
SSCI: Self-Supervised Deep Learning Improves Network Structure for Cancer Driver Gene Identification


Abstract
:1. Introduction
2. Results
2.1. Computational Complexity
2.2. Improved Predictive Performance of SSCI
- GCN [19] is the typical graph neural network that processes graph-structured data by aggregating features from a node’s direct neighbors along with its own features. This aggregation captures the local graph structure, allowing the network to learn node representations enriched with neighborhood information.
- GAT [27] is a method based on GCN that utilizes an attention mechanism to extract node features. The attention mechanism in GAT computes a weight for each neighbor based on the feature vector of the node itself and the feature vectors of the neighbors, which allows GAT to assign different weights to the features of neighboring nodes, resulting in a more nuanced representation that reflects the relative importance of the contribution from each neighbor.
- Chebnet [21] is a variant of GCN that utilizes Chebyshev filters. Chebyshev filters offer the advantage of efficiently capturing the most significant frequencies in graph-structured data, which is particularly useful for modeling large-scale and high-order neighborhood information. Compared to the standard GCN, ChebNet enables a more flexible aggregation of information across broader graph neighborhoods.
- EMOGI [18] leverages pan-cancer multi-omics data and the PPI network to derive more useful gene representations. Consequently, it can identify pan-cancer driver genes more accurately.
- MTGCN [20] integrates biological features and structural features to construct enhanced features for each gene, and it proposes a multi-task learning framework, focusing on optimizing the main task of node prediction and the auxiliary task of link prediction. Meanwhile, the framework incorporates a weight learner to automatically balance the contributions of both tasks. To bolster the model’s generalization and robustness, MTGCN randomly omits a few edges while training.
- SMG [22] addresses the scarcity of labeled data for cancer driver gene identification. It adopts the strategy of EMOGI to construct multi-omics-featured PPI networks and then randomly masks some nodes. Finally, it utilizes a GNN-based autoencoder to reconstruct the masked nodes by referring to the neighborhood information. In this way, SMG effectively captures the complex interaction relationships between nodes while preserving topological information. In the task-specific fine-tuning stage, SMG leverages the pretrained GNN encoder to embed PPI networks into the feature graphs and adopts a task-specific layer to make the prediction.
2.3. Robustness Evaluation
2.4. Analysis of Potential Cancer Driver Genes
2.5. Enrichment Analysis
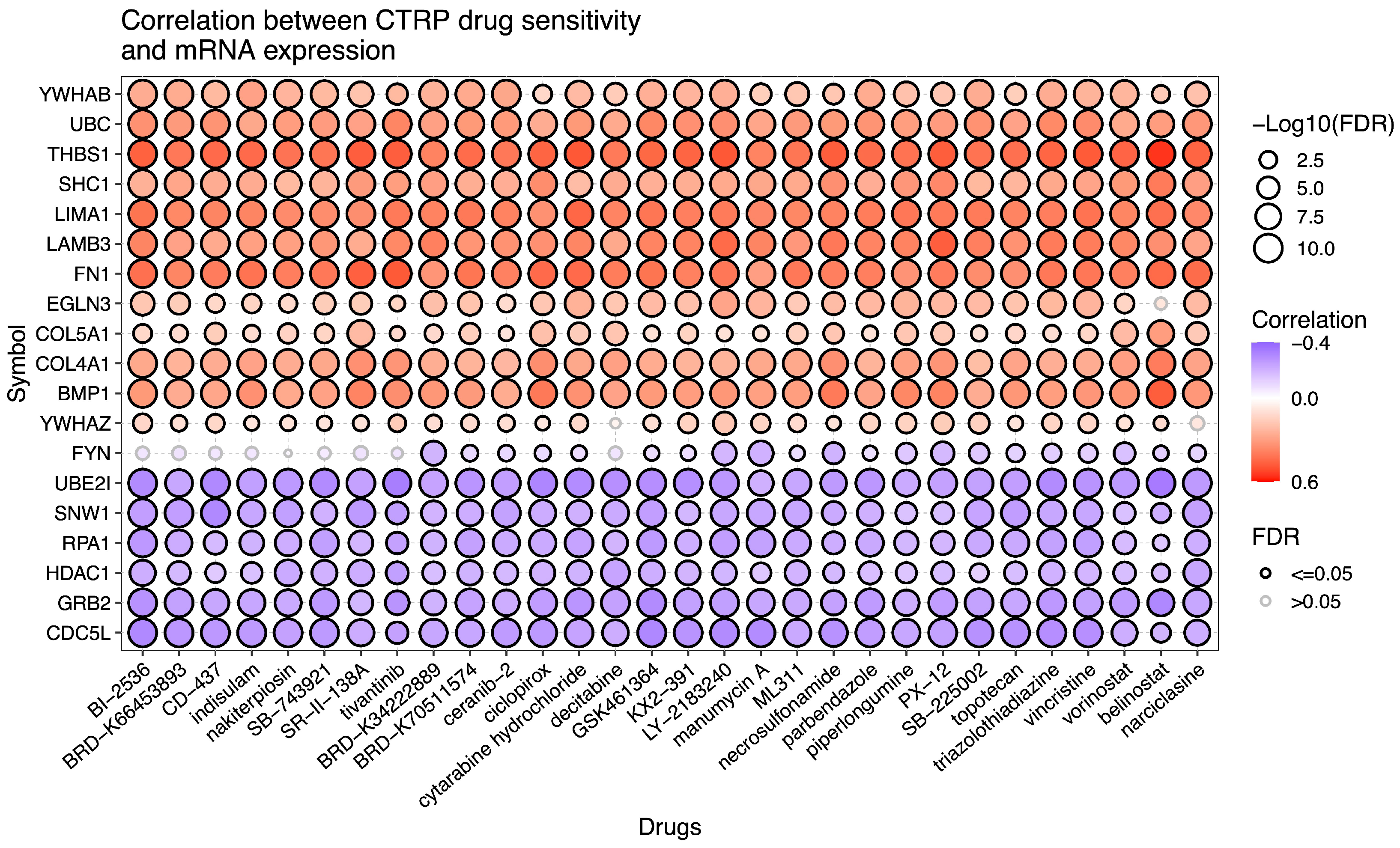
2.6. Drug Sensitivity Analysis
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.2. Negative Sample Inference
| Algorithm 1 Positive unlabeled learning algorithm for negative sample inference |
|
4.3. Network Structure Improvement
4.4. Hyperparameter Setting
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, M.; Xiang, J.; Zhao, Z.; Shang, X. SEPA: Signaling entropy-based algorithm to evaluate personalized pathway activation for survival analysis on pan-cancer data. Bioinformatics 2022, 38, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Dees, N.D.; Zhang, Q.; Kandoth, C.; Wendl, M.C.; Schierding, W.; Koboldt, D.C.; Mooney, T.B.; Callaway, M.B.; Dooling, D.; Mardis, E.R.; et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012, 22, 1589–1598. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Tamborero, D.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUST: Exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 2013, 29, 2238–2244. [Google Scholar] [CrossRef]
- Jiang, R. Walking on multiple disease-gene networks to prioritize candidate genes. J. Mol. Cell Biol. 2015, 7, 214–230. [Google Scholar] [CrossRef]
- Li, Y.; Patra, J.C. Genome-wide inferring gene–phenotype relationship by walking on the heterogeneous network. Bioinformatics 2010, 26, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Hwang, T.; Kuang, R. Prioritizing disease genes by bi-random walk. In Proceedings of the Advances in Knowledge Discovery and Data Mining: 16th Pacific-Asia Conference, PAKDD 2012, Kuala Lumpur, Malaysia, 29 May–1 June 2012; Proceedings, Part II 16. Springer: Berlin/Heidelberg, Germany, 2012; pp. 292–303. [Google Scholar]
- Xiang, J.; Zhang, J.; Zheng, R.; Li, X.; Li, M. NIDM: Network impulsive dynamics on multiplex biological network for disease-gene prediction. Brief. Bioinform. 2021, 22, bbab080. [Google Scholar] [CrossRef]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013, 155, 948–962. [Google Scholar] [CrossRef]
- Collier, O.; Stoven, V.; Vert, J.P. LOTUS: A single-and multitask machine learning algorithm for the prediction of cancer driver genes. PLoS Comput. Biol. 2019, 15, e1007381. [Google Scholar] [CrossRef] [PubMed]
- Mourikis, T.P.; Benedetti, L.; Foxall, E.; Temelkovski, D.; Nulsen, J.; Perner, J.; Cereda, M.; Lagergren, J.; Howell, M.; Yau, C.; et al. Patient-specific cancer genes contribute to recurrently perturbed pathways and establish therapeutic vulnerabilities in esophageal adenocarcinoma. Nat. Commun. 2019, 10, 3101. [Google Scholar] [CrossRef]
- Nulsen, J.; Misetic, H.; Yau, C.; Ciccarelli, F.D. Pan-cancer detection of driver genes at the single-patient resolution. Genome Med. 2021, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Schulte-Sasse, R.; Budach, S.; Hnisz, D.; Marsico, A. Integration of multiomics data with graph convolutional networks to identify new cancer genes and their associated molecular mechanisms. Nat. Mach. Intell. 2021, 3, 513–526. [Google Scholar] [CrossRef]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Peng, W.; Tang, Q.; Dai, W.; Chen, T. Improving cancer driver gene identification using multi-task learning on graph convolutional network. Brief. Bioinform. 2022, 23, bbab432. [Google Scholar] [CrossRef]
- Defferrard, M.; Bresson, X.; Vandergheynst, P. Convolutional neural networks on graphs with fast localized spectral filtering. In Proceedings of the Advances in Neural Information Processing Systems 29: Annual Conference on Neural Information Processing Systems 2016, Barcelona, Spain, 5–10 December 2016; pp. 1–9. [Google Scholar]
- Cui, Y.; Wang, Z.; Wang, X.; Zhang, Y.; Zhang, Y.; Pan, T.; Zhang, Z.; Li, S.; Guo, Y.; Akutsu, T.; et al. SMG: Self-supervised masked graph learning for cancer gene identification. Brief. Bioinform. 2023, 24, bbad406. [Google Scholar] [CrossRef]
- Chen, Z.; Li, W.; Xing, X.; Yuan, Y. Medical federated learning with joint graph purification for noisy label learning. Med. Image Anal. 2023, 90, 102976. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, W.; Liu, J.; Chen, H.; Yuan, Y. GRAB-Net: Graph-based boundary-aware network for medical point cloud segmentation. IEEE Trans. Med Imaging 2023, 42, 2776–2786. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, X.; Yuan, Y. Sigma: Semantic-complete graph matching for domain adaptive object detection. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, New Orleans, LA, USA, 18–24 June 2022; pp. 5291–5300. [Google Scholar]
- Li, W.; Chen, Z.; Li, B.; Zhang, D.; Yuan, Y. Htd: Heterogeneous task decoupling for two-stage object detection. IEEE Trans. Image Process. 2021, 30, 9456–9469. [Google Scholar] [CrossRef]
- Veličković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Lio, P.; Bengio, Y. Graph attention networks. arXiv 2017, arXiv:1710.10903. [Google Scholar]
- Consortium, G.O. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Gala, K.; Jain, M.; Shah, P.; Pandey, A.; Garg, M.; Khattar, E. Role of p53 transcription factor in determining the efficacy of telomerase inhibitors in cancer treatment. Life Sci. 2024, 339, 1–13. [Google Scholar] [CrossRef]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. Apoptosis: Mechanisms and relevance in cancer. Ann. Hematol. 2005, 84, 627–639. [Google Scholar] [CrossRef]
- Slattery, M.L.; Mullany, L.E.; Sakoda, L.C.; Wolff, R.K.; Samowitz, W.S.; Herrick, J.S. Dysregulated genes and miRNAs in the apoptosis pathway in colorectal cancer patients. Apoptosis 2018, 23, 237–250. [Google Scholar] [CrossRef]
- Matsuura, K.; Canfield, K.; Feng, W.; Kurokawa, M. Metabolic regulation of apoptosis in cancer. Int. Rev. Cell Mol. Biol. 2016, 327, 43–87. [Google Scholar] [PubMed]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Hu, F.F.; Xia, M.X.; Han, L.; Zhang, Q.; Guo, A.Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef]
- Liu, C.J.; Hu, F.F.; Xie, G.Y.; Miao, Y.R.; Li, X.W.; Zeng, Y.; Guo, A.Y. GSCA: An integrated platform for gene set cancer analysis at genomic, pharmacogenomic and immunogenomic levels. Brief. Bioinform. 2023, 24, bbac558. [Google Scholar] [CrossRef]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lénárt, P.; Petronczki, M.; Krššák, M.; Gürtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007, 17, 316–322. [Google Scholar] [CrossRef]
- Liang, J.Y.; Fontana, J.; Rao, J.; Ordonez, J.; Dawson, M.; Shroot, B.; Wilber, J.; Feng, P. Synthetic retinoid CD437 induces S-phase arrest and apoptosis in human prostate cancer cells LNCaP and PC-3. Prostate 1999, 38, 228–236. [Google Scholar] [CrossRef]
- You, B.R.; Shin, H.R.; Park, W.H. PX-12 inhibits the growth of A549 lung cancer cells via G2/M phase arrest and ROS-dependent apoptosis. Int. J. Oncol. 2014, 44, 301–308. [Google Scholar] [CrossRef]
- Spencer, D.H.; Sehn, J.K.; Abel, H.J.; Watson, M.A.; Pfeifer, J.D.; Duncavage, E.J. Comparison of clinical targeted next-generation sequence data from formalin-fixed and fresh-frozen tissue specimens. J. Mol. Diagn. 2013, 15, 623–633. [Google Scholar] [CrossRef]
- Wong, S.Q.; Li, J.; Tan, A.Y.; Vedururu, R.; Pang, J.M.B.; Do, H.; Ellul, J.; Doig, K.; Bell, A.; McArthur, G.A.; et al. Sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med. Genom. 2014, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Kamburov, A.; Pentchev, K.; Galicka, H.; Wierling, C.; Lehrach, H.; Herwig, R. ConsensusPathDB: Toward a more complete picture of cell biology. Nucleic Acids Res. 2011, 39, D712–D717. [Google Scholar] [CrossRef] [PubMed]
- Dressler, L.; Bortolomeazzi, M.; Keddar, M.R.; Misetic, H.; Sartini, G.; Acha-Sagredo, A.; Montorsi, L.; Wijewardhane, N.; Repana, D.; Nulsen, J.; et al. Comparative assessment of genes driving cancer and somatic evolution in non-cancer tissues: An update of the Network of Cancer Genes (NCG) resource. Genome Biol. 2022, 23, 35. [Google Scholar] [CrossRef] [PubMed]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Kim, J.; So, S.; Lee, H.J.; Park, J.C.; Kim, J.j.; Lee, H. DigSee: Disease gene search engine with evidence sentences (version cancer). Nucleic Acids Res. 2013, 41, W510–W517. [Google Scholar] [CrossRef]
- Perozzi, B.; Al-Rfou, R.; Skiena, S. Deepwalk: Online learning of social representations. In Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 24–27 August 2014; pp. 701–710. [Google Scholar]
- Zhou, Z.H.; Feng, J. Deep forest. Natl. Sci. Rev. 2019, 6, 74–86. [Google Scholar] [CrossRef]
- Fatemi, B.; El Asri, L.; Kazemi, S.M. Slaps: Self-supervision improves structure learning for graph neural networks. In Proceedings of the Advances in Neural Information Processing Systems 34: Annual Conference on Neural Information Processing Systems 2021, NeurIPS 2021, Virtual, 6–14 December 2021; pp. 22667–22681. [Google Scholar]
- Vincent, P.; Larochelle, H.; Bengio, Y.; Manzagol, P.A. Extracting and composing robust features with denoising autoencoders. In Proceedings of the 25th International Conference on Machine Learning, Helsinki, Finland, 5–9 July 2008; pp. 1096–1103. [Google Scholar]
- Nair, V.; Hinton, G.E. Rectified linear units improve restricted boltzmann machines. In Proceedings of the 27th International Conference on Machine Learning (ICML-10), Haifa, Israel, 21–24 June 2010; pp. 807–814. [Google Scholar]
- Kingma, D.P. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| METHOD | AUPRC | AUROC | F1 Score |
|---|---|---|---|
| GCN | 0.921 | 0.947 | 0.877 |
| GAT | 0.938 | 0.956 | 0.887 |
| Chebnet | 0.940 | 0.945 | 0.866 |
| EMOGI | 0.921 | 0.946 | 0.876 |
| MTGCN | 0.907 | 0.921 | 0.822 |
| SMG | 0.942 | 0.951 | 0.876 |
| SSCI-AN | 0.955 | 0.958 | 0.891 |
| SSCI-2S | 0.956 | 0.958 | 0.891 |
| SSCI | 0.964 | 0.966 | 0.913 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Hao, J.; Liao, X.; Shang, X.; Li, X. SSCI: Self-Supervised Deep Learning Improves Network Structure for Cancer Driver Gene Identification. Int. J. Mol. Sci. 2024, 25, 10351. https://doi.org/10.3390/ijms251910351
Xu J, Hao J, Liao X, Shang X, Li X. SSCI: Self-Supervised Deep Learning Improves Network Structure for Cancer Driver Gene Identification. International Journal of Molecular Sciences. 2024; 25(19):10351. https://doi.org/10.3390/ijms251910351
Chicago/Turabian StyleXu, Jialuo, Jun Hao, Xingyu Liao, Xuequn Shang, and Xingyi Li. 2024. "SSCI: Self-Supervised Deep Learning Improves Network Structure for Cancer Driver Gene Identification" International Journal of Molecular Sciences 25, no. 19: 10351. https://doi.org/10.3390/ijms251910351