
Molecular Study of the Fukutin-Related Protein (FKRP) Gene in Patients from Southern Italy with Duchenne/Becker-like Phenotype

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Sanger Sequencing
2.2. Functional Prediction of the Missense Variant in the FKRP Gene (c.427C > A, p.R143S)
2.3. Multi-Species Alignment
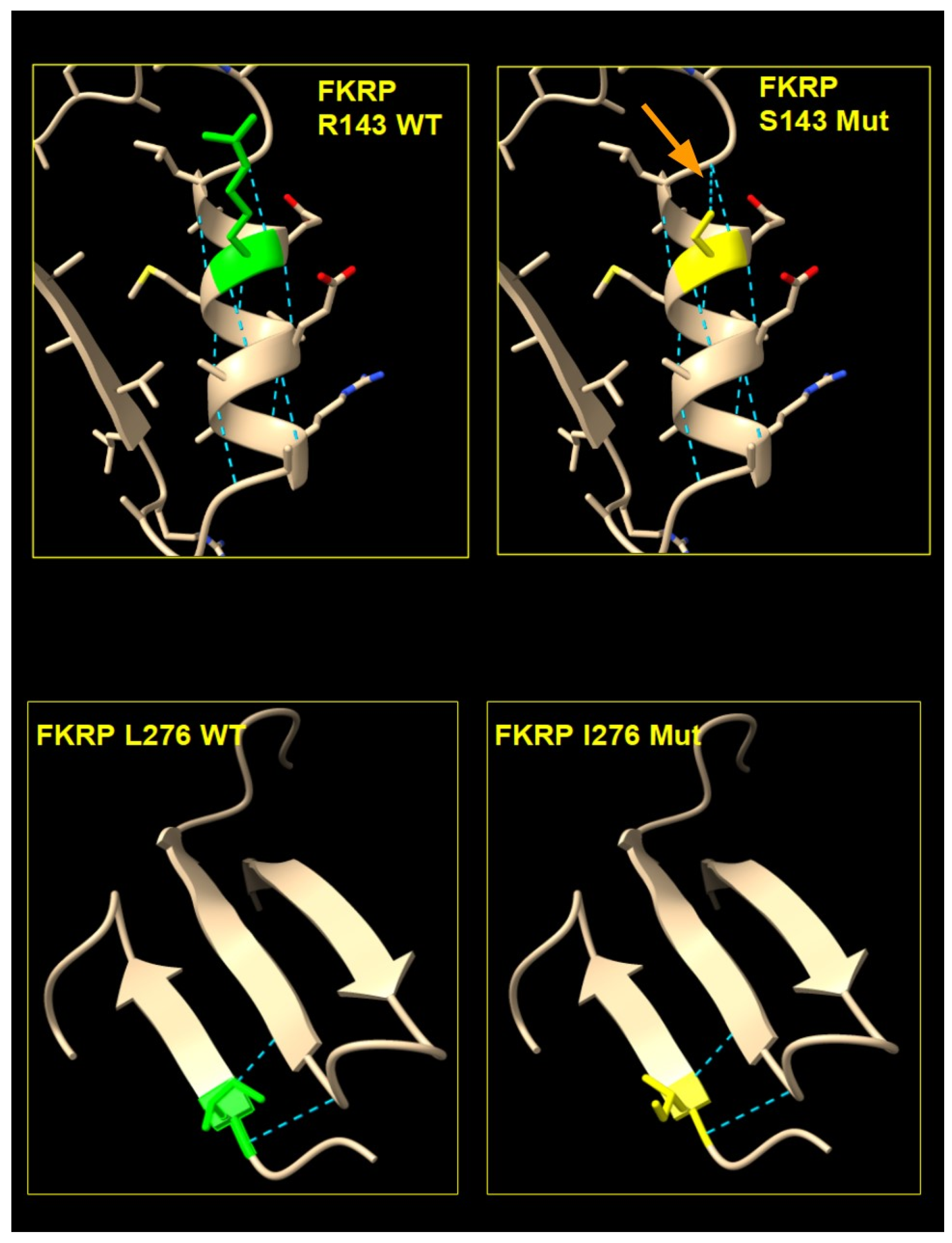
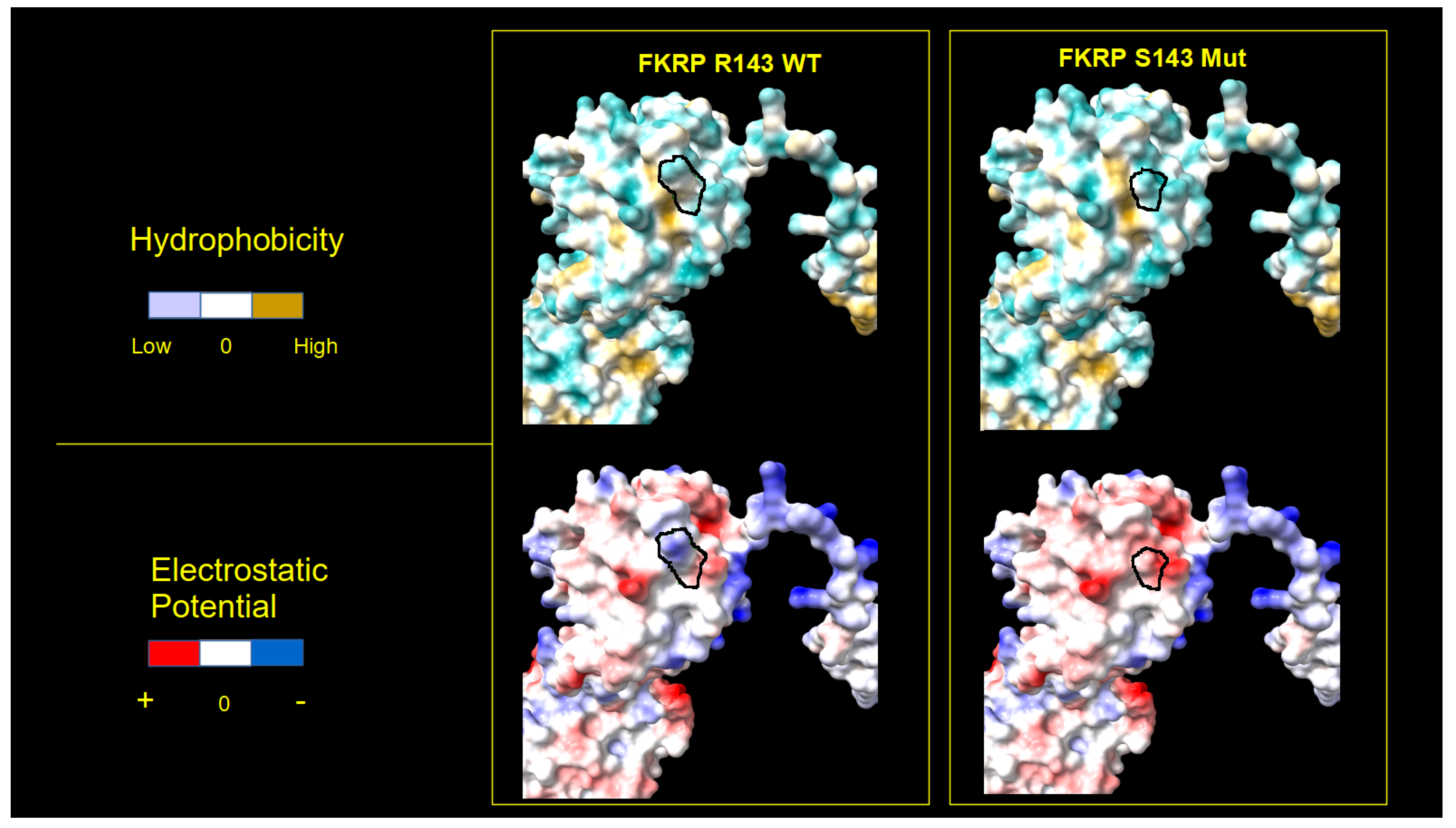
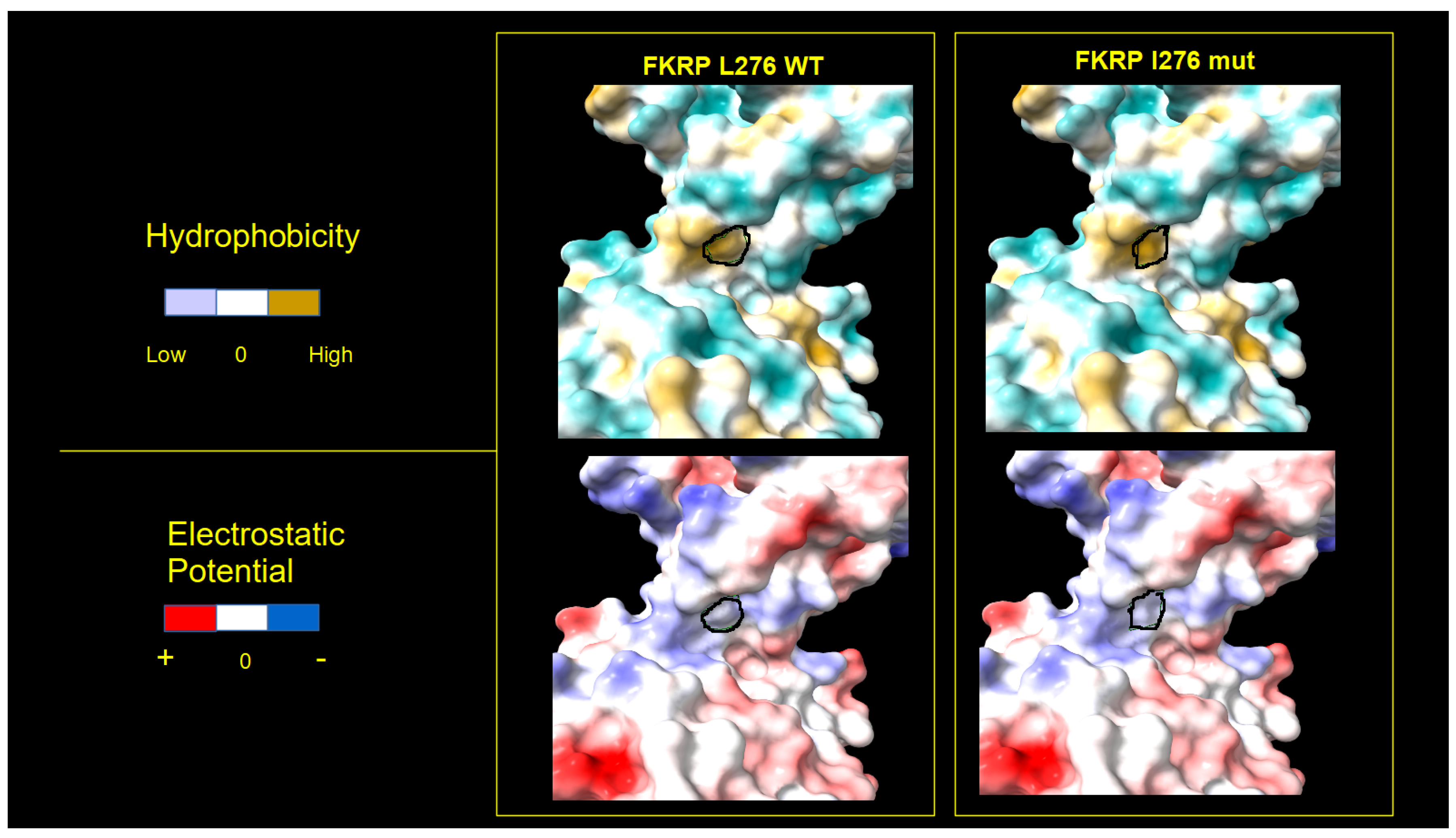
2.4. FKRP 3D Structure Analysis
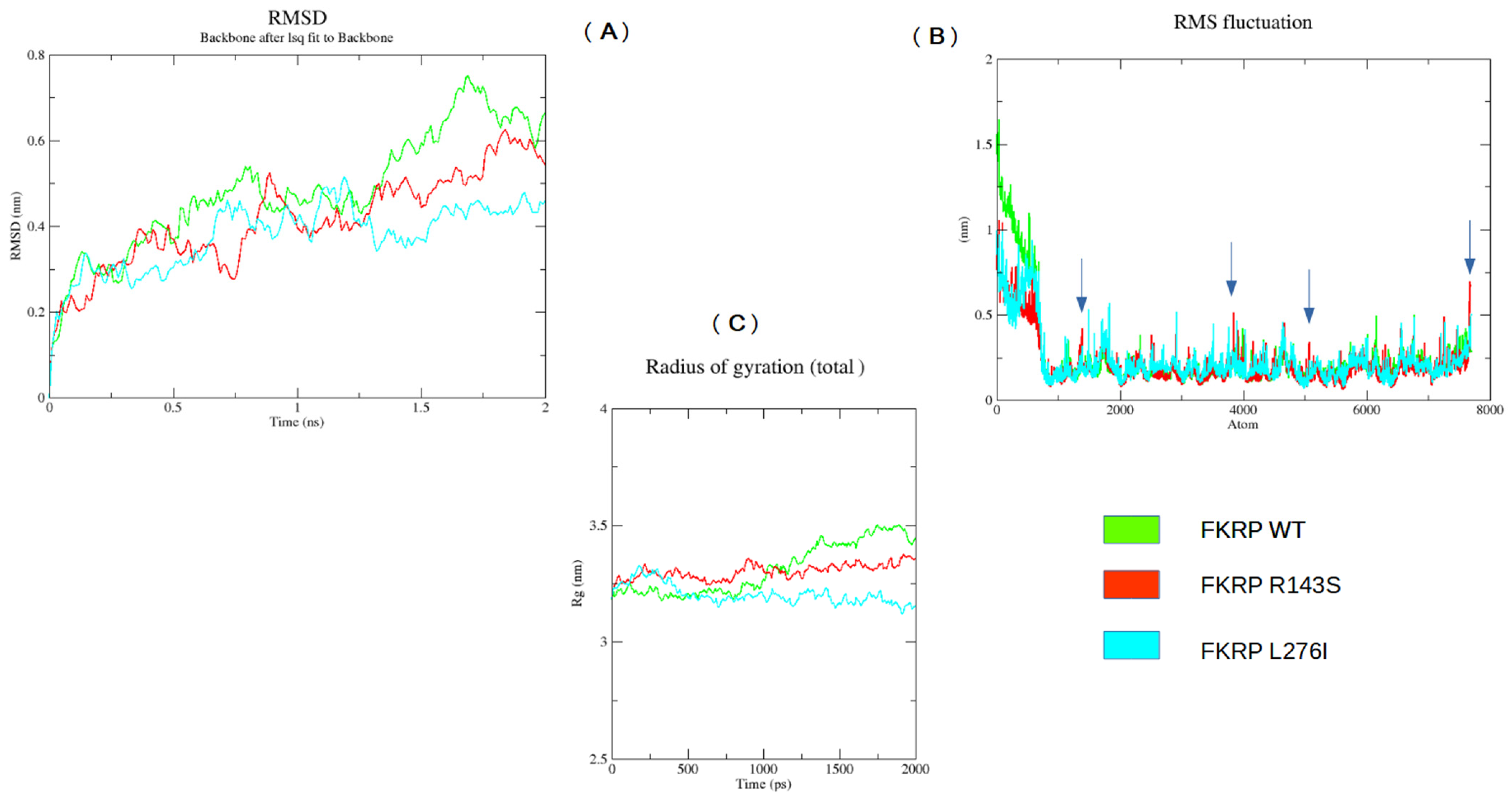
2.5. MD Analysis
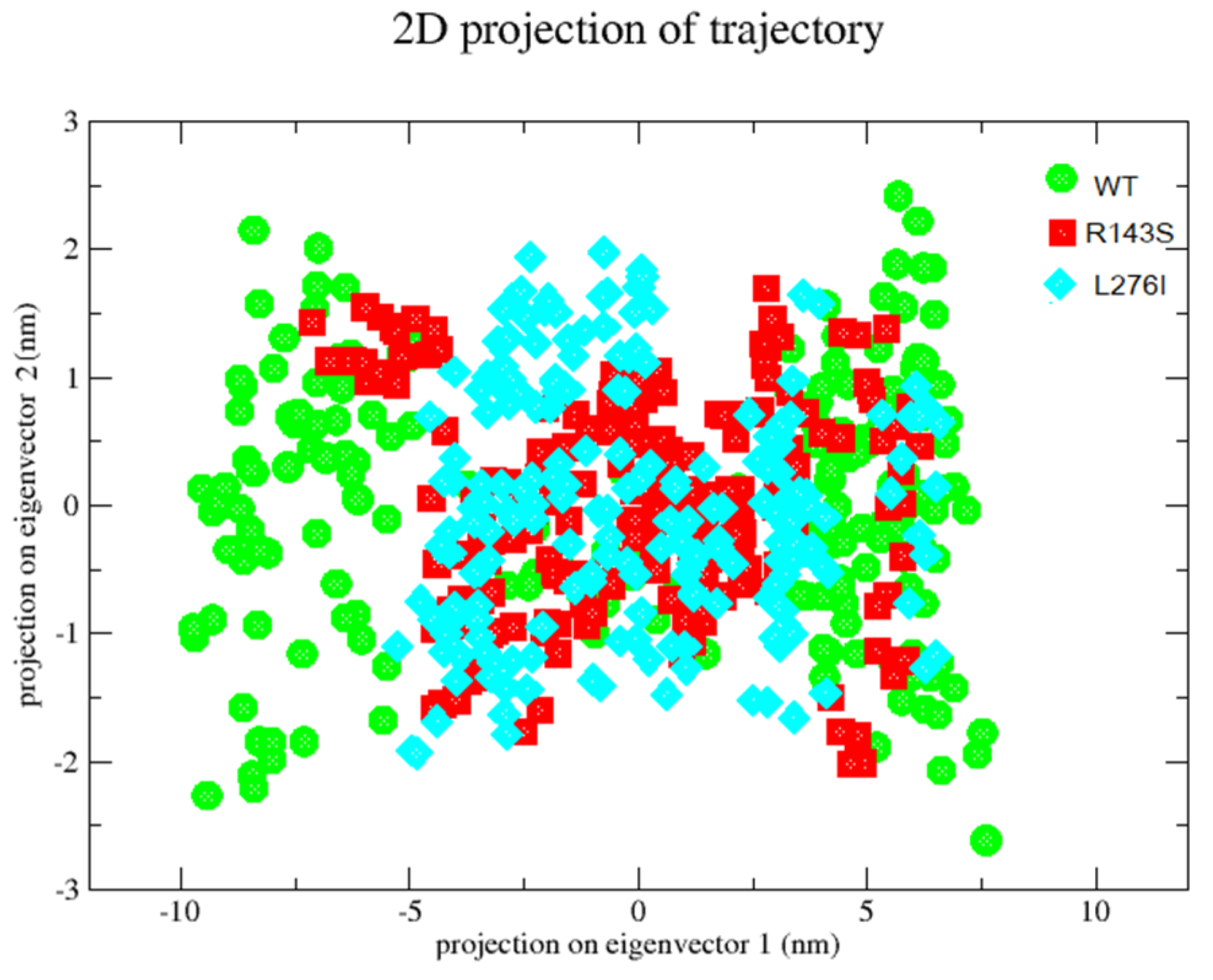
2.6. Principal Component Analysis-PCA
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. DNA Extraction
4.3. DNA Assay
4.4. Molecular Analysis of the FKRP Gene
4.5. PCR Reaction
4.6. Sequence Reaction
4.7. FKRP 3D Structure
4.8. Molecular Dinamic Simulation (MD)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Driss, A.; Amouri, R.; Ben Hamida, C.; Souilem, S.; Gouider-Khouja, N.; Ben Hamida, M.; Hentati, F. A new locus for autosomal recessive limb-girdle muscular dystrophy in a large consanguineous Tunisian family maps to chromosome 19q13.3. Neuromuscul. Disord. 2000, 10, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, P.A. Targeting of proteins to the Golgi apparatus. Histochem. Cell Biol. 1998, 109, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Vieira, N.M.; Schlesinger, D.; de Paula, F.; Vainzof, M.; Zatz, M. Mutation analysis in the FKRP gene provides an explanation for a rare cause of intrafamilial clinical variability in LGMD2I. Neuromuscul. Disord. 2006, 16, 870–873. [Google Scholar] [CrossRef]
- Alhamidi, M.; Buvang, E.K.; Fagerheim, T.; Brox, V.; Lindal, S.; Van Ghelue, M.; Nilssen, Ø. Fukutin-Related Protein Resides in the Golgi Cisternae of Skeletal Muscle Fibres and Forms Disulfide-Linked Homodimers via an N-Terminal Interaction. PLoS ONE 2011, 6, e22968. [Google Scholar] [CrossRef]
- Kuwabara, N.; Imae, R.; Manya, H.; Tanaka, T.; Mizuno, M.; Tsumoto, H.; Kanagawa, M.; Kobayashi, K.; Toda, T.; Senda, T.; et al. Crystal structures of fukutin-related protein (FKRP), a ribitol-phosphate transferase related to muscular dystrophy. Nat. Commun. 2020, 11, 303. [Google Scholar] [CrossRef]
- Munro, S. Localization of proteins to the Golgi apparatus. Trends Cell Biol. 1998, 8, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Richard, I.; Laurent, J.P.; Cirak, S.; Vissing, J. ENMC FKRP Study Group. 216th ENMC international workshop: Clinical readiness in FKRP related myopathies January 15–17, 2016 Naarden, The Netherlands. Neuromuscul. Disord. 2016, 26, 717–724. [Google Scholar] [CrossRef]
- Sheikh, M.O.; Halmo, S.M.; Wells, L. Recent advancements in understanding mammalian O-mannosylation. Glycobiology 2017, 27, 806–819. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef]
- Brown, S.C.; Fernandez-Fuente, M.; Muntoni, F.; Vissing, J. Phenotypic Spectrum of α-Dystroglycanopathies Associated With the c.919T > a Variant in the FKRP Gene in Humans and Mice. J. Neuropathol. Exp. Neurol. 2020, 79, 1257–1264. [Google Scholar] [CrossRef]
- Wilson, D.G.S.; Tinker, A.; Iskratsch, T. The role of the dystrophin glycoprotein complex in muscle cell mechanotransduction. Commun. Biol. 2022, 5, 1022. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef]
- Barresi, R.; Campbell, K.P. Dystroglycan: From biosynthesis to pathogenesis of human disease. J. Cell Sci. 2006, 119, 199–207. [Google Scholar] [CrossRef]
- Brockington, M.; Yuva, Y.; Prandini, P.; Brown, S.C.; Torelli, S.; Benson, M.A.; Herrmann, R.; Anderson, L.V.; Bashir, R.; Burgunder, J.M.; et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum. Mol. Genet. 2001, 10, 2851–2859. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Valero de Bernabé, D.; Voit, T.; Longman, C.; Steinbrecher, A.; Straub, V.; Yuva, Y.; Herrmann, R.; Sperner, J.; Korenke, C.; Diesen, C.; et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker–Warburg syndrome. J. Med. Genet. 2004, 41, e61. [Google Scholar] [CrossRef] [PubMed]
- Louhichi, N.; Triki, C.; Quijano-Roy, S.; Richard, P.; Makri, S.; Méziou, M.; Estournet, B.; Mrad, S.; Romero, N.B.; Ayadi, H.; et al. New FKRP mutations causing congenital muscular dystrophy associated with mental retardation and central nervous system abnormalities. Identification of a founder mutation in Tunisian families. Neurogenetics 2004, 5, 27–34. [Google Scholar] [CrossRef]
- Mercuri, E.; Topaloglu, H.; Brockington, M.; Berardinelli, A.; Pichiecchio, A.; Santorelli, F.; Rutherford, M.; Talim, B.; Ricci, E.; Voit, T.; et al. Spectrum of brain changes in patients with congenital muscular dystrophy and FKRP gene mutations. Arch. Neurol. 2006, 63, 251–257. [Google Scholar] [CrossRef]
- Van Reeuwijk, J.; Olderode-Berends, M.J.; Van den Elzen, C.; Brouwer, O.F.; Roscioli, T.; Van Pampus, M.G.; Scheffer, H.; Brunner, H.G.; Van Bokhoven, H.; Hol, F.A. A homozygous FKRP start codon mutation is associated with Walker-Warburg syndrome, the severe end of the clinical spectrum. Clin. Genet. 2010, 78, 275–281. [Google Scholar] [CrossRef]
- Brockington, M.; Blake, D.J.; Prandini, P.; Brown, S.C.; Torelli, S.; Benson, M.A.; Ponting, C.P.; Estournet, B.; Romero, N.B.; Mercuri, E.; et al. Mutations in the Fukutin-Related Protein Gene (FKRP) Cause a Form of Congenital Muscular Dystrophy with Secondary Laminin a2 Deficiency and Abnormal Glycosylation of a-Dystroglycan. Am. J. Hum. Genet. 2001, 69, 1198–1209. [Google Scholar] [CrossRef]
- Navarro-Cobos, M.J.; González-Del Angel, A.; Estandia-Ortega, B.; Ruiz-Herrera, A.; Becerra, A.; Vargas-Ramírez, G.; Bermúdez-López, C.; Alcántara-Ortigoza, M.A. Molecular analysis confirms that FKRP-Related Disorders are Underdiagnosed in Mexican Patients with Neuromuscular Diseases. Neuropediatrics 2017, 48, 442–450. [Google Scholar] [CrossRef]
- Magri, F.; Nigro, V.; Angelini, C.; Mongini, T.; Mora, M.; Moroni, I.; Toscano, A.; D’angelo, M.G.; Tomelleri, G.; Siciliano, G.; et al. The italian limb girdle muscular dystrophy registry: Relative frequency, clinical features, and differential diagnosis. Muscle Nerve 2017, 55, 55–68. [Google Scholar] [CrossRef] [PubMed]
- de Paula, F.; Vieira, N.; Starling, A.; Yamamoto, L.U.; Lima, B.; de Cássia Pavanello, R.; Vainzof, M.; Nigro, V.; Zatz, M. Asymptomatic carriers for homozygous novel mutations in the FKRP gene: The other end of the spectrum. Eur. J. Hum. Genet. 2003, 11, 923–930. [Google Scholar] [CrossRef]
- Boito, C.A.; Melacini, P.; Vianello, A.; Prandini, P.; Gavassini, B.F.; Bagattin, A.; Siciliano, G.; Angelini, C.; Pegoraro, E. Clinical and molecular characterization of patients with limb-girdle muscular dystrophy type 2I. Arch. Neurol. 2005, 62, 1894–1899. [Google Scholar] [CrossRef] [PubMed]
- Sveen, M.L.; Schwartz, M.; Vissing, J. High prevalence and phenotype-genotype correlations of limb girdle muscular dystrophy type 2I in Denmark. Ann. Neurol. 2006, 59, 808–815. [Google Scholar] [CrossRef]
- Rasmussen, M.; Scheie, D.; Breivik, N.; Mork, M.; Lindal, S. Clinical and muscle biopsy findings in Norwegian paediatric patients with limb girdle muscular dystrophy 2I. Acta Paediatr. 2014, 103, 553–558. [Google Scholar] [CrossRef]
- Frosk, P.; Greenberg, C.R.; Tennese, A.A.; Lamont, R.; Nylen, E.; Hirst, C.; Frappier, D.; Roslin, N.M.; Zaik, M.; Bushby, K.; et al. The most common mutation in FKRP causing limb girdle muscular dystrophy type 2I (LGMD2I) may have occurred only once and is present in Hutterites and other populations. Hum. Mutat. 2005, 25, 38–44. [Google Scholar] [CrossRef]
- Murphy, L.B.; Schreiber-Katz, O.; Rafferty, K.; Robertson, A.; Topf, A.; Willis, T.A.; Heidemann, M.; Thiele, S.; Bindoff, L.; Laurent, J.P.; et al. Global FKRP Registry: Observations in more than 300 patients with Limb Girdle Muscular Dystrophy R9. Ann. Clin. Transl. Neurol. 2020, 7, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.; Mayhew, A.; Muni-Lofra, R.; Murphy, L.B.; Straub, V. Prevalence of Pain within Limb Girdle Muscular Dystrophy R9 and Implications for Other Degenerative Diseases. J. Clin. Med. 2021, 10, 5517. [Google Scholar] [CrossRef]
- Mercuri, E.; Brockington, M.; Straub, V.; Quijano-Roy, S.; Yuva, Y.; Herrmann, R.; Brown, S.C.; Torelli, S.; Dubowitz, V.; Blake, D.J.; et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann. Neurol. 2003, 53, 537–542. [Google Scholar] [CrossRef]
- Poppe, M.; Cree, L.; Bourke, J.; Eagle, M.; Anderson, L.V.; Birchall, D.; Brockington, M.; Buddles, M.; Busby, M.; Muntoni, F.; et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology 2003, 60, 1246–1251. [Google Scholar] [CrossRef]
- Bushby, K.M.; Beckmann, J.S. The 105th ENMC sponsored workshop: Pathogenesis in the non-sarcoglycan limb-girdle muscular dystrophies, Naarden, April 12–14, 2002. Neuromuscul. Disord. 2003, 13, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Cordero, C.; Azzag, K.; Perlingeiro, R.C.R. Fukutin-Related Protein: From Pathology to Treatments. Trends Cell Biol. 2021, 31, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Qiao, C.; Wang, C.H.; Zhao, C.; Lu, P.; Awano, H.; Xiao, B.; Li, J.; Yuan, Z.; Dai, Y.; Martin, C.B.; et al. Muscle and heart function restoration in a limb girdle muscular dystrophy 2I (LGMD2I) mouse model by systemic FKRP gene delivery. Mol. Ther. 2014, 22, 1890–1899. [Google Scholar] [CrossRef] [PubMed]
- Krag, T.O.; Vissing, J. A New Mouse Model of Limb-Girdle Muscular Dystrophy Type 2I Homozygous for the Common L276I Mutation Mimicking the Mild Phenotype in Humans. J. Neuropathol. Exp. Neurol. 2015, 74, 1137–1146. [Google Scholar] [CrossRef]
- Serafini, P.R.; Feyder, M.J.; Hightower, R.M.; Garcia-Perez, D.; Vieira, N.M.; Lek, A.; Gibbs, D.E.; Moukha-Chafiq, O.; Augelli-Szafran, C.E.; Kawahara, G.; et al. A limb-girdle muscular dystrophy 2I model of muscular dystrophy identifies corrective drug compounds for dystroglycanopathies. JCI Insight 2018, 3, e120493. [Google Scholar] [CrossRef]
- Keramaris-Vrantsis, E.; Lu, P.J.; Doran, T.; Zillmer, A.; Ashar, J.; Esapa, C.T.; Benson, M.A.; Blake, D.J.; Rosenfeld, J.; Lu, Q.L. Fukutin-related protein localizes to the Golgi apparatus and mutations lead to mislocalization in muscle in vivo. Muscle Nerve 2007, 36, 455–465. [Google Scholar] [CrossRef]
- Boito, C.A.; Fanin, M.; Gavassini, B.F.; Cenacchi, G.; Angelini, C.; Pegoraro, E. Biochemical and ultrastructural evidence of endoplasmic reticulum stress in LGMD2I. Virchows Arch. 2007, 451, 1047–1055. [Google Scholar] [CrossRef]
- Poppe, M.; Bourke, J.; Eagle, M.; Frosk, P.; Wrogemann, K.; Greenberg, C.; Muntoni, F.; Voit, T.; Straub, V.; Hilton-Jones, D.; et al. Cardiac and respiratory failure in limb-girdle muscular dystrophy 2I. Ann. Neurol. 2004, 56, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Sveen, M.L.; Thune, J.J.; Køber, L.; Vissing, J. Cardiac involvement in patients with limb-girdle muscular dystrophy type 2 and Becker muscular dystrophy. Arch. Neurol. 2008, 65, 1196–1201. [Google Scholar] [CrossRef]
- Libell, E.M.; Richardson, J.A.; Lutz, K.L.; Ng, B.Y.; Mockler, S.R.H.; Laubscher, K.M.; Stephan, C.M.; Zimmerman, B.M.; Edens, E.R.; Reinking, B.E.; et al. Cardiomyopathy in limb girdle muscular dystrophy R9, FKRP related. Muscle Nerve 2020, 62, 626–632. [Google Scholar] [CrossRef]
- Schwartz, M.; Hertz, J.M.; Sveen, M.L.; Vissing, J. LGMD2I presenting with a characteristic Duchenne or Becker muscular dystrophy phenotype. Neurology 2005, 64, 1635–1637. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Matsuura, K.; Kasagi, N.; Adachi, K.; Kai, M.; Okubo, M.; Nishino, I.; Nanba, E.; Maegaki, Y. Duchenne muscular dystrophy-like phenotype in an LGMD2I patient with novel FKRP gene variants. Hum. Genome Var. 2020, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Wahbi, K.; Meune, C.; Hamouda, E.H.; Stojkovic, T.; Laforêt, P.; Bécane, H.M.; Eymard, B.; Duboc, D. Cardiac assessment of limb-girdle muscular dystrophy 2I patients: An echography, Holter ECG and magnetic resonance imaging study. Neuromuscul. Disord. 2008, 18, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Manzini, M.C.; Gleason, D.; Chang, B.S.; Hill, R.S.; Barry, B.J.; Partlow, J.N.; Poduri, A.; Currier, S.; Galvin-Parton, P.; Shapiro, L.R.; et al. Ethnically diverse causes of Walker-Warburg syndrome (WWS): FCMD mutations are a more common cause of WWS outside of the Middle East. Hum. Mutat. 2008, 29, E231–E241. [Google Scholar] [CrossRef] [PubMed]
- Guglieri, M.; Magri, F.; D’Angelo, M.G.; Prelle, A.; Morandi, L.; Rodolico, C.; Cagliani, R.; Mora, M.; Fortunato, F.; Bordoni, A.; et al. Clinical, molecular, and protein correlations in a large sample of genetically diagnosed Italian limb girdle muscular dystrophy patients. Hum. Mutat. 2008, 29, 258–266. [Google Scholar] [CrossRef]
- Mercuri, E.; Messina, S.; Bruno, C.; Mora, M.; Pegoraro, E.; Comi, G.P.; D’Amico, A.; Aiello, C.; Biancheri, R.; Berardinelli, A.; et al. Congenital muscular dystrophies with defective glycosylation of dystroglycan: A population study. Neurology 2009, 72, 1802–1809. [Google Scholar] [CrossRef]
- Gerin, I.; Ury, B.; Breloy, I.; Bouchet-Seraphin, C.; Bolsée, J.; Halbout, M.; Graff, J.; Vertommen, D.; Muccioli, G.G.; Seta, N.; et al. ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto α-dystroglycan. Nat. Commun. 2016, 7, 11534. [Google Scholar] [CrossRef]
- Angelini, C. LGMD. Identification, description and classification. Acta Myol. 2020, 39, 207–217. [Google Scholar] [CrossRef]
- Awano, H.; Saito, Y.; Shimizu, M.; Sekiguchi, K.; Niijima, S.; Matsuo, M.; Maegaki, Y.; Izumi, I.; Kikuchi, C.; Ishibashi, M.; et al. FKRP mutations cause congenital muscular dystrophy 1C and limb-girdle muscular dystrophy 2I in Asian patients. J. Clin. Neurosci. 2021, 92, 215–221. [Google Scholar] [CrossRef]
- Gonzalez-Quereda, L.; Rodriguez, M.J.; Diaz-Manera, J.; Alonso-Perez, J.; Gallardo, E.; Nascimento, A.; Ortez, C.; Natera-de Benito, D.; Olive, M.; Gonzalez-Mera, L.; et al. Targeted Next-Generation Sequencing in a Large Cohort of Genetically Undiagnosed Patients with Neuromuscular Disorders in Spain. Genes 2020, 11, 539. [Google Scholar] [CrossRef]
- Kesari, A.; Fukuda, M.; Knoblach, S.; Bashir, R.; Nader, G.A.; Rao, D.; Nagaraju, K.; Hoffman, E.P. Dysferlin deficiency shows compensatory induction of Rab27A/Slp2a that may contribute to inflammatory onset. Am. J. Pathol. 2008, 173, 1476–1487. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Mamelona, J.; Crapoulet, N.; Marrero, A. A new case of spastic paraplegia type 64 due to a missense mutation in the ENTPD1 gene. Hum. Genome Var. 2019, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Spadafora, P.; Qualtieri, A.; Cavalcanti, F.; Di Palma, G.; Gallo, O.; De Benedittis, S.; Cerantonio, A.; Citrigno, L. A Novel Homozygous Variant in DYSF Gene Is Associated with Autosomal Recessive Limb Girdle Muscular Dystrophy R2/2B. Int. J. Mol. Sci. 2022, 23, 8932. [Google Scholar] [CrossRef]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Boito et al., 2005 [23] | Magri et al., 2005 [21] | Guglieri et al., 2008 [45] | Mercuri et al., 2009 [46] | Our Work |
|---|---|---|---|---|
| L276I | L276I | L276I | W231C | L276I |
| R143S | L172I | R143S | P315S | R143S |
| P462S | L319R | L319R | P89L | |
| P358L | V160 | A114G | ||
| R244H | P104S | S115L |
| Variants | dbSNP |
|---|---|
| −34C > T | rs3201779 |
| A83A | rs149030303 |
| A45A | rs2287717 |
| P64P | rs111754012 |
| Tool | Effect | Score/Reliability Index | Link |
|---|---|---|---|
| PolyPhen2 | Deleterious | 0.93 | http://genetics.bwh.harvard.edu/pph2/ (accessed on 28 September 2022) |
| Panther | Deleterious | 0.5 | http://www.pantherdb.org/tools/ (accessed on 28 September 2022) |
| Mutation Taster | Deleterious | 0.99 | https://www.mutationtaster.org/ (accessed on 28 September 2022) |
| EXON | PRIMER |
|---|---|
| FKRP-EX4-1F | 5′ AAAGGGAATTGAGAAAGAGC 3′ |
| FKRP-EX4-2R | 5′ CCGAGAGGTTGAAGAGGT 3′ |
| FKRP-EX4-3F | 5′ AGTTTGTGGCCCTAGTACCT 3′ |
| FKRP-EX4-4R | 5′ CCTTCTCCCATACGAAGC 3′ |
| FKRP-EX4-5F | 5′ TGGAGGCTGCGGGCGTGCGCTACTG 3′ |
| FKRP-EX4-5R | 5′ GCTCACACAGAGCTTCTCC 3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qualtieri, A.; De Benedittis, S.; Cerantonio, A.; Citrigno, L.; Di Palma, G.; Gallo, O.; Cavalcanti, F.; Spadafora, P. Molecular Study of the Fukutin-Related Protein (FKRP) Gene in Patients from Southern Italy with Duchenne/Becker-like Phenotype. Int. J. Mol. Sci. 2024, 25, 10356. https://doi.org/10.3390/ijms251910356
Qualtieri A, De Benedittis S, Cerantonio A, Citrigno L, Di Palma G, Gallo O, Cavalcanti F, Spadafora P. Molecular Study of the Fukutin-Related Protein (FKRP) Gene in Patients from Southern Italy with Duchenne/Becker-like Phenotype. International Journal of Molecular Sciences. 2024; 25(19):10356. https://doi.org/10.3390/ijms251910356
Chicago/Turabian StyleQualtieri, Antonio, Selene De Benedittis, Annamaria Cerantonio, Luigi Citrigno, Gemma Di Palma, Olivier Gallo, Francesca Cavalcanti, and Patrizia Spadafora. 2024. "Molecular Study of the Fukutin-Related Protein (FKRP) Gene in Patients from Southern Italy with Duchenne/Becker-like Phenotype" International Journal of Molecular Sciences 25, no. 19: 10356. https://doi.org/10.3390/ijms251910356