Cells Is Reproducibly Associated with 7q21 Chromosomal Anomalies. Cancer Genet. Cytogenet. 1996, 86, 116–119. [Google Scholar] [CrossRef]
- Asano, T.; Tsutsuda-Asano, A.; Fukunaga, Y. Indomethacin Overcomes Doxorubicin Resistance by Decreasing Intracellular Content of Glutathione and Its Conjugates with Decreasing Expression of γ-Glutamylcysteine Synthetase via Promoter Activity in Doxorubicin-Resistant Leukemia Cells. Cancer Chemother. Pharmacol. 2009, 64, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Yan, S. Piperlongumine Reverses Doxorubicin Resistance through the PI3K/Akt Signaling Pathway in K562/A02 Human Leukemia Cells. Exp. Ther. Med. 2015, 9, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Toma, S.; Maselli, G.; Dastoli, G.; De Francisci, E.; Raffo, P. Synergistic Effect between Doxorubicin and a Low Dose of All-Trans-Retinoic Acid in MCF-7 Breast Cancer Cell Line. Cancer Lett. 1997, 116, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Czeczuga-Semeniuk, E.; Wołczyński, S.; Dabrowska, M.; Dziecioł, J.; Anchim, T. The Effect of Doxorubicin and Retinoids on Proliferation, Necrosis and Apoptosis in MCF-7 Breast Cancer Cells. Folia Histochem. Cytobiol. 2004, 42, 221–227. [Google Scholar] [PubMed]
- Liu, X.; Zhong, S.; Qiu, K.; Chen, X.; Wu, W.; Zheng, J.; Liu, Y.; Wu, H.; Fan, S.; Nie, D.; et al. Targeting NRF2 Uncovered an Intrinsic Susceptibility of Acute Myeloid Leukemia Cells to Ferroptosis. Exp. Hematol. Oncol. 2023, 12, 47. [Google Scholar] [CrossRef]
- Praharaj, P.P.; Singh, A.; Patra, S.; Bhutia, S.K. Co-Targeting Autophagy and NRF2 Signaling Triggers Mitochondrial Superoxide to Sensitize Oral Cancer Stem Cells for Cisplatin-Induced Apoptosis. Free Radic. Biol. Med. 2023, 207, 72–88. [Google Scholar] [CrossRef]
- Cucci, M.A.; Grattarola, M.; Dianzani, C.; Damia, G.; Ricci, F.; Roetto, A.; Trotta, F.; Barrera, G.; Pizzimenti, S. Ailanthone Increases Oxidative Stress in CDDP-Resistant Ovarian and Bladder Cancer Cells by Inhibiting of Nrf2 and YAP Expression through a Post-Translational Mechanism. Free Radic. Biol. Med. 2020, 150, 125–135. [Google Scholar] [CrossRef]
- Fouzder, C.; Mukhuty, A.; Kundu, R. Kaempferol Inhibits Nrf2 Signalling Pathway via Downregulation of Nrf2 mRNA and Induces Apoptosis in NSCLC Cells. Arch. Biochem. Biophys. 2021, 697, 108700. [Google Scholar] [CrossRef]
- Yao, J.; Wei, X.; Lu, Y. Chaetominine Reduces MRP1-Mediated Drug Resistance via Inhibiting PI3K/Akt/Nrf2 Signaling Pathway in K562/Adr Human Leukemia Cells. Biochem. Biophys. Res. Commun. 2016, 473, 867–873. [Google Scholar] [CrossRef]
- Datta, S.; Bishayee, A.; Sinha, D. Black Tea Bioactive Phytoconstituents Realign NRF2 for Anticancer Activity in Lung Adenocarcinoma. Front. Pharmacol. 2023, 14, 1176819. [Google Scholar] [CrossRef]
- Xian, P.; Hei, Y.; Wang, R.; Wang, T.; Yang, J.; Li, J.; Di, Z.; Liu, Z.; Baskys, A.; Liu, W.; et al. Mesenchymal Stem Cell-Derived Exosomes as a Nanotherapeutic Agent for Amelioration of Inflammation-Induced Astrocyte Alterations in Mice. Theranostics 2019, 9, 5956–5975. [Google Scholar] [CrossRef] [PubMed]
- Kopacz, A.; Rojo, A.I.; Patibandla, C.; Lastra-Martínez, D.; Piechota-Polanczyk, A.; Kloska, D.; Jozkowicz, A.; Sutherland, C.; Cuadrado, A.; Grochot-Przeczek, A. Overlooked and Valuable Facts to Know in the NRF2/KEAP1 Field. Free Radic. Biol. Med. 2022, 192, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Kopacz, A.; Kloska, D.; Forman, H.J.; Jozkowicz, A.; Grochot-Przeczek, A. Beyond Repression of Nrf2: An Update on Keap1. Free Radic. Biol. Med. 2020, 157, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Fu, X.; Jia, Y.; Ma, X.; Tao, J.; Yang, T.; Ma, H.; Liang, X.; Liu, X.; Yang, J.; et al. Nrf2/Keap1/ARE Signaling Mediated an Antioxidative Protection of Human Placental Mesenchymal Stem Cells of Fetal Origin in Alveolar Epithelial Cells. Oxidative Med. Cell. Longev. 2019, 2019, 2654910. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.-J.; Choi, J.J.; Lee, S.Y.; Kim, Y.S. The Effects of ML385 on Head and Neck Squamous Cell Carcinoma: Implications for NRF2 Inhibition as a Therapeutic Strategy. Int. J. Mol. Sci. 2024, 25, 7011. [Google Scholar] [CrossRef]
- Hannon Barroeta, P.; O’Sullivan, M.J.; Zisterer, D.M. The Role of the Nrf2/GSH Antioxidant System in Cisplatin Resistance in Malignant Rhabdoid Tumours. J. Cancer Res. Clin. Oncol. 2023, 149, 8379–8391. [Google Scholar] [CrossRef]
- Kim, H.G.; Kim, C.W.; Lee, D.H.; Lee, J.-S.; Oh, E.-T.; Park, H.J. Quinacrine-Mediated Inhibition of Nrf2 Reverses Hypoxia-Induced 5-Fluorouracil Resistance in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 4366. [Google Scholar] [CrossRef]
- Chian, S.; Li, Y.-Y.; Wang, X.-J.; Tang, X.-W. Luteolin Sensitizes Two Oxaliplatin-Resistant Colorectal Cancer Cell Lines to Chemotherapeutic Drugs Via Inhibition of the Nrf2 Pathway. Asian Pac. J. Cancer Prev. 2014, 15, 2911–2916. [Google Scholar] [CrossRef]
- Pouremamali, F.; Jeddi, F.; Samadi, N. Nrf2-ME-1 Axis Is Associated with 5-FU Resistance in Gastric Cancer Cell Line. Process Biochem. 2022, 114, 174–184. [Google Scholar] [CrossRef]
- Kamińska, K.; Cudnoch-Jędrzejewska, A. A Review on the Neurotoxic Effects of Doxorubicin. Neurotox. Res. 2023, 41, 383–397. [Google Scholar] [CrossRef]
- Nordgren, K.K.S.; Wallace, K.B. Disruption of the Keap1/Nrf2-Antioxidant Response System After Chronic Doxorubicin Exposure In Vivo. Cardiovasc. Toxicol. 2020, 20, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Tian, Z.; Sun, M.; Dong, D. Nrf2: A Dark Horse in Doxorubicin-Induced Cardiotoxicity. Cell Death Discov. 2023, 9, 261. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Xia, L.; Xu, D.; Liu, Y.; Jin, P.; Zhai, M.; Mao, Y.; Wang, Y.; Wen, A.; Yang, J.; et al. Cardioprotective Effects of Asiaticoside against Diabetic Cardiomyopathy: Activation of the AMPK/Nrf2 Pathway. J. Cell. Mol. Medi 2024, 28, e18055. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, W.; Fu, H.; Wang, X.; Tang, Y.; Huang, C. DL-3-n-Butylphthalide Attenuates Doxorubicin-Induced Acute Cardiotoxicity via Nrf2/HO-1 Signaling Pathway. Heliyon 2024, 10, e27644. [Google Scholar] [CrossRef]
- Wang, W.; Shen, Q. Tranilast Reduces Cardiomyocyte Injury Induced by Ischemia-reperfusion via Nrf2/HO-1/NF-κB Signaling. Exp. Ther. Med. 2023, 25, 160. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, J.; Yao, Z.-M.; Sun, X.-R.; Tong, X.-H.; Hu, M.; Zhang, Y.; Dong, S.-Y. Procyanidins Alleviated Cerebral Ischemia/Reperfusion Injury by Inhibiting Ferroptosis via the Nrf2/HO-1 Signaling Pathway. Molecules 2023, 28, 3582. [Google Scholar] [CrossRef]
- Shen, J.; Chen, S.; Li, X.; Wu, L.; Mao, X.; Jiang, J.; Zhu, D. Salidroside Mediated the Nrf2/GPX4 Pathway to Attenuates Ferroptosis in Parkinson’s Disease. Neurochem. Res. 2024, 49, 1291–1305. [Google Scholar] [CrossRef]
- Jiang, Z.; Liu, D.; Li, T.; Gai, C.; Xin, D.; Zhao, Y.; Song, Y.; Cheng, Y.; Li, T.; Wang, Z. Hydrogen Sulfide Reduces Oxidative Stress in Huntington’s Disease via Nrf2. Neural Regen. Res. 2025, 20, 1776–1788. [Google Scholar] [CrossRef]
- Juszczak, M.; Das, S.; Kosińska, A.; Rybarczyk-Pirek, A.J.; Wzgarda-Raj, K.; Tokarz, P.; Vasudevan, S.; Chworos, A.; Woźniak, K.; Rudolf, B. Piano-Stool Ruthenium(ii) Complexes with Maleimide and Phosphine or Phosphite Ligands: Synthesis and Activity against Normal and Cancer Cells. Dalton Trans. 2023, 52, 4237–4250. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (Resazurin) Fluorescent Dye for the Assessment of Mammalian Cell Cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juszczak, M.; Tokarz, P.; Woźniak, K. Potential of NRF2 Inhibitors—Retinoic Acid, K67, and ML-385—In Overcoming Doxorubicin Resistance in Promyelocytic Leukemia Cells. Int. J. Mol. Sci. 2024, 25, 10257. https://doi.org/10.3390/ijms251910257
Juszczak M, Tokarz P, Woźniak K. Potential of NRF2 Inhibitors—Retinoic Acid, K67, and ML-385—In Overcoming Doxorubicin Resistance in Promyelocytic Leukemia Cells. International Journal of Molecular Sciences. 2024; 25(19):10257. https://doi.org/10.3390/ijms251910257
Chicago/Turabian StyleJuszczak, Michał, Paulina Tokarz, and Katarzyna Woźniak. 2024. "Potential of NRF2 Inhibitors—Retinoic Acid, K67, and ML-385—In Overcoming Doxorubicin Resistance in Promyelocytic Leukemia Cells" International Journal of Molecular Sciences 25, no. 19: 10257. https://doi.org/10.3390/ijms251910257