

Computational Study of Molecular Mechanism for the Involvement of Human Serum Albumin in the Renin–Angiotensin–Aldosterone System


Abstract
:1. Introduction
2. Results
2.1. Macromolecular Docking of HSA onto the Surface of ACE
2.2. Molecular Dynamics of HSA–ACE Complexes
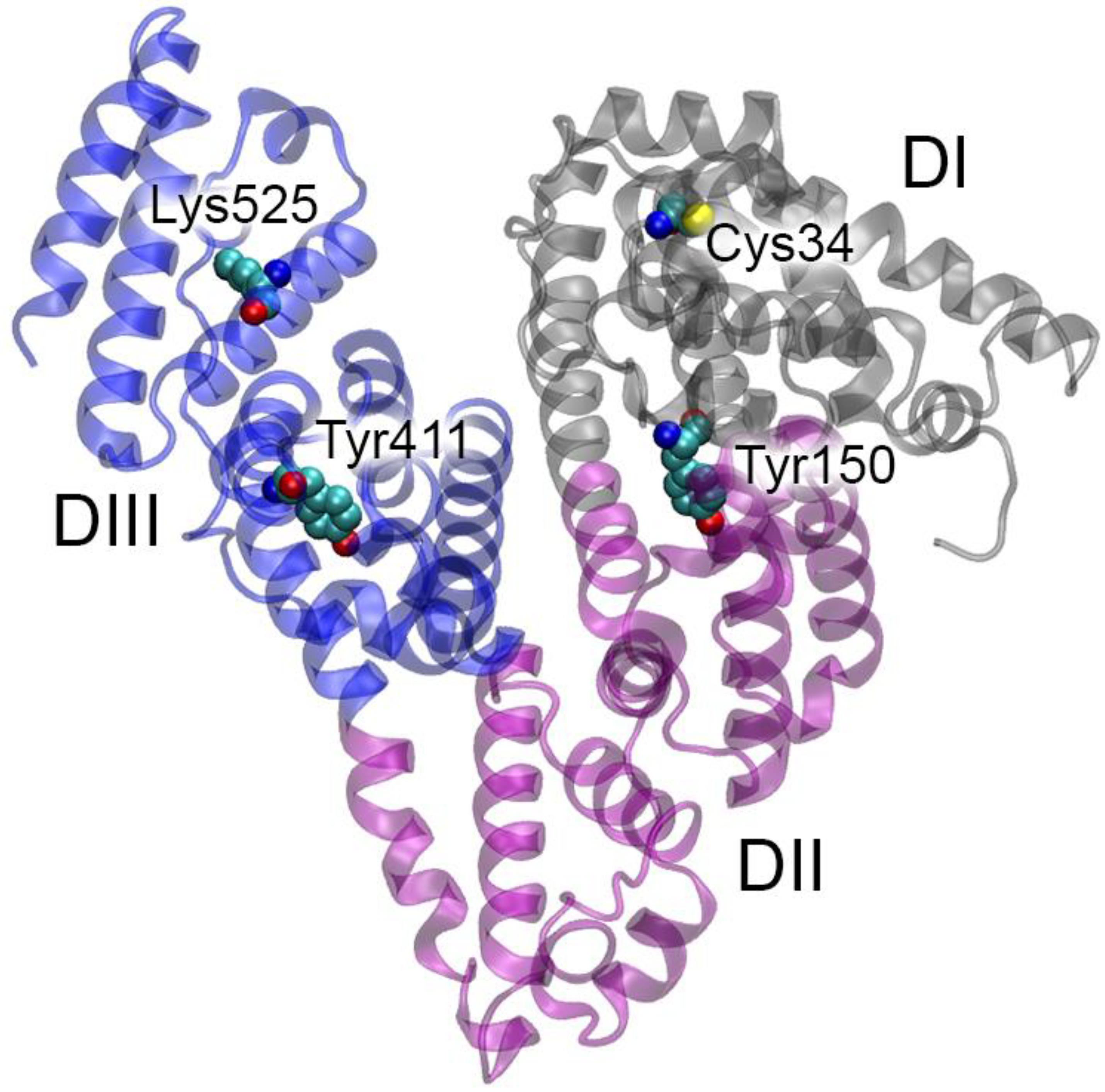
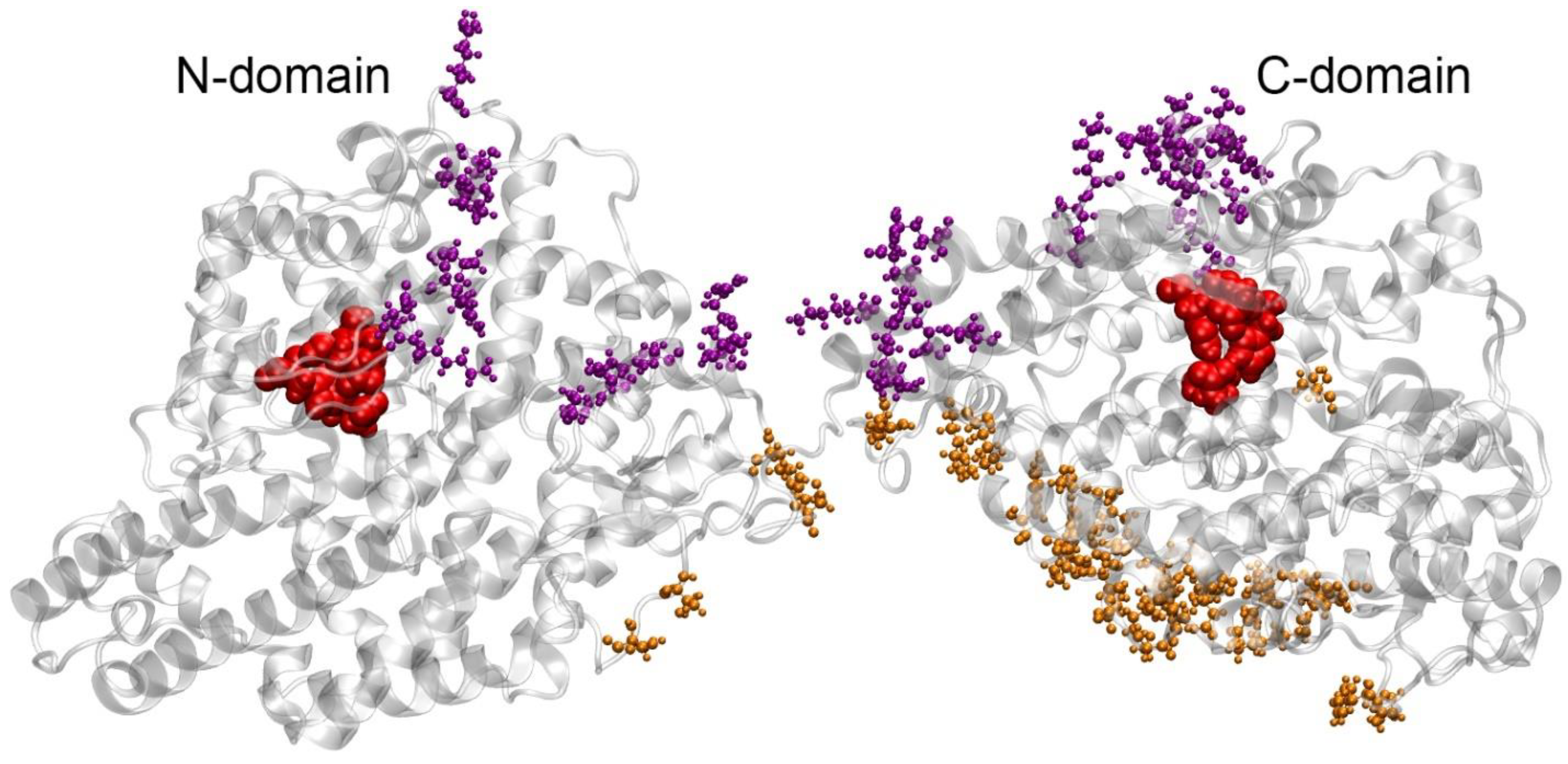
2.3. How Binding to ACE Can Affect HSA Activity According to Molecular Modeling
2.4. How Binding to HSA Can Affect ACE Activity According to Molecular Modeling
2.5. How Point Mutations in HSA and ACE Can Affect Their Interaction According to Molecular Modeling
2.6. HSA–ACE Complexes Acording to AlphaFold 3
2.7. Limitations of the Research
3. Discussion
4. Materials and Methods
4.1. Building of Three-Dimensional Models
4.2. Macromolecular Docking
4.3. Molecular Dynamics
4.4. Constructing HSA–ACE Complexes Using AlphaFold 3
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE | angiotensin I-converting enzyme; |
| AD | Alzheimer’s Disease; |
| AF3 | AlphaFold 3; |
| FA | fatty acids; |
| FcRn | neonatal Fc receptor; |
| HB | hydrogen bond; |
| HSA | human serum albumin; |
| MD | molecular dynamics; |
| RAAS | renin–angiotensin–aldosterone system; |
| RMSD | root means square deviation; |
| SB | salt bridge; |
| VMD | Visual Molecular Dynamics |
References
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2018, 71, e127–e248. [Google Scholar] [CrossRef] [PubMed]
- Harrison, G.A.; Robinson, M.; Stacey, R.V.; McCulloch, C.H.; Torda, T.A.; Wright, J.S. Hypotensive effects of stable plasma protein solution (SPPS): A preliminary communication. Med. J. Aust. 1971, 2, 1040–1041. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Nilsson, B.; Modig, J.; Acosta, R.; Johansson, B.; Larsson, A. A case report. Hypotensive reaction caused by albumin infusion. Lakartidningen 2001, 98, 439–442. (In Swedish) [Google Scholar] [PubMed]
- Howard, G.; Downward, G.; Bowie, D. Human serum albumin induced hypotension in the postoperative phase of cardiac surgery. Anaesth. Intensive Care 2001, 29, 591–594. [Google Scholar] [CrossRef]
- Oda, E. Decreased serum albumin predicts hypertension in a Japanese health screening population. Intern. Med. 2014, 53, 655–660. [Google Scholar] [CrossRef]
- Klauser, R.J.; Robinson, C.J.; Marinkovic, D.V.; Erdös, E.G. Inhibition of human peptidyl dipeptidase (angiotensin I converting enzyme: Kininase II) by human serum albumin and its fragments. Hypertension 1979, 1, 281–286. [Google Scholar] [CrossRef]
- Fagyas, M.; Úri, K.; Siket, I.M.; Fülöp, G.Á.; Csató, V.; Daragó, A.; Boczán, J.; Bányai, E.; Szentkirályi, I.E.; Maros, T.M.; et al. New perspectives in the renin-angiotensin-aldosterone system (RAAS) II: Albumin suppresses angiotensin converting enzyme (ACE) activity in human. PLoS ONE 2014, 9, e87844. [Google Scholar] [CrossRef]
- Danilov, S.M.; Jain, M.S.; Petukhov, P.A.; Kurilova, O.V.; Ilinsky, V.V.; Trakhtman, P.E.; Dadali, E.L.; Samokhodskaya, L.M.; Kamalov, A.A.; Kost, O.A. Blood ACE Phenotyping for Personalized Medicine: Revelation of Patients with Conformationally Altered ACE. Biomedicines 2023, 11, 534. [Google Scholar] [CrossRef]
- Kozuch, A.J.; Petukhov, P.A.; Fagyas, M.; Popova, I.A.; Lindeblad, M.O.; Bobkov, A.P.; Kamalov, A.A.; Toth, A.; Dudek, S.M.; Danilov, S.M. Urinary ACE Phenotyping as a Research and Diagnostic Tool: Identification of Sex-Dependent ACE Immunoreactivity. Biomedicines 2023, 11, 953. [Google Scholar] [CrossRef]
- Danilov, S.M.; Adzhubei, I.A.; Kozuch, A.J.; Petukhov, P.A.; Popova, I.A.; Choudhury, A.; Sengupta, D.; Dudek, S.M. Carriers of Heterozygous Loss-of-Function ACE Mutations Are at Risk for Alzheimer’s Disease. Biomedicines 2024, 12, 162. [Google Scholar] [CrossRef]
- Enyedi, E.E.; Petukhov, P.A.; Kozuch, A.J.; Dudek, S.M.; Toth, A.; Fagyas, M.; Danilov, S.M. ACE Phenotyping in Human Blood and Tissues: Revelation of ACE Outliers and Sex Differences in ACE Sialylation. Biomedicines 2024, 12, 940. [Google Scholar] [CrossRef] [PubMed]
- Kragh-Hansen, U. Structure and ligand binding properties of human serum albumin. Dan. Med. Bull. 1990, 37, 57–84. [Google Scholar] [PubMed]
- Kragh-Hansen, U.; Brennan, S.O.; Minchiotti, L.; Galliano, M. Modified high-affinity binding of Ni2+, Ca2+ and Zn2+ to natural mutants of human serum albumin and proalbumin. Biochem. J. 1994, 301 Pt 1, 217–223. [Google Scholar] [CrossRef]
- Kragh-Hansen, U.; Saito, S.; Nishi, K.; Anraku, M.; Otagiri, M. Effect of genetic variation on the thermal stability of human serum albumin. Biochim. Biophys. Acta 2005, 1747, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Kragh-Hansen, U.; Minchiotti, L.; Galliano, M.; Peters, T., Jr. Human serum albumin isoforms: Genetic and molecular aspects and functional consequences. Biochim. Biophys. Acta 2013, 1830, 5405–5417. [Google Scholar] [CrossRef]
- Caridi, G.; Lugani, F.; Angeletti, A.; Campagnoli, M.; Galliano, M.; Minchiotti, L. Variations in the Human Serum Albumin Gene: Molecular and Functional Aspects. Int. J. Mol. Sci. 2022, 23, 1159. [Google Scholar] [CrossRef]
- Na Takuathung, M.; Sakuludomkan, W.; Khatsri, R.; Dukaew, N.; Kraivisitkul, N.; Ahmadmusa, B.; Mahakkanukrauh, C.; Wangthaweesap, K.; Onin, J.; Srichai, S.; et al. Adverse Effects of Angiotensin-Converting Enzyme Inhibitors in Humans: A Systematic Review and Meta-Analysis of 378 Randomized Controlled Trials. Int. J. Environ. Res. Public Health 2022, 19, 8373. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.H.; Pan, D.Q.; Jiang, M.; Liu, T.T.; Wang, Q. Binding interaction of ramipril with bovine serum albumin (BSA): Insights from multi-spectroscopy and molecular docking methods. J. Photochem. Photobiol. B 2016, 164, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.L.; Pan, D.Q.; Zhou, K.L.; Lou, Y.Y.; Shi, J.H. Multi-spectroscopic approaches and molecular simulation research of the intermolecular interaction between the angiotensin-converting enzyme inhibitor (ACE inhibitor) benazepril and bovine serum albumin (BSA). Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 212, 15–24. [Google Scholar] [CrossRef]
- Alhumaydhi, F.A.; Aljasir, M.A.; Aljohani, A.S.; Alsagaby, S.A.; Alwashmi, A.S.; Shahwan, M.; Hassan, M.I.; Islam, A.; Shamsi, A. Probing the interaction of memantine, an important Alzheimer’s drug, with human serum albumin: In silico and in vitro approach. J. Mol. Liq. 2021, 340, 116888. [Google Scholar] [CrossRef]
- Shamsi, A.; Shahwan, M.; Khan, M.S.; Alhumaydhi, F.A.; Alsagaby, S.A.; Al Abdulmonem, W.; Abdullaev, B.; Yadav, D.K. Mechanistic Insight into Binding of Huperzine A with Human Serum Albumin: Computational and Spectroscopic Approaches. Molecules 2022, 27, 797. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- He, X.M.; Carter, D.C. Atomic structure and chemistry of human serum albumin. Nature 1992, 358, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef] [PubMed]
- van der Vusse, G.J. Albumin as fatty acid transporter. Drug Metab. Pharmacokinet. 2009, 24, 300–307. [Google Scholar] [CrossRef]
- Petitpas, I.; Grüne, T.; Bhattacharya, A.A.; Curry, S. Crystal structures of human serum albumin complexed with monounsaturated and polyunsaturated fatty acids. J. Mol. Biol. 2001, 314, 955–960. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; di Masi, A.; Ascenzi, P. Serum Albumin: A Multifaced Enzyme. Int. J. Mol. Sci. 2021, 22, 10086. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol. 1976, 12, 1052–1061. [Google Scholar]
- Zunszain, P.A.; Ghuman, J.; Komatsu, T.; Tsuchida, E.; Curry, S. Crystal structural analysis of human serum albumin complexed with hemin and fatty acid. BMC Struct. Biol. 2003, 3, 6. [Google Scholar] [CrossRef]
- Zsila, F. Subdomain IB is the third major drug binding region of human serum albumin: Toward the three-sites model. Mol. Pharm. 2013, 10, 1668–1682. [Google Scholar] [CrossRef] [PubMed]
- Petitpas, I.; Petersen, C.E.; Ha, C.E.; Bhattacharya, A.A.; Zunszain, P.A.; Ghuman, J.; Bhagavan, N.V.; Curry, S. Structural basis of albumin-thyroxine interactions and familial dysalbuminemic hyperthyroxinemia. Proc. Natl. Acad. Sci. USA 2003, 100, 6440–6445. [Google Scholar] [CrossRef] [PubMed]
- Hein, K.L.; Kragh-Hansen, U.; Morth, J.P.; Jeppesen, M.D.; Otzen, D.; Møller, J.V.; Nissen, P. Crystallographic analysis reveals a unique lidocaine binding site on human serum albumin. J. Struct. Biol. 2010, 171, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Lockridge, O.; Xue, W.; Gaydess, A.; Grigoryan, H.; Ding, S.J.; Schopfer, L.M.; Hinrichs, S.H.; Masson, P. Pseudo-esterase activity of human albumin: Slow turnover on tyrosine 411 and stable acetylation of 82 residues including 59 lysines. J. Biol. Chem. 2008, 283, 22582–22590. [Google Scholar] [CrossRef] [PubMed]
- Belinskaia, D.A.; Voronina, P.A.; Vovk, M.A.; Shmurak, V.I.; Batalova, A.A.; Jenkins, R.O.; Goncharov, N.V. Esterase Activity of Serum Albumin Studied by 1H NMR Spectroscopy and Molecular Modelling. Int. J. Mol. Sci. 2021, 22, 10593. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, F.; Shibata, T.; Kamiya, K.; Yoshitake, J.; Kikuchi, R.; Matsushita, T.; Ishii, I.; Giménez-Bastida, J.A.; Schneider, C.; Uchida, K. Structural and functional insights into S-thiolation of human serum albumins. Sci. Rep. 2018, 8, 932. [Google Scholar] [CrossRef]
- Qiu, H.Y.; Hou, N.N.; Shi, J.F.; Liu, Y.P.; Kan, C.X.; Han, F.; Sun, X.D. Comprehensive overview of human serum albumin glycation in diabetes mellitus. World J. Diabetes 2021, 12, 1057–1069. [Google Scholar] [CrossRef]
- Lubbe, L.; Sewell, B.T.; Woodward, J.D.; Sturrock, E.D. Cryo-EM reveals mechanisms of angiotensin I-converting enzyme allostery and dimerization. EMBO J. 2022, 41, e110550. [Google Scholar] [CrossRef]
- Wei, L.; Alhenc-Gelas, F.; Corvol, P.; Clauser, E. The two homologous domains of human angiotensin I-converting enzyme are both catalytically active. J. Biol. Chem. 1991, 266, 9002–9008. [Google Scholar] [CrossRef]
- Wei, L.; Clauser, E.; Alhenc-Gelas, F.; Corvol, P. The two homologous domains of human angiotensin I-converting enzyme interact differently with competitive inhibitors. J. Biol. Chem. 1992, 267, 13398–13405. [Google Scholar] [CrossRef]
- Jaspard, E.; Wei, L.; Alhenc-Gelas, F. Differences in the properties and enzymatic specificities of the two active sites of angiotensin I-converting enzyme (kininase II). Studies with bradykinin and other natural peptides. J. Biol. Chem. 1993, 268, 9496–9503. [Google Scholar] [CrossRef]
- Cozier, G.E.; Lubbe, L.; Sturrock, E.D.; Acharya, K.R. ACE-domain selectivity extends beyond direct interacting residues at the active site. Biochem. J. 2020, 477, 1241–1259. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Copeland, M.M.; Kundrotas, P.J.; Vakser, I.A. GRAMM Web Server for Protein Docking. Methods Mol. Biol. 2024, 2714, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.J.; Blindauer, C.A.; Berezenko, S.; Sleep, D.; Tooth, D.; Sadler, P.J. Role of Tyr84 in controlling the reactivity of Cys34 of human albumin. FEBS J. 2005, 272, 353–362. [Google Scholar] [CrossRef] [PubMed]
- VCV000904661.4—ClinVar—NCBI. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/904661 (accessed on 13 August 2024).
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, V.; Damschroder, M.M.; Cook, K.E.; Li, Q.; Gao, C.; Wu, H.; Dall’Acqua, W.F. Structural insights into neonatal Fc receptor-based recycling mechanisms. J. Biol. Chem. 2014, 289, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Zunszain, P.A.; Ghuman, J.; McDonagh, A.F.; Curry, S. Crystallographic analysis of human serum albumin complexed with 4Z,15E-bilirubin-IXalpha. J. Mol. Biol. 2008, 381, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Paar, M.; Fengler, V.H.; Rosenberg, D.J.; Krebs, A.; Stauber, R.E.; Oettl, K.; Hammel, M. Albumin in patients with liver disease shows an altered conformation. Commun. Biol. 2021, 4, 731. [Google Scholar] [CrossRef]
- Fujiwara, S.; Amisaki, T. Molecular dynamics study of conformational changes in human serum albumin by binding of fatty acids. Proteins 2006, 64, 730–739. [Google Scholar] [CrossRef]
- Ketrat, S.; Japrung, D.; Pongprayoon, P. Exploring how structural and dynamic properties of bovine and canine serum albumins differ from human serum albumin. J. Mol. Graph. Model. 2020, 98, 107601. [Google Scholar] [CrossRef]
- Leblanc, Y.; Berger, M.; Seifert, A.; Bihoreau, N.; Chevreux, G. Human serum albumin presents isoform variants with altered neonatal Fc receptor interactions. Protein Sci. 2019, 28, 1982–1992. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.C.; Myslinski, J.; Pratap, S.; Flores, B.; Rhodes, G.; Campos-Bilderback, S.B.; Sandoval, R.M.; Kumar, S.; Patel, M.; Ashish; et al. Mechanism of increased clearance of glycated albumin by proximal tubule cells. Am. J. Physiol. Renal Physiol. 2016, 310, F1089–F1102. [Google Scholar] [CrossRef] [PubMed]
- Sand, K.M.; Bern, M.; Nilsen, J.; Dalhus, B.; Gunnarsen, K.S.; Cameron, J.; Grevys, A.; Bunting, K.; Sandlie, I.; Andersen, J.T. Interaction with both domain I and III of albumin is required for optimal pH-dependent binding to the neonatal Fc receptor (FcRn). J. Biol. Chem. 2014, 289, 34583–34594. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Bocedi, A.; Notari, S.; Fanali, G.; Fesce, R.; Fasano, M. Allosteric modulation of drug binding to human serum albumin. Mini Rev. Med. Chem. 2006, 6, 483–489. [Google Scholar] [CrossRef]
- Ascenzi, P.; Fasano, M. Allostery in a monomeric protein: The case of human serum albumin. Biophys. Chem. 2010, 48, 16–22. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, S.; Xu, D. Catalytic mechanism of angiotensin-converting enzyme and effects of the chloride ion. J. Phys. Chem. B 2013, 117, 6635–6645. [Google Scholar] [CrossRef]
- Foloppe, N.; MacKerell, A.D., Jr. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- Jorgensen, W.L. Quantum and statistical mechanical studies of liquids. 10. Transferable intermolecular potential functions for water, alcohols, and ethers. Application to liquid water. J. Am. Chem. Soc. 1981, 103, 335–340. [Google Scholar] [CrossRef]
- Bussi, G.; Zykova-Timan, T.; Parrinello, M. Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J. Chem. Phys. 2009, 130, 074101. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 3, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comp. Chem. 1997, 18, 1463–1473. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex # Quantity of Interacting Atoms, HSA/ACE | HSA | ACE | Specific Interactions HSA–ACE |
|---|---|---|---|
| 1 372/396 | Ala172, Asp173, Lys174, Ala176, Pro180, Lys181, Asp183, Glu184, Asp187, Phe228, Lys274, Leu275, Lys276, Glu277, Cys278, Glu294, Asp296, Glu297, Ala300, Asp301, Pro303, Ala306, Ala307, Asp308, Glu311, Asp314, Lys317, Asn318, Met329, Tyr332, Arg336, Pro339, Glu393, Glu396, Gln397, Leu398, Gly399, Glu400, Lys402, Lys439, His440, Pro441, Lys444, Arg445, Lys519 | Ala232, Arg235, Met267, Val269, Phe271, Pro272, Asp273, Asn416, Asp417, Thr418, Gln579, Trp580, Gln584, Gln587, Glu617, Ala618, Ser621, Lys622, Glu626, Val633, Glu637, Glu640, Asn644, Asn648, Ile656, Asn685, Ile688, Leu905, Pro906, Pro909, Lys914, Asn937, Lys939, Gln969, Lys971, Asp972, Lys1132, Lys1143, Leu1144 | Lys174-Asn648 (HB) Lys181-Glu637 (SB) Asp187-Lys939 (SB) Lys274-Ala618O (HB) Glu294-Asn685 (HB) Pro303O-Gln584 (HB) Glu311-Gln587 (SB) Lys317-Asp417 (SB) Tyr332-Asp273 (SB) Pro339O-Asn685 (HB) Glu396-Gln969 (HB) Glu396-Lys1143 (SB) Gln397-Lys1132 (HB) Lys439-Asp972 (SB) |
| 2 319/287 | Phe11, Lys12, Asp13, Gly15, Glu16, Glu17, Asn18, Glu48, Lys51, Val54, Ala55, Asp56, Glu57, Lys159, Glu167, Ala171, Ala172, Asp173, Ala175, Cys177, Pro180, Lys181, Glu184, Pro282, Leu284, Glu294, Glu297, Glu396, Gln397, Gly399, Glu400, Tyr401, Lys402, Ser435, Lys436, Cys437, Cys438, Lys439, His440, Pro441, Lys444, Arg445 | Arg245, Arg453, Arg458, Phe461, Tyr465, Lys469, Val477, Thr478, Asn480, Gly589, Val591, Trp594, Pro595, Glu596, Tyr597, His600, Leu603, Asp605, Pro608, Glu609, Pro803, Ser804, Gln807, Glu810, Arg811, Gln814, Glu815, Gln817, Leu821, Lys1067, Asn1162, Leu1190 | Ala172HN-Glu609 (HB) Asp173-Asn480 (HB) Glu294-Arg245 (SB) Glu396-Tyr465 (HB) Glu396-Lys469 (SB) |
| 3 278/267 | Ala2, His3, His67, Pro96, Glu208, Lys212, Ala229, Ser232, Lys233, Thr236, Thr239, Lys240, Thr243, His247, Ala320, Lys323, Asp324, Val325, Thr355, Glu358, Lys359, Cys361, Ala362, Ala363, Pro366, Glu376, Pro379, Asp471, Arg472, Lys475, Glu479, Asn483, Ala490, Glu492, Glu495, Thr496, Lys538 | Glu212, Gln216, Glu219, Arg235, Arg245, Arg458, Gln586, Gln587, Asn588, Gly589, Glu590, Val591, Glu596, Tyr597, Gln598, Asn685, Thr686, Thr687, Arg811, Gln814, Glu815, Pro818, Asp972, Leu973, Arg1137, Leu1144, Arg1148, Leu1156, Gln1160, Pro1161, Asn1162, Ser1166 | Glu208-Arg245 (SB) Lys212-Gly589O (HB) Ser232-Gln587O (HB) Thr236-Glu590O (HB) Asp324-Arg235 (SB) Glu358-Thr686 (HB) Cys361O-Asn685 (HB) Asp471-Arg811 (SB) Arg472-Glu815 (SB) |
| 4 540/543 | His3, Ser5, His9, Lys12, Asp13, Leu14, Gly15, Glu16, Glu17, Asn18, Lys20, Gln33, Cys34, Pro35, Phe36, Glu37, Asp38, His39, Lys41, Asn44, Glu45, Glu48, Lys51, Thr52, Val54, Glu57, Thr79, Leu80, Glu82, Thr83, Tyr84, Asn111, Pro113, Val116, His128, Asp129, Asn130, Glu131, Glu132, Leu135, Leu155, Ala158, Lys159, Lys162, Thr166, Asp259, Lys262, Cys265, Glu266, Cys279, Glu280, Lys281, Pro282, Leu283, Leu284, Lys286, Glu505, Thr508, His510, Ala511, Asp512, Asp563, Lys564, Glu565, Thr566 | Asn145, Ala148, Ser149, Arg151, Arg231, Ala232, Arg235, Arg236, Asn263, Tyr265, Asp266, Met267, Val268, Val269, Phe271, Pro272, Asp273, Lys274, Pro275, Asn276, Leu277, Asp278, Thr280, Ser281, Thr282, Leu284, Cys330, Arg350, Thr352, Leu410, Leu411, Asp412, Arg413, Thr415, Asp417, Glu419, Asn423, Lys427, Asn588, Asp616, Glu617, Ala618, Glu619, Ser621, Lys622, Phe623, Glu625, Glu626, Arg629, Lys670, Tyr671, Gln674, Asn685, Lys893, Asp896, Thr900, Pro906, Pro909, Asn913, Lys939, Lys971, Lys1132 | Asp13-Ser281 (HB) Glu16-Lys427 (SB) Glu17-Tyr265 (HB) Glu17-Asn423 (HB) Asn18-Asn276O, NH (HB) Lys20-Asp266 (SB) Gln33-Glu625 (HB) Asp38-Glu619HN (HB) Asp38-Ala618HN (HB) Lys41-Asp616 (SB) Lys51-Asn263 (HB) Glu57O-Arg350 (HB) Thr79-Glu619 (HB) Glu82-Gln674 (HB) Thr83-Lys622 (HB) Tyr84-Lys622 (HB) Asn111-Glu625 (HB) His128O-Asn588 (HB) Asp129-Arg235 (SB) Asn130-Met267O (HB) Glu131-Phe271HN (HB) Lys159-Glu419 (SB) Lys159-Asp273O(HB) Lys162-Glu419 (SB) Lys262-Leu410O (HB) Glu280-Arg413 (SB) Lys286-Asp412 (SB) Glu505-Lys939 (SB) Asp512-Lys971 (SB) Asp563-Lys1132 (SB) Glu565-Thr900 (HB) |
| 5 347/327 | Ala2, His3, Ala8, Phe11, Lys12, Cys53, Val54, Ala55, Asp56, Ser58, His128, Asp129, Phe156, Arg160, Ala163, Ala164, Thr166, Glu167, Cys168, Gln170, Ala172, Asp173, Leu178, Pro180, Lys181, Asp183, Glu184, Asp187, Glu188, Glu266, Asn267, Lys274, Glu277, Lys281, Pro282, His288, Cys289, Glu292, Glu294, Glu297, Cys438, Lys439, His440, Glu442 | Asn131, Ala134, Arg151, Arg236, Pro272, Asp273, Leu284, Gln285, Glu320, Lys321, Ala323, Arg350, Thr352, Glu609, Asp612, Leu613, Val614, Asp616, Glu617, Ala618, Glu619, Lys622, Arg629, Thr630, Gln632, Val633, Lys670, Thr673, Gln674, Lys677, Phe678, Asn685, Leu786, Lys939 | Cys53O-Arg350 (HB) His128O-Ala134HN (HB) Arg160-Glu619 (SB) Ala164O-Lys677 (HB) Gln170-Glu609O (HB) Leu178O-Lys677 (HB) Glu188-Lys622 (SB) Glu294-Asn685 (HB) Glu297- Asn685 (HB) Lys439-Arg629O (HB) Glu442-Lys939 (SB) |
| 6 472/432 | Asp13, Glu16, Glu17, His128, Glu131, Lys159, Lys162, Thr166, Ala172, Glu184, Ala258, Asp259, Lys262, Glu266, Ser273, Lys274, Lys276, Glu277, Lys281, Pro282, Leu283, Leu284, Glu294, Glu297, Met298, Pro299, Leu302, Ser304, Leu305, Ala306, Tyr334, Arg337, His338, Pro339, Asp340, Tyr341, Lys372, Phe374, Asp375, Phe377, Lys378, Pro379, Val381, Glu382, Glu383, Gln385, Lys389, Lys439, Pro441, Glu442, Ala443, Lys444, Met446 | Asn45, Ile46, Thr47, Ala48, Glu49, Asn117, Asn131, Ala134, Thr135, Pro141, Thr144, Asn145, Ile146, Ala148, Ser149, Arg151, Arg236, Asn263, Asp266, Asn276, Thr280, Leu284, Lys321, Ala323, Asp324, Gly325, Arg326, Glu327, Cys330, Arg350, Asp612, Leu613, Val614, Asp616, Ala618, Glu619, Ser621, Lys622, Phe623, Glu625, Glu626, Arg629, Thr630, Ser631, Val633, Val634, Asn636, Asn666, His667, Leu669, Lys670, Tyr671, Thr673, Gln674, Lys677, Asn685 | Glu17HN-Ala323O (HB) Glu184-Thr135 (HB) Lys276-Asn145 (SB) Glu277-Thr144 (HB) Glu297-Arg236 (SB) Ser304-Asn685 (HB) Ala306HN-Glu625 (HB) Tyr334-Glu626 (HB) Tyr334-Arg629 (HB) Asp340HN-Glu619 (HB) Tyr341-Glu626 (HB) Lys372-Asn636 (SB) Phe377O-Lys670 (HB) Gln385-Lys677 (HB) |
| 7 447/444 | Gln33, Cys34, Pro35, Phe36, Glu37, Lys41, Val77, Thr79, Leu80, Arg81, Glu82, Thr83, Gly85, Glu86, Met87, Asp89, Cys90, Lys93, Gln94, Glu97, Glu100, Gln104, Asn111, Pro113, Leu115, Val116, Pro118, Glu119, Asp121, Val122, Thr125, Ala126, Asn130, Thr133, Tyr140, Glu141, Arg144, Lys205, His464, Glu465, Pro468, Ser470, Asp471, Thr474, Thr478, His510, Ala511, Asp512, Thr515, Leu516, Glu565, Thr566 | Arg240, Tyr241, Leu244, Arg245, Val477, Thr478, Asn480, Gly589, Val591, Pro595, Glu596, Trp599, His600, Pro602, Leu603, Asp605, Asn681, Gln682, Lys693, Tyr789, Val790, Asp794, Ser798, Pro803, Ser804, Glu806, Gln807, Asp808, Glu810, Arg811, Gln814, Glu815, Pro818, Leu821, Gln1042, Asp1049, Lys1067, Gln1115, Ala1116, Ser1131, Lys1132, Glu1133, Arg1137, Asn1162, Ser1164, Ser1166, Leu1169, Lys1173, Arg1180 | Cys34O-Gln807 (HB) Lys41-Asp1049 (SB) Thr79-Asp794 (HB) Arg81-Asp605 (SB) Glu82-Ser798 (HB) Glu82O-Lys693 (HB) Glu86-Tyr241 (HB) Glu86-Asn681 (HB) Glu86-Gln682 (HB) Glu97-Thr478HN (HB) Gln104-His600O (HB) Glu119-Lys1173 (SB) Glu141-Arg811 (SB) Lys205-Glu596 (SB) Ala511HN-Glu1133 (HB) Asp512-Arg1137 (SB) Glu565-Lys1132 (SB) |
| 8 362/390 | Gln33, Thr83, Asn111, Leu112, Pro113, Val116, Arg117, Pro118, Glu119, Asp121, Val122, Glu167, Gln170, Ala171, Ala172, Ala176, Cys177, Pro180, Lys181, Glu184, Asp187, Glu277, Lys281, Pro421, Lys432, Lys439, Asn503, Ala504, Glu505, His510, Asp512, Cys514, Thr515, Leu516, Ser517, Lys519, Arg521, Lys524, Glu556, Cys559, Lys560, Lys573 | Asn145, Ala148, Ser149, Arg231, His234, Arg235, Gly238, Leu244, Glu262, Asn263, Asp266, Pro270, Pro272, Asp273, Pro275, Asn276, Asp278, Thr280, Ser281, Leu284, Gln285, Arg350, Val351, Thr352, Asp354, Gln355, Asp417, Lys427, Asn588, Gly589, Glu590, Val591, Ala618, Glu619, Ser621, Lys622, Glu625, Arg629, Thr630, Lys670, Gln684, Asn685, Thr686, Thr1140, Ser1147, Arg1148, Gln1155, Leu1156 | Gln33-Lys622 (HB) Gln33-Glu625 (HB) Arg117-Asp266 (SB) Glu119-Ser149 (HB) Glu167-Thr280 (HB) Gln170-Arg350 (HB) Lys181-Thr280 (HB) Lys281-Gln285 (HB) Lys432-Asp273 (SB) Lys439-Asp417 (SB) Glu505-Ser1147 (HB) Lys524-Asn685 (HB) Cys559O-Arg231 (HB) Lys560-Glu590 (SB) Lys560-Leu244O (HB) Lys573-Gln1155 (HB) |
| 9 351/352 | Gln33, Glu82, Thr83, Asn111, Pro113, Arg117, Glu119, Asp121, Val122, Glu167, Gln170, Ala172, Lys174, Pro180, Lys181, Glu184, Asp187, Lys190, Glu277, Lys281, Phe395, Glu400, Tyr401, Lys402, Lys432, Ser435, Lys436, Lys439, His440, Ala504, Glu505, His510, Asp512, Ile513, Cys514, Thr515, Leu516, Ser517, Glu518, Lys519, Arg521, Lys524, Lys525, Glu556, Cys559, Lys560, Asp562, Asp563 | Asn145, Ala148, Ser149, Arg151, Phe228, Arg230, Arg231, His234, Arg235, Gly238, Asp239, Asn243, Leu244, Arg245, Glu262, Asp266, Val269, Phe271, Pro272, Asp273, Pro275, Asn276, Asp278, Thr280, Ser281, Gln285, His331, Arg350, Thr352, Thr415, Asp417, Thr418, Glu419, Lys427, Glu583, Gln587, Asn588, Gly589, Glu590, Val591, Asp616, Ala618, Lys622, Glu625, Arg629, Val633, Gln684, Asn685, Thr686, Arg1148, Leu1156 | Asn111-Asn685 (HB) Arg117-Asp266 (SB) Glu119-Arg151 (SB) Asp121-Ser149 (HB) Lys190-Asp273 (SB) Glu277-Gln285 (HB) Lys281-Gln285 (HB) Lys432-Asp273 (SB) Ser435-Glu419 (HB) Lys436-Asp273O (HB) Lys439-Asp417 (SB) Ile513O-Arg235 (HB) Lys519-Val269O (HB) Lys560-Glu590 (SB) Lys560-Leu244O (HB) Asp562O-Asn243 (HB) |
| 10 267/231 | Ala78, Thr79, Arg81, Glu82, Glu86, Cys91, Ala92, Lys93, Pro96, Glu97, Glu100, Leu203, Gln204, Lys205, Phe206, Gly207, Glu208, Arg209, Lys212, Lys240, Thr243, Glu244, Lys317, Ala320, Glu321, Ala322, Lys323, Asp324, Val325, Met329, Glu358, Ala363, Pro366, Lys573 | Leu129, Asn131, Thr133, Ala134, Thr135, Cys136, Trp137, Ile146, Gln285, Asn289, Asp612, Leu613, Glu626, Arg629, Thr630, Val633, Glu640, Asn644, Asn648, Lys660, Gln663, Asn666, Lys670, Thr673, Gln674, Lys677, Asn913, Lys914, Asn937, Lys939, Asp940, Arg942 | Arg81-Thr135O (HB) Lys93-Asp612 (SB) Glu97O-Lys677 (HB) Glu100-Gln674 (HB) Lys205O-Arg629 (HB) Glu244-Asn666 (HB) Lys317-Asn648 (HB) Lys323-Asp940 (SB) Glu358-Lys939 (SB) |
| Complex # Quantity of Interacting Atoms, HSA/ACE | HSA | ACE | Specific Interactions HSA–ACE and Their Lifetime |
|---|---|---|---|
| 4 278/259 | Asp1, His9, Lys12, Asp13, Glu16, Glu17, Asn18, Lys20, Gln33, Pro35, Glu37, Asp38, Lys41, Asn44, Glu45, Glu48, Thr52, Val54, Ala55, Asp56, Glu57, Leu80, Thr83, Tyr84, Asn111, Leu112, Pro113, Asp129, Asn130, Glu131, Glu132, Leu135, Lys159, Lys162, Glu280, Pro282, Ala504, Glu505, Thr508, His510, Asp512, Glu565, Thr566, Ala569, Gly572, Lys573, Gln580 | Pro141, Asn145, Ala148, Ser149, Arg235, Arg236, Thr280, Ser281, Leu284, Glu320, Lys321, Arg350, Thr352, Arg413, Lys622, Glu625, Arg629, Asn666, Lys670, Thr673, Gln674, Lys893, Thr900, Pro906, Pro908, Pro909, Glu910, Trp912, Asn913, Lys914, Lys939, Lys971, Lys1132 | Asp1NH-Glu320 (SB, 29%) Asp13-Ser281 (HB, 75%) Glu17-Tyr265 (HB, 65%) Glu17-Asn423 (HB, 18%) Lys20-Asp266 (SB, 86%) Glu37-Arg236 (SB, 66%) Glu37-Lys622 (SB, 19%) Asp38-Lys622 (SB, 91%) Lys41-Asp616 (SB, 17%) Ala55O-Arg350 (HB, 67%) Glu57-Arg350 (SB, 76%) Thr79-Glu619 (HB, 5%) Asn111O-Arg629 (HB, 5%) His128O-Asn588 (HB, 30%) Asp129-Arg235 (SB, 96%) Lys159-Asp273 (SB, 63%) Lys162-Asp273 (SB, 89%) Glu280-Arg413 (SB, 67%) Lys286-Asp412 (SB, 11%) Glu505-Lys914 (SB, 71%) Glu505O-Lys939 (HB, 43%) Asp512-Lys971 (SB, 86%) Glu565-Lys1132 (SB, 34%) |
| 7 198/202 | Gln33, Pro35, Phe36, Glu37, Asp38, Lys41, Thr68, Asp72, Thr79, Leu80, Arg81, Glu82, Thr83, Tyr84, Glu86, Asp89, Ala92, Lys93, Gln94, Pro113, Arg114, Val116, Arg117, Glu119, Thr125, Asp129, Lys137, Arg144, Ala569 | Arg240, Thr478, Asn480, Asp605, Arg676, Lys693, Lys689, Tyr789, Asp791, Asp794, Arg797, Ser798, Thr802, Pro803, Ser804, Glu806, Gln807, Glu810, Arg811, Gln814, Glu815, Gln817, Pro818, Arg1046, Asp1049, Ser1051, Lys1067, Thr1120, Pro1193, Tyr1195 | Gln33O-Ser804 (HB, 80%) Cys34O-Gln807 (HB, 27%) Phe36HN-Glu806 (SB, 22%) Glu37-Arg1046 (SB, 83%) Asp38-Arg797 (SB, 98%) Lys41-Asp1049 (SB, 99%) Thr79-Asp794 (HB, 6%) Arg81-Asp605 (SB, 85%) Glu82-Lys689 (SB, 61%) Thr83-Ser798 (HB, 23%) Glu86-Arg240 (SB, 98%) Asp89-Arg240 (SB, 80%) Glu97-Thr478HN (HB, 36%) Arg114-Glu815 (SB, 39%) Arg117O-Gln814 (HB, 46%) Asp129-Lys1067 (SB, 48%) Lys137-Glu810 (SB, 68%) Lys205-Glu596 (SB, 11%) Asp512-Arg1137 (SB, 7%) Glu565-Lys1132 (SB, 6%) |
| 6 120/90 | Asn130, Glu131, Glu132, Lys159, Arg160, Lys162, Thr166, Glu167, Gln170, Lys174, Lys181, Glu184, Leu185, Glu277, Glu280, Pro282, Lys432, Lys439 | Asn45, Ile46, Thr47, Asn117, Arg120, Ile121, Thr124, Lys126, Asn131, Lys132, Thr133, Ala134, Thr135, Gly325, Glu327, Arg350 | Lys159-Glu327 (SB, 74%) Lys162-Ile46O (HB, 58%) Glu167-Lys126 (SB, 94%) Glu184-Lys132 (SB, 37%) Glu277-Thr144 (HB, 7%) Glu277-Arg350 (SB, 44%) Glu280-Arg350 (SB, 74%) Glu297-Arg236 (SB, 8%) Tyr334-Glu626 (HB, 14%) Tyr334-Arg629 (HB, 5%) Tyr341-Glu626 (HB, 16%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belinskaia, D.A.; Shestakova, N.N.; Samodurova, K.V.; Goncharov, N.V. Computational Study of Molecular Mechanism for the Involvement of Human Serum Albumin in the Renin–Angiotensin–Aldosterone System. Int. J. Mol. Sci. 2024, 25, 10260. https://doi.org/10.3390/ijms251910260
Belinskaia DA, Shestakova NN, Samodurova KV, Goncharov NV. Computational Study of Molecular Mechanism for the Involvement of Human Serum Albumin in the Renin–Angiotensin–Aldosterone System. International Journal of Molecular Sciences. 2024; 25(19):10260. https://doi.org/10.3390/ijms251910260
Chicago/Turabian StyleBelinskaia, Daria A., Natalia N. Shestakova, Kamila V. Samodurova, and Nikolay V. Goncharov. 2024. "Computational Study of Molecular Mechanism for the Involvement of Human Serum Albumin in the Renin–Angiotensin–Aldosterone System" International Journal of Molecular Sciences 25, no. 19: 10260. https://doi.org/10.3390/ijms251910260