
Delineating Molecular Regulatory of Flavonoids Indicated by Transcriptomic and Metabolomics Analysis during Flower Development in Chrysanthemum morifolium ‘Boju’

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
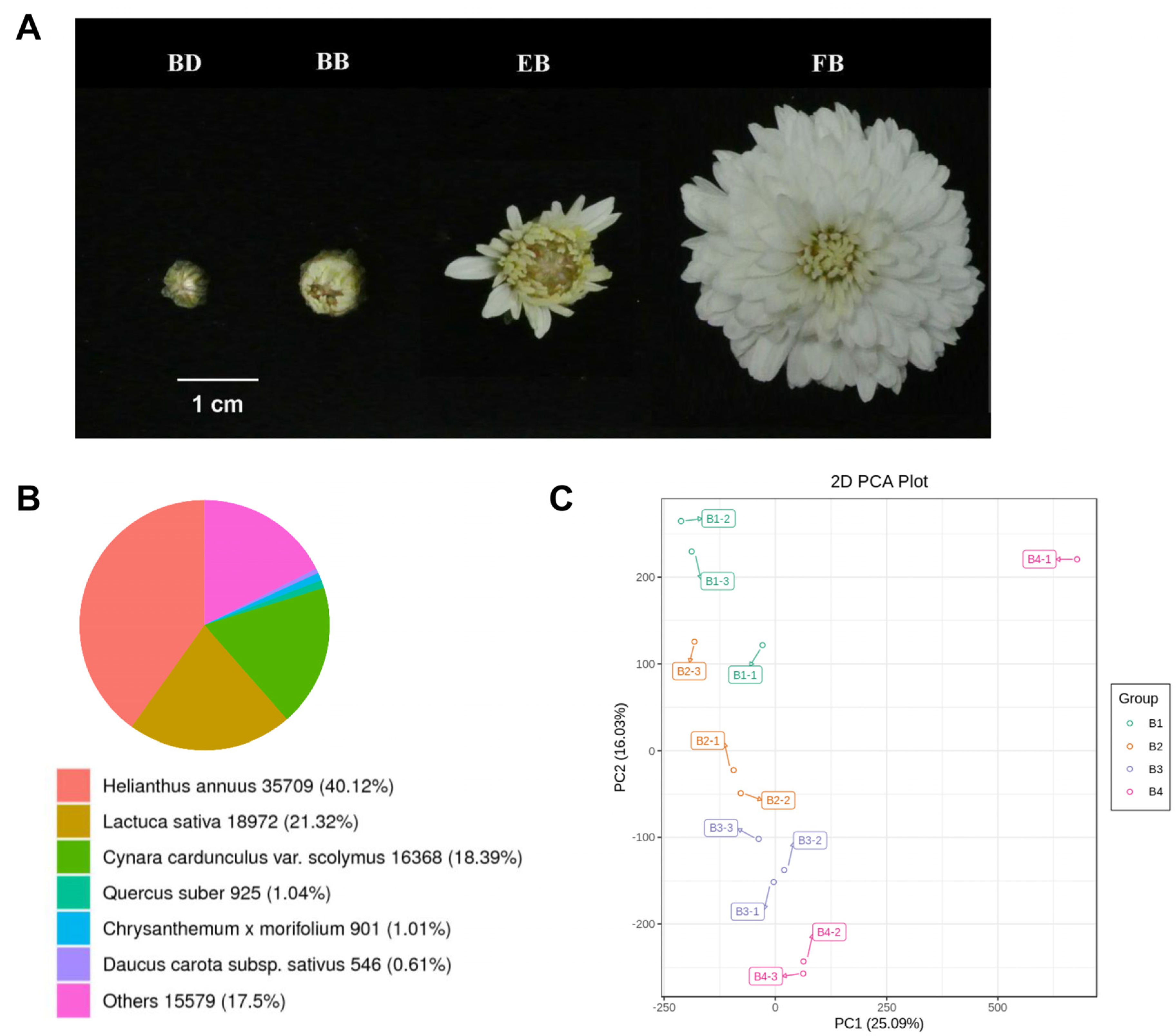
2.1. Transcriptomic Landscape of Flower of C. morifolium at Different Four Stages
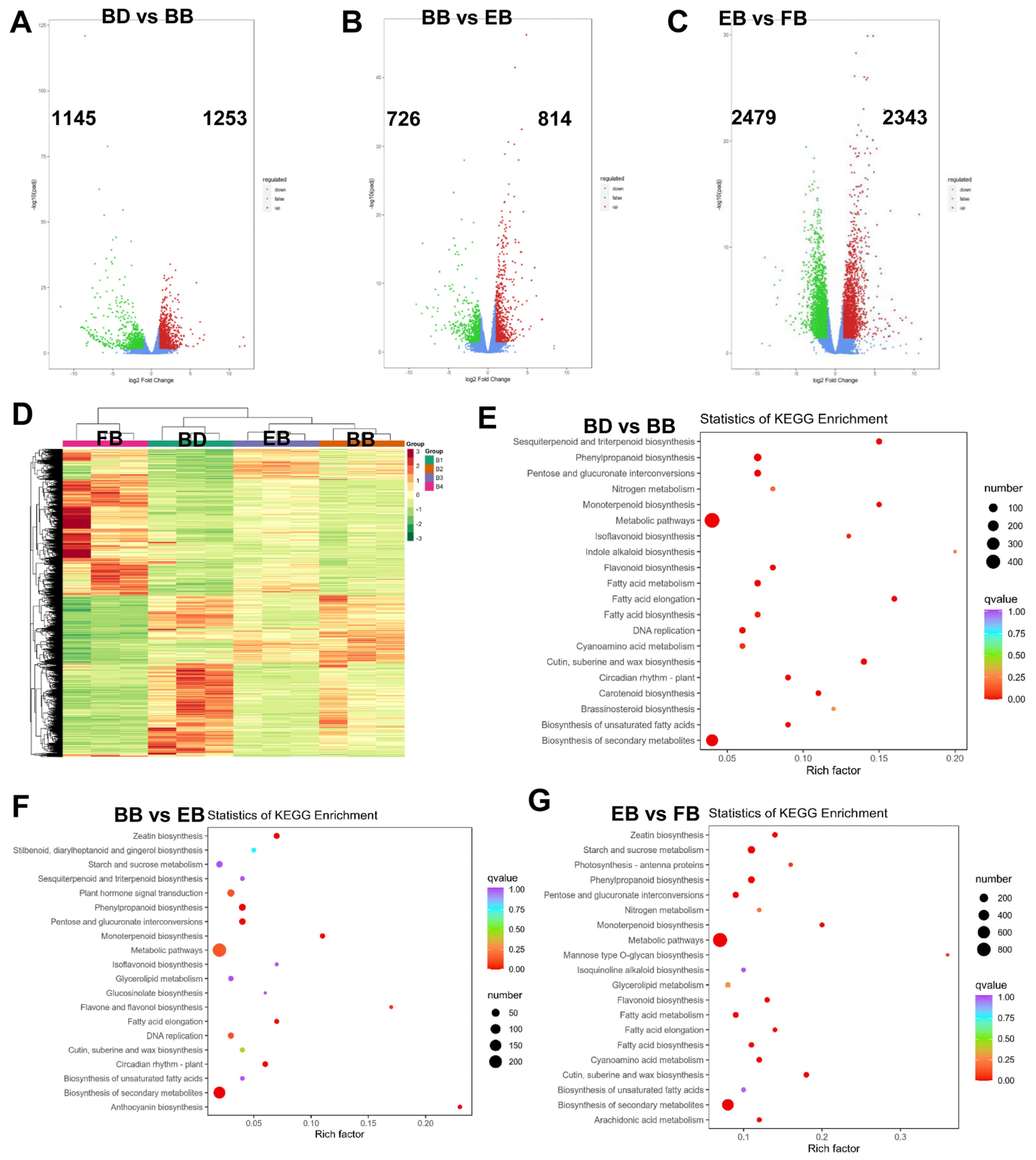
2.2. Flowers of C. morifolium Exhibit Differential Transcriptomic Profiles at Different Four Stages
2.3. Identification of Common Differentially Expressed Unigenes
2.4. Quality Control of the Flavonoid-Targeted Metabolome across the Four Stages of Flower Development
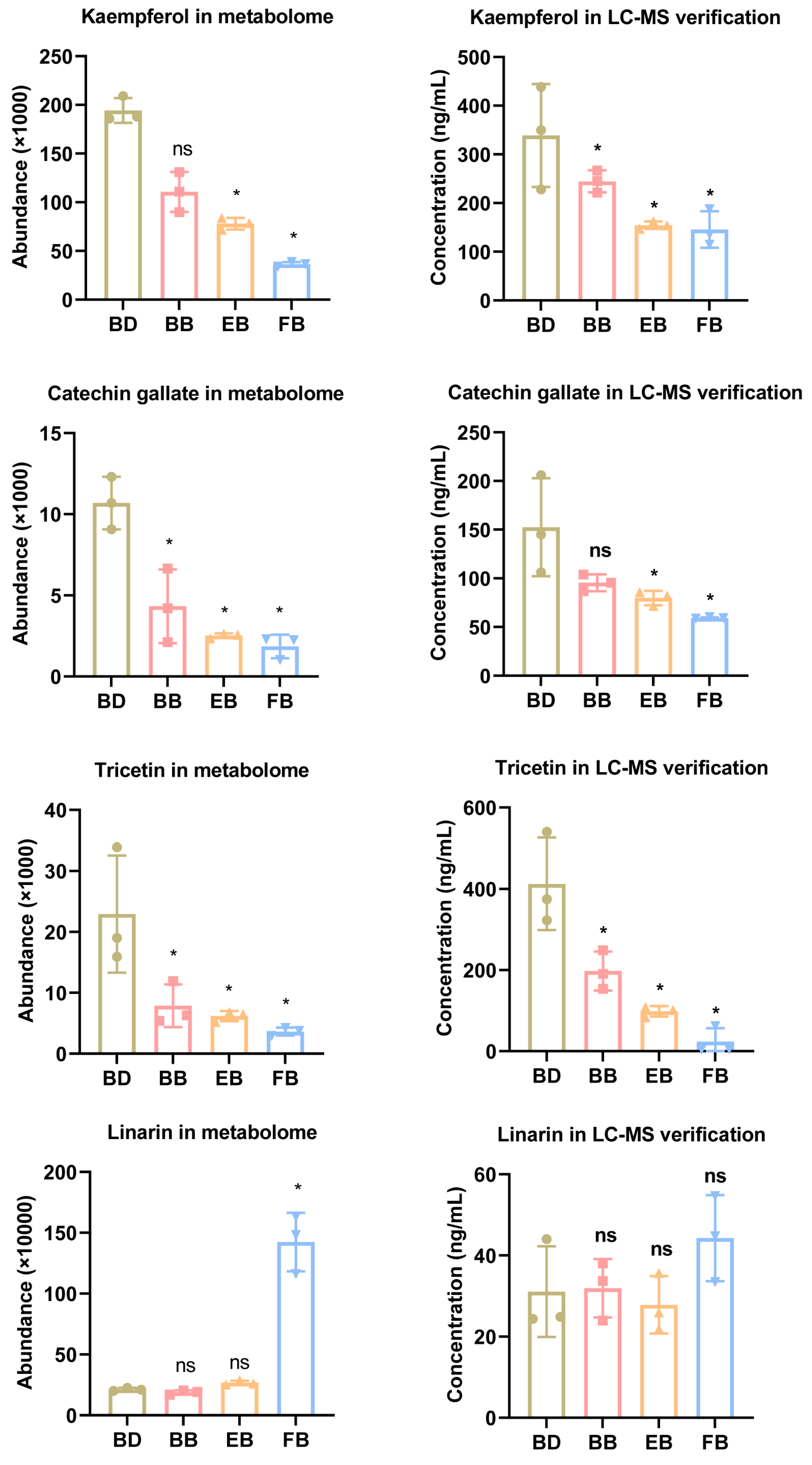
2.5. Metabonomics Landscape of Flavonoid Compounds in the Flowers Undergoes Significant Changes, Particularly in the Early and Mature Stages
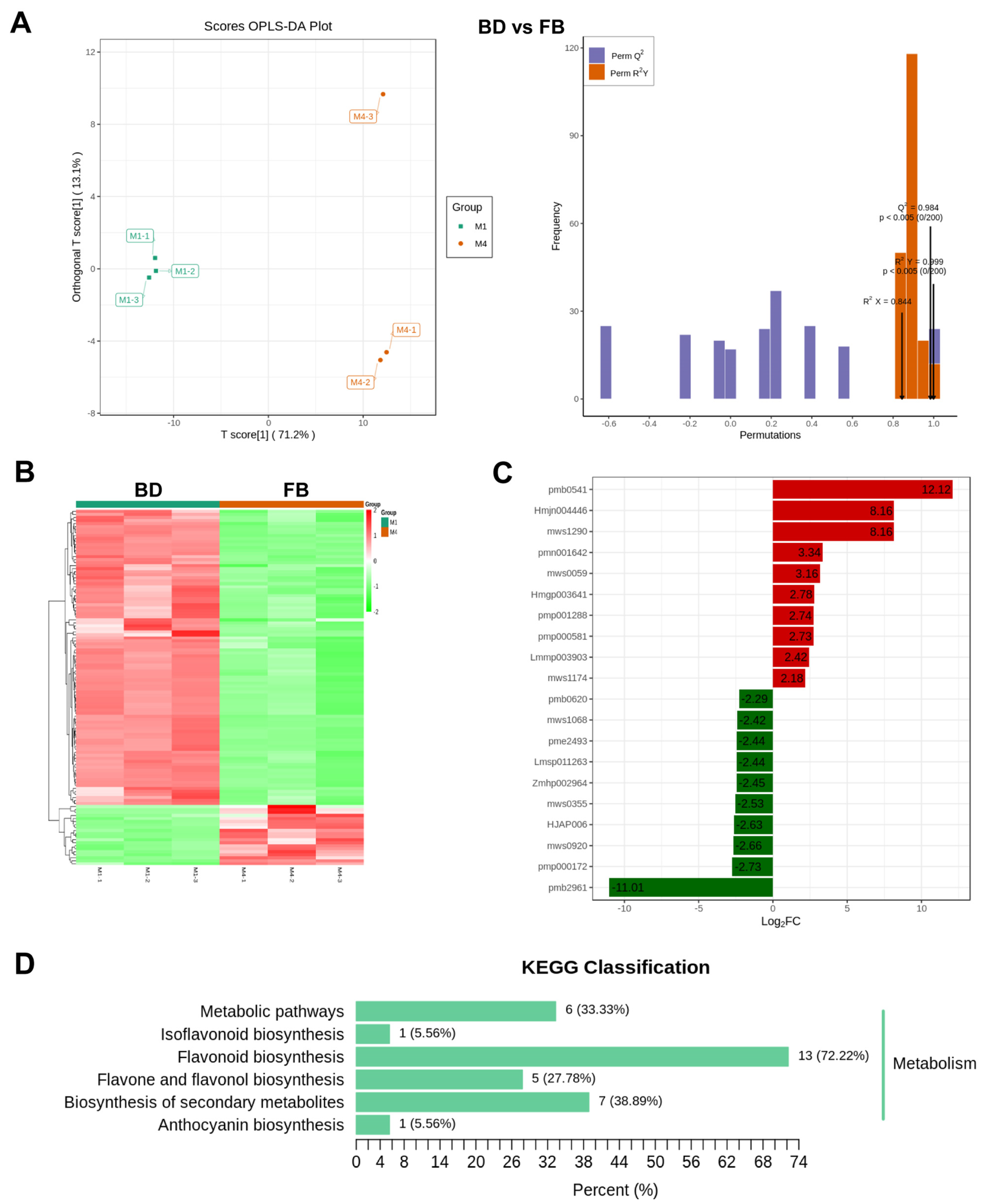
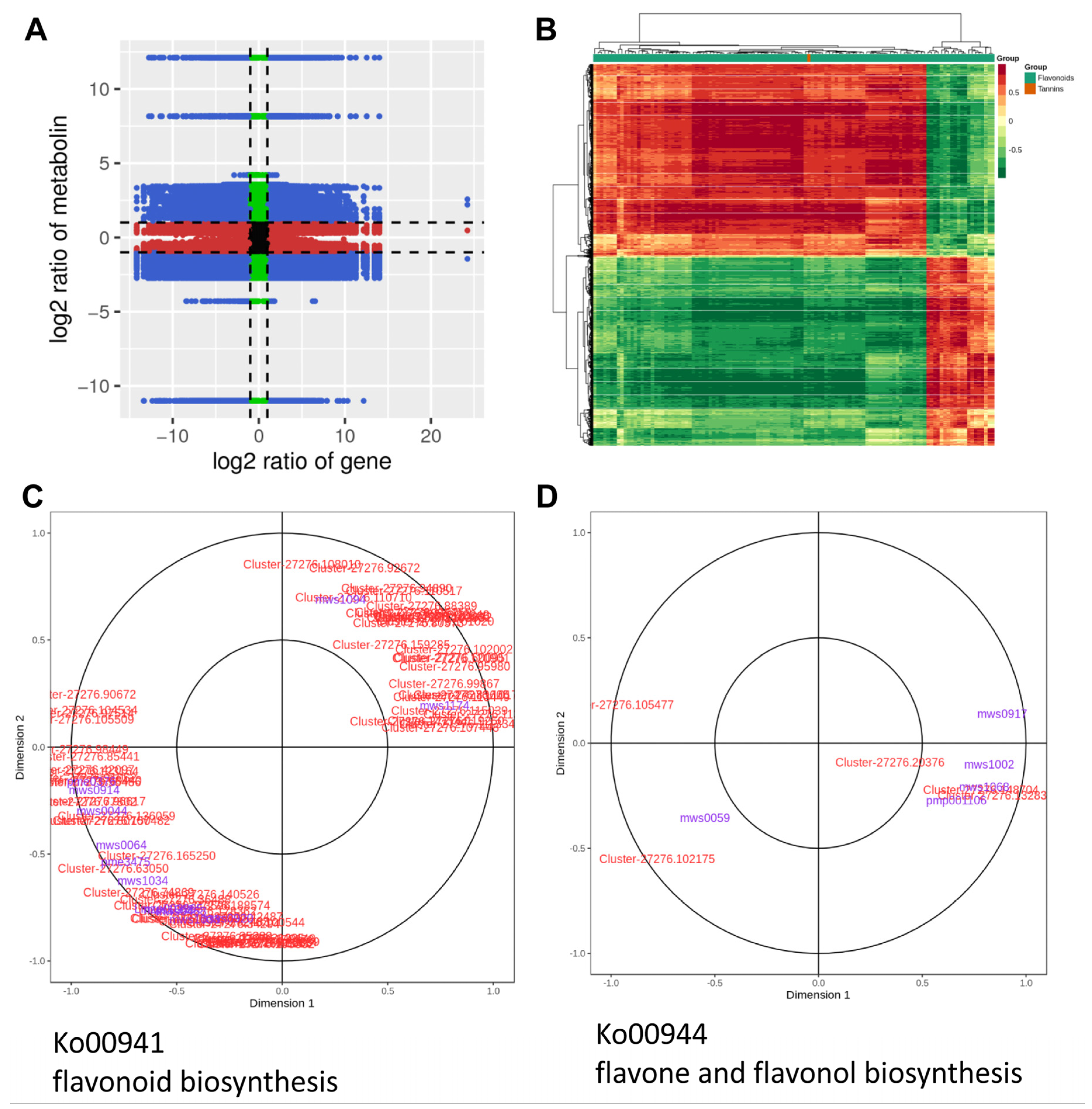
2.6. Integrative Analyses of Transcriptome and Metabolome Reveal the Molecular Mechanism Underlying Variation in Flavonoid during Flower Development of C. morifolium
3. Discussion
4. Materials and Methods
4.1. Plant Material and Sample Collection
4.2. RNA Isolation and Transcriptome Sequencing
4.3. Flavonoid-Targeted Metabolomics Analysis
4.3.1. Flower Sample Extraction
4.3.2. UPLC Conditions
4.3.3. ESI-Q TRAP-MS/MS
4.4. Integrated Analysis of Transcriptomic and Metabolomic Data
4.5. Metabolite Confirmation Using LC-MS
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, X.; Xu, H.; Shang, Y.; Zhu, R.; Hong, X.; Song, Z.; Yang, Z. Development of the general chapters of the Chinese Pharmacopoeia 2020 edition: A review. J. Pharm. Anal. 2021, 11, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Shahrajabian, M.H.; Sun, W.; Zandi, P.; Cheng, Q. A Review of Chrysanthemum, the Eastern Queen in Traditional Chinese Medicine with Healing Power in Modern Pharmaceutical Sciences. Appl. Ecol. Environ. Res. 2019, 17, 13355–13369. [Google Scholar] [CrossRef]
- Hou, X.; Huang, X.; Li, J.; Jiang, G.; Shen, G.; Li, S.; Luo, Q.; Wu, H.; Li, M.; Liu, X.; et al. Extraction Optimization and Evaluation of the Antioxidant and α-Glucosidase Inhibitory Activity of Polysaccharides from Chrysanthemum morifolium cv. Hangju. Antioxidants 2020, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Sun, Y.; Li, D.; Chen, Y. Chrysanthemum indicum L.: A Comprehensive Review of its Botany, Phytochemistry and Pharmacology. Am. J. Chin. Med. 2020, 48, 871–897. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.-L.; Hua, S.; Ye, J.-H.; Zheng, X.-Q.; Liang, Y.-R. Flavonoids and volatiles in Chrysanthemum morifolium Ramat flower from Tongxiang County in China. Afr. J. Biotechnol. 2010, 9, 3817–3821. [Google Scholar]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- Yue, J.; Zhu, C.; Zhou, Y.; Niu, X.; Miao, M.; Tang, X.; Chen, F.; Zhao, W.; Liu, Y. Transcriptome analysis of differentially expressed unigenes involved in flavonoid biosynthesis during flower development of Chrysanthemum morifolium ‘Chuju’. Sci. Rep. 2018, 8, 13414. [Google Scholar] [CrossRef]
- Wang, T.; Zou, Q.; Guo, Q.; Yang, F.; Wu, L.; Zhang, W. Widely Targeted Metabolomics Analysis Reveals the Effect of Flooding Stress on the Synthesis of Flavonoids in Chrysanthemum morifolium. Molecules 2019, 24, 3695. [Google Scholar] [CrossRef]
- Alseekh, S.; Perez de Souza, L.; Benina, M.; Fernie, A.R. The style and substance of plant flavonoid decoration; towards defining both structure and function. Phytochemistry 2020, 174, 112347. [Google Scholar] [CrossRef]
- Wen, W.; Alseekh, S.; Fernie, A.R. Conservation and diversification of flavonoid metabolism in the plant kingdom. Curr. Opin. Plant Biol. 2020, 55, 100–108. [Google Scholar] [CrossRef]
- De Vries, S.; Fürst-Jansen, J.M.R.; Irisarri, I.; Dhabalia Ashok, A.; Ischebeck, T.; Feussner, K.; Abreu, I.N.; Petersen, M.; Feussner, I.; de Vries, J. The evolution of the phenylpropanoid pathway entailed pronounced radiations and divergences of enzyme families. Plant J. 2021, 107, 975–1002. [Google Scholar] [CrossRef] [PubMed]
- Dadras, A.; Rieseberg, T.P.; Zegers, J.M.S.; Fürst-Jansen, J.M.R.; Irisarri, I.; de Vries, J.; de Vries, S. Accessible versatility underpins the deep evolution of plant specialized metabolism. Phytochem. Rev. 2023. [Google Scholar] [CrossRef]
- Yu, S.; Yang, F.; Pu, J.; Guo, Q.; Zou, Q.; Zhang, H.; Liu, M.; Zheng, Y.; Wang, T. Integrated transcriptomic and metabolomic analyses reveal the characteristics of volatile oils in Chrysanthemum morifolium. Sci. Hortic. 2023, 315, 111983. [Google Scholar] [CrossRef]
- Zhu, L.; Liao, J.; Liu, Y.; Zhou, C.; Wang, X.; Hu, Z.; Huang, B.; Zhang, J. Integrative metabolome and transcriptome analyses reveal the molecular mechanism underlying variation in floral scent during flower development of Chrysanthemum indicum var. aromaticum. Front. Plant Sci. 2022, 13, 919151. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Wang, T.; Guo, Q.; Yang, F.; Chen, J.; Zhang, W. Combined metabolomic and transcriptomic analysis reveals redirection of the phenylpropanoid metabolic flux in different colored medicinal Chrysanthemum morifolium. Ind. Crop. Prod. 2021, 164, 113343. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, H.; Jiang, X.; Zhang, L.; Qin, Y. Integrated physiological, transcriptomic, and metabolomic analyses reveal that low-nitrogen conditions improve the accumulation of flavonoids in snow chrysanthemum. Ind. Crop. Prod. 2023, 197, 116574. [Google Scholar] [CrossRef]
- Kanani, M.; Chamani, E.; Shokouhian, A.A.; Torabi-Giglou, M. Plant secondary metabolism and flower color changes in damask rose at different flowering development stages. Acta Physiol. Plant. 2021, 43, 55. [Google Scholar] [CrossRef]
- Liu, J.-C.; Jiao, Z.-G.; Yang, W.-B.; Zhang, C.-L.; Liu, H.; Lv, Z.-Z. Variation in Phenolics, Flavanoids, Antioxidant and Tyrosinase Inhibitory Activity of Peach Blossoms at Different Developmental Stages. Molecules 2015, 20, 20460–20472. [Google Scholar] [CrossRef]
- Chen, W.; Xiao, Z.; Wang, Y.; Wang, J.; Zhai, R.; Lin-Wang, K.; Espley, R.; Ma, F.; Li, P. Competition between anthocyanin and kaempferol glycosides biosynthesis affects pollen tube growth and seed set of Malus. Hortic. Res. 2021, 8, 173. [Google Scholar] [CrossRef]
- Wee, S.P.; Loh, K.E.; Tan, S.P.; Ong, G.H. Interaction between Apigenin, Kaempferol and Trans-Cinnamic Acid: The Compounds Contributed to the Xanthine Oxidase Inhibitory Activity of Chrysanthemum morifolium. In Proceedings of the 2023 13th International Conference on Biomedical Engineering and Technology, Tokyo, Japan, 15–18 June 2023; pp. 250–255. [Google Scholar]
- Wee, S.P.; Loh, K.E.; Lam, K.W.; Ismail, I.S. A Study of the Interaction between Xanthine Oxidase and Its Inhibitors from Chrysanthemum morifolium Using Computational Simulation and Multispectroscopic Methods. Metabolites 2023, 13, 113. [Google Scholar] [CrossRef]
- Xu, Z.; Nambara, E. Family Members and Their Individual Roles: An Arabidopsis Arogenate Dehydratase ADT2 and its Role in Seed Development. Plant Cell Physiol. 2018, 59, 2395–2397. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-G.; Kim, H.-j.; Kim, J.; Lim, Y.; Ahn, J.H. Synthesis of Ermanin, 5,7-Dihydroxy-3,4′-dimethoxyflavone from Kaempferol, 3,5,7,4′-Tetrahydroxyflavone with Two O-Methyltransferases Expressed in E. coli. Bull. Korean Chem. Soc. 2006, 27, 357–358. [Google Scholar] [CrossRef]
- Jianyu, L.; Ahmad, N.; Yingqi, H.; Wenkai, W.; Haiyan, L. Unprecedented Evolution and Transcriptional Reprogramming of CYP81E Subfamily in Carthamus Tinctorius during Flavonoid Biosynthesis. 2020. Available online: https://www.researchgate.net/publication/346761856_Unprecedented_evolution_and_transcriptional_reprogramming_of_CYP81E_subfamily_in_Carthamus_tinctorius_during_flavonoid_biosynthesis (accessed on 17 September 2024). [CrossRef]
- Tholl, D.; Lee, S. Terpene Specialized Metabolism in Arabidopsis thaliana. Arab. Book 2011, 9, e0143. [Google Scholar] [CrossRef]
- Tounekti, T.; Hernández, I.; Munné-Bosch, S. Salicylic Acid Biosynthesis and Role in Modulating Terpenoid and Flavonoid Metabolism in Plant Responses to Abiotic Stress. In Salicylic Acid: Plant Growth and Development; Hayat, S., Ahmad, A., Alyemeni, M.N., Eds.; Springer: Dordrecht, The Netherlands, 2013; pp. 141–162. [Google Scholar]
- Sugimoto, K.; Zager, J.J.; Aubin, B.S.; Lange, B.M.; Howe, G.A. Flavonoid deficiency disrupts redox homeostasis and terpenoid biosynthesis in glandular trichomes of tomato. Plant Physiol. 2021, 188, 1450–1468. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, Z.; Xiong, R.; Peng, X.; Cui, G.; Dong, L.; Li, W. Delineating Molecular Regulatory of Flavonoids Indicated by Transcriptomic and Metabolomics Analysis during Flower Development in Chrysanthemum morifolium ‘Boju’. Int. J. Mol. Sci. 2024, 25, 10261. https://doi.org/10.3390/ijms251910261
Chu Z, Xiong R, Peng X, Cui G, Dong L, Li W. Delineating Molecular Regulatory of Flavonoids Indicated by Transcriptomic and Metabolomics Analysis during Flower Development in Chrysanthemum morifolium ‘Boju’. International Journal of Molecular Sciences. 2024; 25(19):10261. https://doi.org/10.3390/ijms251910261
Chicago/Turabian StyleChu, Zhuannan, Rui Xiong, Xingxing Peng, Guangsheng Cui, Ling Dong, and Weiwen Li. 2024. "Delineating Molecular Regulatory of Flavonoids Indicated by Transcriptomic and Metabolomics Analysis during Flower Development in Chrysanthemum morifolium ‘Boju’" International Journal of Molecular Sciences 25, no. 19: 10261. https://doi.org/10.3390/ijms251910261