
Lysine 473 Regulates the Progression of SLC7A11, the Cystine/Glutamate Exchanger, through the Secretory Pathway


Abstract
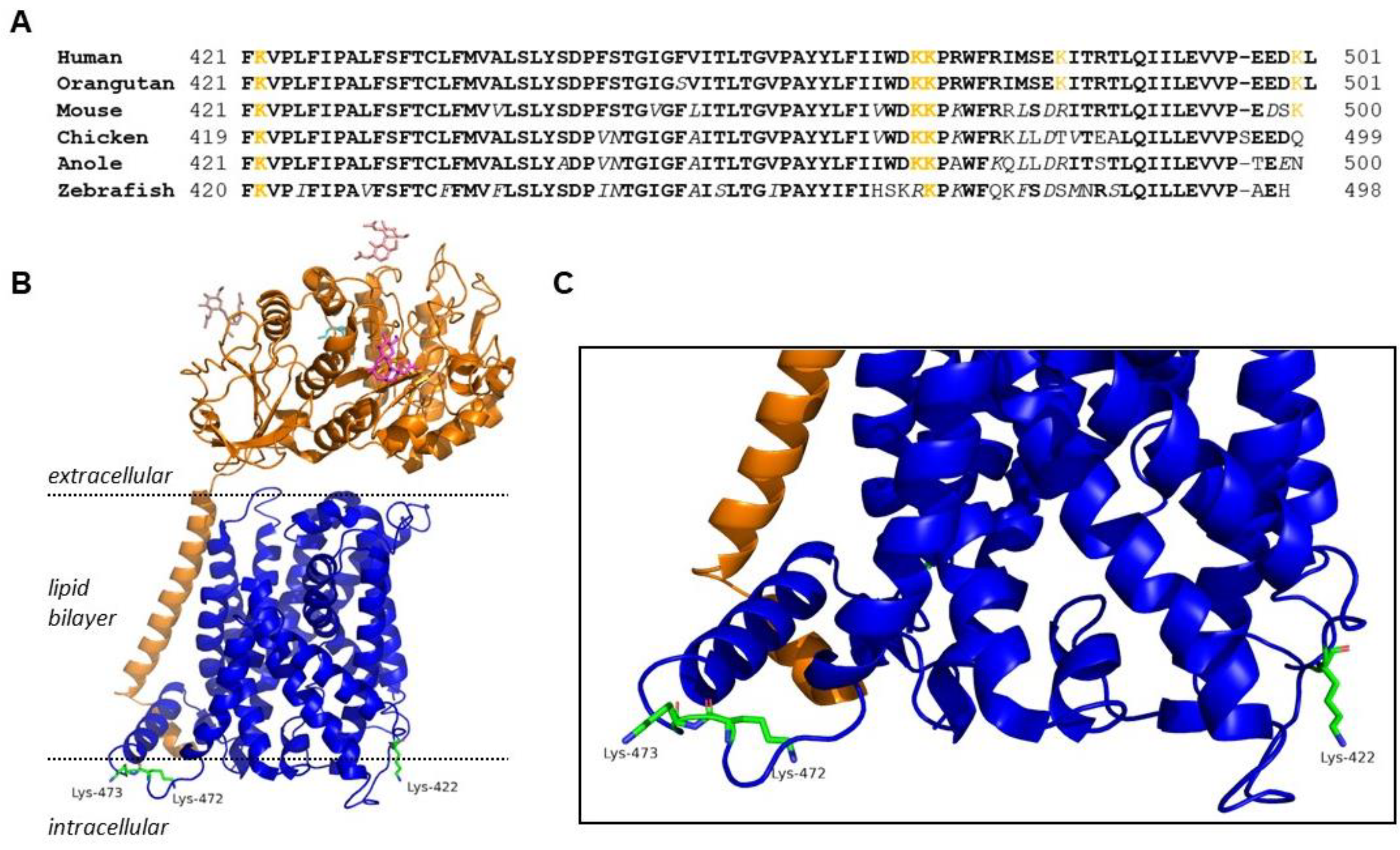
:1. Introduction
2. Results
2.1. Effect of C-Terminal Lysine Mutations on System xc− Activity and Molecular Weight of xCT
2.2. K473R Affects xCT Localization to the Plasma Membrane
2.3. K473R Does Not Impact the Ubiquitination of the Monomer Form of xCT
2.4. Neddylation Status of xCT
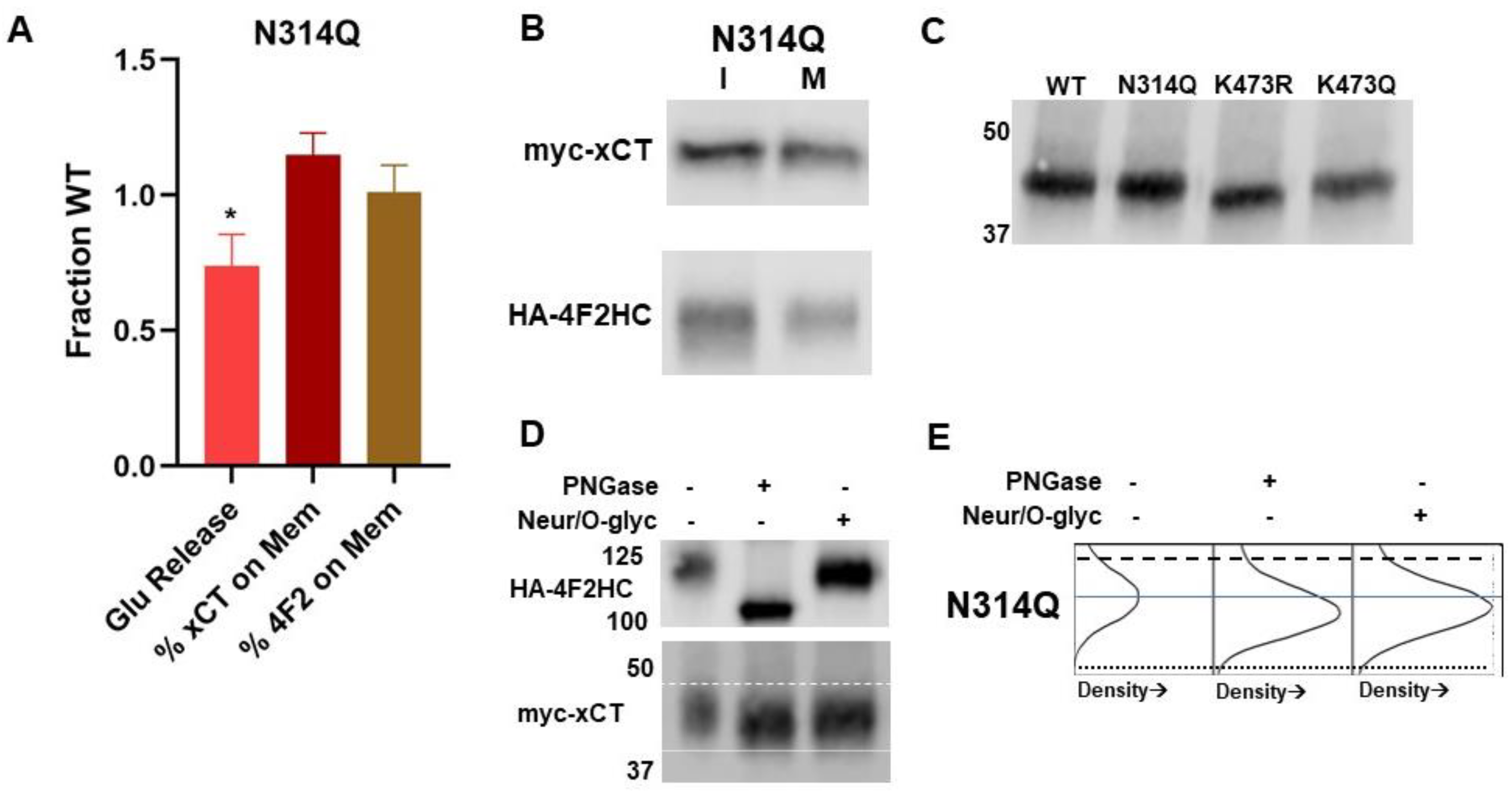
2.5. Glycosylation Status of xCT and 4F2HC
2.6. Activity and Glycosylation Status of K473Q, an Acetylation Mimic Mutant of xCT
2.7. Evaluation of Putative Glycosylation Site N314
2.8. xCT Localization in ER and Golgi Apparatus
2.9. Effect of K473 Mutation on Association of xCT with 4F2HC
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Human myc-xCT and HA-4F2HC Constructs
4.3. Creation of Mutants Using Site-Directed Mutagenesis
4.4. Heterologous Expression of Human myc-xCT and HA-4F2HC in COS-7 Cells
4.5. Glutamate Oxidase and HRP Assay for Measuring xCT Activity
4.6. Immunoprecipitation for Isolating myc-xCT
4.7. Biotinylation of Cell Surface Proteins
4.8. PNGase F and EndoH to Cleave N-Linked Oligosaccharides
4.9. O-Glycosidase to Cleave O-Linked Oligosaccharides
4.10. Western Blot Analysis
4.11. Immunocytochemistry
4.12. Immunocytochemistry Co-Localization Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chase, L.A.; VerHeulen Kleyn, M.; Schiller, N.; King, A.G.; Flores, G.; Engelsman, S.B.; Bowles, C.; Smith, S.L.; Robinson, A.E.; Rothstein, J. Hydrogen Peroxide Triggers an Increase in Cell Surface Expression of System Xc− in Cultured Human Glioma Cells. Neurochem. Int. 2020, 134, 104648. [Google Scholar] [CrossRef] [PubMed]
- Bridges, R.; Lutgen, V.; Lobner, D.; Baker, D.A. Thinking Outside the Cleft to Understand Synaptic Activity: Contribution of the Cystine-Glutamate Antiporter (System Xc−) to Normal and Pathological Glutamatergic Signaling. Pharmacol. Rev. 2012, 64, 780–802. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Black, J.L. The Voltage-Gated Calcium Channel Gamma Subunits: A Review of the Literature. J. Bioenerg. Biomembr. 2003, 35, 649–660. [Google Scholar] [CrossRef]
- Miranda, M.; Dionne, K.R.; Sorkina, T.; Sorkin, A. Three Ubiquitin Conjugation Sites in the Amino Terminus of the Dopamine Transporter Mediate Protein Kinase C–Dependent Endocytosis of the Transporter. Mol. Biol. Cell 2007, 18, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Alvi, F.; Idkowiak-Baldys, J.; Baldys, A.; Raymond, J.R.; Hannun, Y.A. Regulation of Membrane Trafficking and Endocytosis by Protein Kinase C: Emerging Role of the Pericentrion, a Novel Protein Kinase C-Dependent Subset of Recycling Endosomes. Cell. Mol. Life Sci. 2007, 64, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Sher, E.; Rosa, P.; Francolini, M.; Codignola, A.; Morlacchi, E.; Taverna, E.; Giovannini, F.; Brioschi, A.; Clementi, F.; McEnery, M.W.; et al. Metabolism and Trafficking of N-Type Voltage-Operated Calcium Channels in Neurosecretory Cells. J. Bioenerg. Biomembr. 1998, 30, 399–407. [Google Scholar] [CrossRef]
- Braithwaite, S.P.; Meyer, G.; Henley, J.M. Interactions between AMPA Receptors and Intracellular Proteins. Neuropharmacology 2000, 39, 919–930. [Google Scholar] [CrossRef]
- Marchand, S.; Cartaud, J. Targeted Trafficking of Neurotransmitter Receptors to Synaptic Sites. Mol. Neurobiol. 2002, 26, 117–135. [Google Scholar] [CrossRef]
- Shepherd, J.D.; Huganir, R.L. The Cell Biology of Synaptic Plasticity: AMPA Receptor Trafficking. Annu. Rev. Cell Dev. Biol. 2007, 23, 613–643. [Google Scholar] [CrossRef]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein—Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef] [PubMed]
- Rotin, D.; Staub, O. Function and Regulation of the Epithelial Na+ Channel ENaC. Compr. Physiol. 2021, 11, 2017–2045. [Google Scholar] [CrossRef] [PubMed]
- Bannai, S.; Kitamura, E. Adaptive Enhancement of Cystine and Glutamate Uptake in Human Diploid Fibroblasts in Culture. Biochim. Biophys. Acta 1982, 721, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Habib, E.; Linher-Melville, K.; Lin, H.-X.; Singh, G. Expression of xCT and Activity of System Xc− Are Regulated by NRF2 in Human Breast Cancer Cells in Response to Oxidative Stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef]
- Liu, X.; Albano, R.; Lobner, D. FGF-2 Induces Neuronal Death through Upregulation of System Xc−. Brain Res. 2014, 1547, 25–33. [Google Scholar] [CrossRef]
- Albrecht, P.; Lewerenz, J.; Dittmer, S.; Noack, R.; Maher, P.; Methner, A. Mechanisms of Oxidative Glutamate Toxicity: The Glutamate/Cystine Antiporter System Xc− as a Neuroprotective Drug Target. CNS Neurol. Disord. Drug Targets 2010, 9, 373–382. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P.; Methner, A. Regulation of xCT Expression and System Xc− function in Neuronal Cells. Amino Acids 2012, 42, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The Cystine/Glutamate Antiporter System Xc− in Health and Disease: From Molecular Mechanisms to Novel Therapeutic Opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Verrey, F.; Closs, E.I.; Wagner, C.A.; Palacin, M.; Endou, H.; Kanai, Y. CATs and HATs: The SLC7 Family of Amino Acid Transporters. Pflüg. Arch. 2004, 447, 532–542. [Google Scholar] [CrossRef]
- Chillarón, J.; Roca, R.; Valencia, A.; Zorzano, A.; Palacín, M. Heteromeric Amino Acid Transporters: Biochemistry, Genetics, and Physiology. Am. J. Physiol. Renal Physiol. 2001, 281, F995–F1018. [Google Scholar] [CrossRef]
- Palacín, M.; Kanai, Y. The Ancillary Proteins of HATs: SLC3 Family of Amino Acid Transporters. Pflüg. Arch. 2004, 447, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and Expression of a Plasma Membrane Cystine/Glutamate Exchange Transporter Composed of Two Distinct Proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef] [PubMed]
- Gasol, E.; Jiménez-Vidal, M.; Chillarón, J.; Zorzano, A.; Palacín, M. Membrane Topology of System Xc− Light Subunit Reveals a Re-Entrant Loop with Substrate-Restricted Accessibility. J. Biol. Chem. 2004, 279, 31228–31236. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Li, Y.; Shi, Y.; Zhou, J.; Lei, J.; Huang, J.; Zhou, Q. Cryo-EM Structure of the Human Heteromeric Amino Acid Transporter B0,+AT-rBAT. Sci. Adv. 2020, 6, eaay6379. [Google Scholar] [CrossRef]
- Chubb, S.; Kingsland, A.L.; Bröer, A.; Bröer, S. Mutation of the 4F2 Heavy-Chain Carboxy Terminus Causes y+LAT2 Light-Chain Dysfunction. Mol. Membr. Biol. 2006, 23, 255–267. [Google Scholar] [CrossRef]
- Console, L.; Scalise, M.; Tarmakova, Z.; Coe, I.R.; Indiveri, C. N-Linked Glycosylation of Human SLC1A5 (ASCT2) Transporter Is Critical for Trafficking to Membrane. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2015, 1853, 1636–1645. [Google Scholar] [CrossRef]
- Devés, R.; Boyd, C.A. Surface Antigen CD98(4F2): Not a Single Membrane Protein, but a Family of Proteins with Multiple Functions. J. Membr. Biol. 2000, 173, 165–177. [Google Scholar] [CrossRef]
- Gu, Y.; Albuquerque, C.P.; Braas, D.; Zhang, W.; Villa, G.R.; Bi, J.; Ikegami, S.; Masui, K.; Gini, B.; Yang, H.; et al. mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine-Glutamate Antiporter xCT. Mol. Cell 2017, 67, 128–138.e7. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Y.; Shen, N.; Liu, Y.; Xu, X.; Zhu, R.; Jiang, H.; Wu, X.; Wei, Y.; Tang, J. AMPKα1-Mediated ZDHHC8 Phosphorylation Promotes the Palmitoylation of SLC7A11 to Facilitate Ferroptosis Resistance in Glioblastoma. Cancer Lett. 2024, 584, 216619. [Google Scholar] [CrossRef]
- Nguyen, H.T.T.; Dalmasso, G.; Yan, Y.; Obertone, T.S.; Sitaraman, S.V.; Merlin, D. Ecto-Phosphorylation of CD98 Regulates Cell-Cell Interactions. PLoS ONE 2008, 3, e3895. [Google Scholar] [CrossRef]
- Guntupalli, S.; Jang, S.E.; Zhu, T.; Huganir, R.L.; Widagdo, J.; Anggono, V. GluA1 Subunit Ubiquitination Mediates Amyloid-β-Induced Loss of Surface α-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid (AMPA) Receptors. J. Biol. Chem. 2017, 292, 8186–8194. [Google Scholar] [CrossRef] [PubMed]
- Cartier, E.; Garcia-Olivares, J.; Janezic, E.; Viana, J.; Moore, M.; Lin, M.L.; Caplan, J.L.; Torres, G.; Kim, Y.-H. The SUMO-Conjugase Ubc9 Prevents the Degradation of the Dopamine Transporter, Enhancing Its Cell Surface Level and Dopamine Uptake. Front. Cell. Neurosci. 2019, 13, 35. [Google Scholar] [CrossRef]
- Kang, M.; Lee, D.; Song, J.; Park, S.; Park, D.; Lee, S.; Suh, Y.H. Neddylation Is Required for Presynaptic Clustering of mGlu7 and Maturation of Presynaptic Terminals. Exp. Mol. Med. 2021, 53, 457–467. [Google Scholar] [CrossRef]
- Farrugia, M.A.; Puglielli, L. Nε-Lysine Acetylation in the Endoplasmic Reticulum—A Novel Cellular Mechanism That Regulates Proteostasis and Autophagy. J. Cell Sci. 2018, 131, jcs221747. [Google Scholar] [CrossRef] [PubMed]
- Staub, O. Regulation of Stability and Function of the Epithelial Na+ Channel (ENaC) by Ubiquitination. EMBO J. 1997, 16, 6325–6336. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.L.; Deme, J.C.; Kolokouris, D.; Kuteyi, G.; Biggin, P.C.; Lea, S.M.; Newstead, S. Molecular Basis for Redox Control by the Human Cystine/Glutamate Antiporter System Xc−. Nat. Commun. 2021, 12, 7147. [Google Scholar] [CrossRef]
- Brockmann, M.M.; Döngi, M.; Einsfelder, U.; Körber, N.; Refojo, D.; Stein, V. Neddylation Regulates Excitatory Synaptic Transmission and Plasticity. Sci. Rep. 2019, 9, 17935. [Google Scholar] [CrossRef]
- Fernández-Tejada, A.; Brailsford, J.; Zhang, Q.; Shieh, J.-H.; Moore, M.A.S.; Danishefsky, S.J. Total Synthesis of Glycosylated Proteins. Top. Curr. Chem. 2015, 362, 1–26. [Google Scholar] [CrossRef]
- Weng, T.-Y.; Chiu, W.-T.; Liu, H.-S.; Cheng, H.-C.; Shen, M.-R.; Mount, D.B.; Chou, C.-Y. Glycosylation Regulates the Function and Membrane Localization of KCC4. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2013, 1833, 1133–1146. [Google Scholar] [CrossRef]
- Li, J.V.; Ng, C.-A.; Cheng, D.; Zhou, Z.; Yao, M.; Guo, Y.; Yu, Z.-Y.; Ramaswamy, Y.; Ju, L.A.; Kuchel, P.W.; et al. Modified N-Linked Glycosylation Status Predicts Trafficking Defective Human Piezo1 Channel Mutations. Commun. Biol. 2021, 4, 1038. [Google Scholar] [CrossRef]
- Thomson, R.B.; Thomson, C.L.; Aronson, P.S. N-Glycosylation Critically Regulates Function of Oxalate Transporter SLC26A6. Am. J. Physiol.—Cell Physiol. 2016, 311, C866–C873. [Google Scholar] [CrossRef] [PubMed]
- Kandel, M.B.; Yamamoto, S.; Midorikawa, R.; Morise, J.; Wakazono, Y.; Oka, S.; Takamiya, K. N-Glycosylation of the AMPA-Type Glutamate Receptor Regulates Cell Surface Expression and Tetramer Formation Affecting Channel Function. J. Neurochem. 2018, 147, 730–747. [Google Scholar] [CrossRef] [PubMed]
- Tachida, Y.; Iijima, J.; Takahashi, K.; Suzuki, H.; Kizuka, Y.; Yamaguchi, Y.; Tanaka, K.; Nakano, M.; Takakura, D.; Kawasaki, N.; et al. O-GalNAc Glycosylation Determines Intracellular Trafficking of APP and Aβ Production. J. Biol. Chem. 2023, 299, 104905. [Google Scholar] [CrossRef] [PubMed]
- Costantini, C.; Ko, M.H.; Jonas, M.C.; Puglielli, L. A Reversible Form of Lysine Acetylation in the ER and Golgi Lumen Controls the Molecular Stabilization of BACE1. Biochem. J. 2007, 407, 383–395. [Google Scholar] [CrossRef]
- Mak, A.B.; Pehar, M.; Nixon, A.M.L.; Williams, R.A.; Uetrecht, A.C.; Puglielli, L.; Moffat, J. Post-Translational Regulation of CD133 by ATase1/ATase2 Mediated Lysine Acetylation. J. Mol. Biol. 2014, 426, 2175–2182. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Byambaragchaa, M.; Choi, S.-H.; Kang, H.-J.; Kang, M.-H.; Min, K.-S. Roles of N-Linked and O-Linked Glycosylation Sites in the Activity of Equine Chorionic Gonadotropin in Cells Expressing Rat Luteinizing Hormone/Chorionic Gonadotropin Receptor and Follicle-Stimulating Hormone Receptor. BMC Biotechnol. 2021, 21, 52. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Wiriyasermkul, P.; Jin, C.; Quan, L.; Ohgaki, R.; Okuda, S.; Kusakizako, T.; Nishizawa, T.; Oda, K.; Ishitani, R.; et al. Cryo-EM Structure of the Human L-Type Amino Acid Transporter 1 in Complex with Glycoprotein CD98hc. Nat. Struct. Mol. Biol. 2019, 26, 510–517. [Google Scholar] [CrossRef]
- Console, L.; Scalise, M.; Salerno, S.; Scanga, R.; Giudice, D.; De Bartolo, L.; Tonazzi, A.; Indiveri, C. N-Glycosylation Is Crucial for Trafficking and Stability of SLC3A2 (CD98). Sci. Rep. 2022, 12, 14570. [Google Scholar] [CrossRef]
- Frappaolo, A.; Karimpour-Ghahnavieh, A.; Sechi, S.; Giansanti, M.G. The Close Relationship between the Golgi Trafficking Machinery and Protein Glycosylation. Cells 2020, 9, 2652. [Google Scholar] [CrossRef]
- Stanley, P. Golgi Glycosylation. Cold Spring Harb. Perspect. Biol. 2011, 3, a005199. [Google Scholar] [CrossRef]
- Guo, W.; Shi, L.; Filizola, M.; Weinstein, H.; Javitch, J.A. Crosstalk in G Protein-Coupled Receptors: Changes at the Transmembrane Homodimer Interface Determine Activation. Proc. Natl. Acad. Sci. USA 2005, 102, 17495–17500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zeng, L. Crosstalk between Ubiquitination and Other Post-Translational Protein Modifications in Plant Immunity. Plant Commun. 2020, 1, 100041. [Google Scholar] [CrossRef] [PubMed]
- Leney, A.C.; El Atmioui, D.; Wu, W.; Ovaa, H.; Heck, A.J.R. Elucidating Crosstalk Mechanisms between Phosphorylation and O-GlcNAcylation. Proc. Natl. Acad. Sci. USA 2017, 114, E7255–E7261. [Google Scholar] [CrossRef]
- Dieterich, I.A.; Cui, Y.; Braun, M.M.; Lawton, A.J.; Robinson, N.H.; Peotter, J.L.; Yu, Q.; Casler, J.C.; Glick, B.S.; Audhya, A.; et al. Acetyl-CoA Flux from the Cytosol to the ER Regulates Engagement and Quality of the Secretory Pathway. Sci. Rep. 2021, 11, 2013. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; Lehnus, M.; Karst, A.; Puglielli, L. Proteomic Assessment Shows That Many Endoplasmic Reticulum (ER)-Resident Proteins Are Targeted by Nϵ-Lysine Acetylation in the Lumen of the Organelle and Predicts Broad Biological Impact. J. Biol. Chem. 2012, 287, 22436–22440. [Google Scholar] [CrossRef]
- Okada, A.K.; Teranishi, K.; Ambroso, M.R.; Isas, J.M.; Vazquez-Sarandeses, E.; Lee, J.-Y.; Melo, A.A.; Pandey, P.; Merken, D.; Berndt, L.; et al. Lysine Acetylation Regulates the Interaction between Proteins and Membranes. Nat. Commun. 2021, 12, 6466. [Google Scholar] [CrossRef]
- McMahon, H.T.; Gallop, J.L. Membrane Curvature and Mechanisms of Dynamic Cell Membrane Remodelling. Nature 2005, 438, 590–596. [Google Scholar] [CrossRef]
- Koppin, A. Available online: www.bioRender.com/a26c151 (accessed on 20 March 2023).
- Ding, Y.; Dellisanti, C.D.; Ko, M.H.; Czajkowski, C.; Puglielli, L. The Endoplasmic Reticulum-Based Acetyltransferases, ATase1 and ATase2, Associate with the Oligosaccharyltransferase to Acetylate Correctly Folded Polypeptides. J. Biol. Chem. 2014, 289, 32044–32055. [Google Scholar] [CrossRef]
- Shrimal, S.; Cherepanova, N.A.; Gilmore, R. Cotranslational and Posttranslocational N-Glycosylation of Proteins in the Endoplasmic Reticulum. Semin. Cell Dev. Biol. 2015, 41, 71–78. [Google Scholar] [CrossRef]
- Wang, Z.A.; Cole, P.A. The Chemical Biology of Reversible Lysine Post-Translational Modifications. Cell Chem. Biol. 2020, 27, 953–969. [Google Scholar] [CrossRef]
- Shi, L.; Tu, B.P. Acetyl-CoA and the Regulation of Metabolism: Mechanisms and Consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Herr, D.J.; Singh, T.; Dhammu, T.; Menick, D.R. Regulation of Metabolism by Mitochondrial Enzyme Acetylation in Cardiac Ischemia-Reperfusion Injury. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2020, 1866, 165728. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; Ball, L.E.; Sharma, D.R.; Harlan, B.A.; Comte-Walters, S.; Neely, B.A.; Vargas, M.R. Changes in Protein Expression and Lysine Acetylation Induced by Decreased Glutathione Levels in Astrocytes. Mol. Cell. Proteom. 2016, 15, 493–505. [Google Scholar] [CrossRef]
- Bah, A.; Forman-Kay, J.D. Modulation of Intrinsically Disordered Protein Function by Post-Translational Modifications. J. Biol. Chem. 2016, 291, 6696–6705. [Google Scholar] [CrossRef] [PubMed]
- SLC7A11—Cystine/Glutamate Transporter—Homo Sapiens (Human)|UniProtKB|UniProt. Available online: https://www.uniprot.org/uniprotkb/Q9UPY5/entry#ptm_processing (accessed on 1 August 2024).
- Jensen, P.H.; Kolarich, D.; Packer, N.H. Mucin-Type O-Glycosylation—Putting the Pieces Together. FEBS J. 2010, 277, 81–94. [Google Scholar] [CrossRef]
- Thomas, A.G.; Sattler, R.; Tendyke, K.; Loiacono, K.A.; Hansen, H.; Sahni, V.; Hashizume, Y.; Rojas, C.; Slusher, B.S. High-Throughput Assay Development for Cystine-Glutamate Antiporter (Xc-) Highlights Faster Cystine Uptake than Glutamate Release in Glioma Cells. PLoS ONE 2015, 10, e0127785. [Google Scholar] [CrossRef]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and Quantitative Measurement of Protein-Protein Colocalization in Live Cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant | Forward | Reverse |
|---|---|---|
| K422R | 5′-aacagtggcaccctgaaaggacgatgcatatctggg-3′ | 5′-cccagatatgcatcgtcctttcagggtgccactgtt-3′ |
| K472R | 5′-ttatctctttattatatgggacaggaaacccaggtggtttagaataa-3′ | 5′-aatagagaaataatataccctgttctttgggtccaccaaatcttatt-3′ |
| K473R | 5′-tctaaaccacctgggtctcttgtcccatataataaagagataatac-3′ | 5′-gtattatctctttattatatgggacaagagacccaggtggtttaga-3′ |
| K473Q | 5′-tgacattattctaaaccacctgggctgcttgtcccatataataaagagata-3′ | 5′-tatctctttattatatgggacaagcagcccaggtggtttagaataatgtca-3′ |
| N314Q | 5′-cggaactgctaatgagaactgtcccagtagccgctcaga-3′ | 5′-tctgagcggctactgggacagttctcattagcagttccg-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koppin, A.; Chase, L. Lysine 473 Regulates the Progression of SLC7A11, the Cystine/Glutamate Exchanger, through the Secretory Pathway. Int. J. Mol. Sci. 2024, 25, 10271. https://doi.org/10.3390/ijms251910271
Koppin A, Chase L. Lysine 473 Regulates the Progression of SLC7A11, the Cystine/Glutamate Exchanger, through the Secretory Pathway. International Journal of Molecular Sciences. 2024; 25(19):10271. https://doi.org/10.3390/ijms251910271
Chicago/Turabian StyleKoppin, Anna, and Leah Chase. 2024. "Lysine 473 Regulates the Progression of SLC7A11, the Cystine/Glutamate Exchanger, through the Secretory Pathway" International Journal of Molecular Sciences 25, no. 19: 10271. https://doi.org/10.3390/ijms251910271