
Mesenchymal Stem Cells Derived from Human Urine-Derived iPSCs Exhibit Low Immunogenicity and Reduced Immunomodulatory Profile
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Differentiation and Characterization of iMSCs
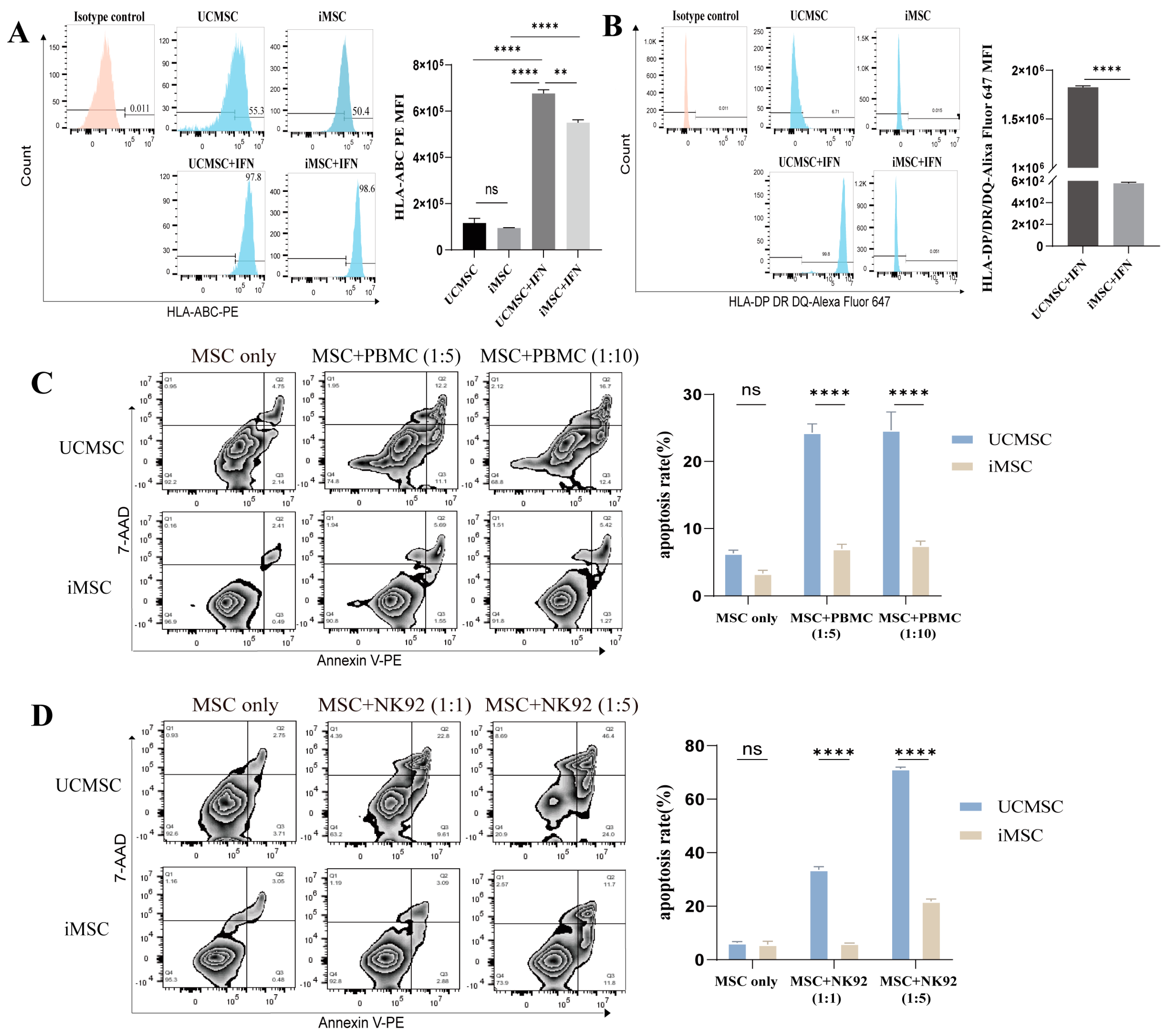
2.2. Low Immunogenicity of iMSCs
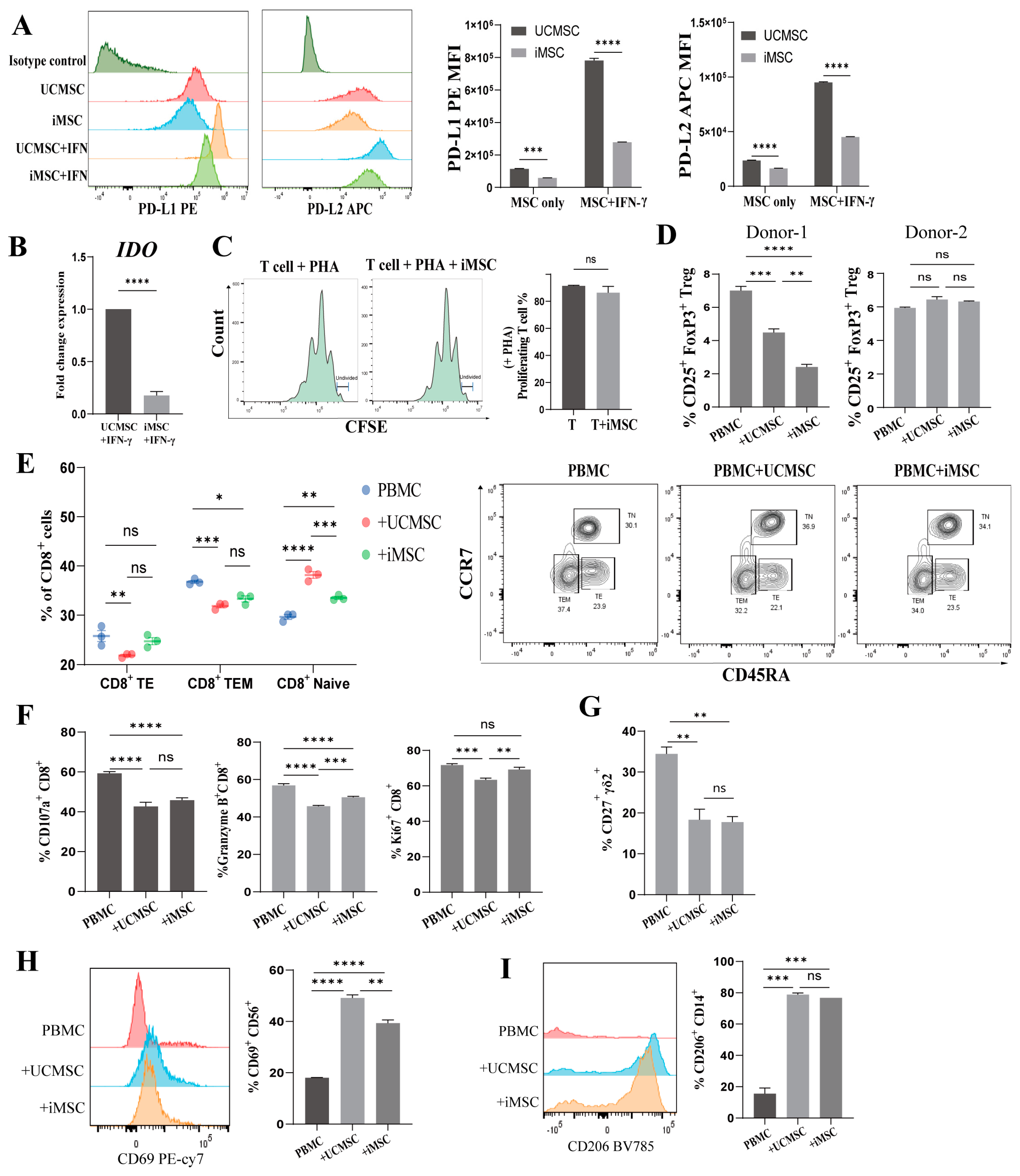
2.3. Immunomodulatory Capacity of iMSCs In Vitro
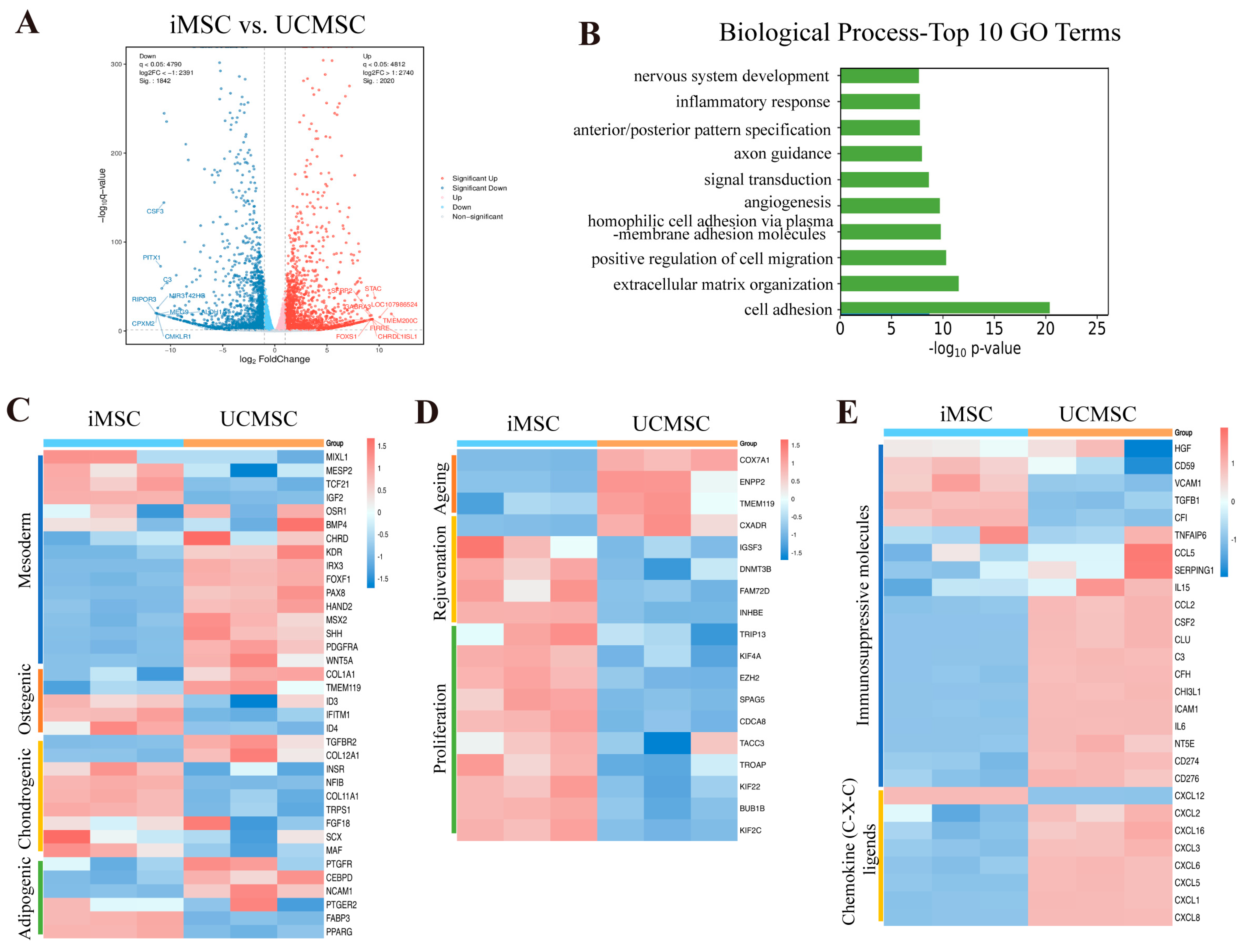
2.4. Transcriptomic Analysis of iMSCs and UCMSCs
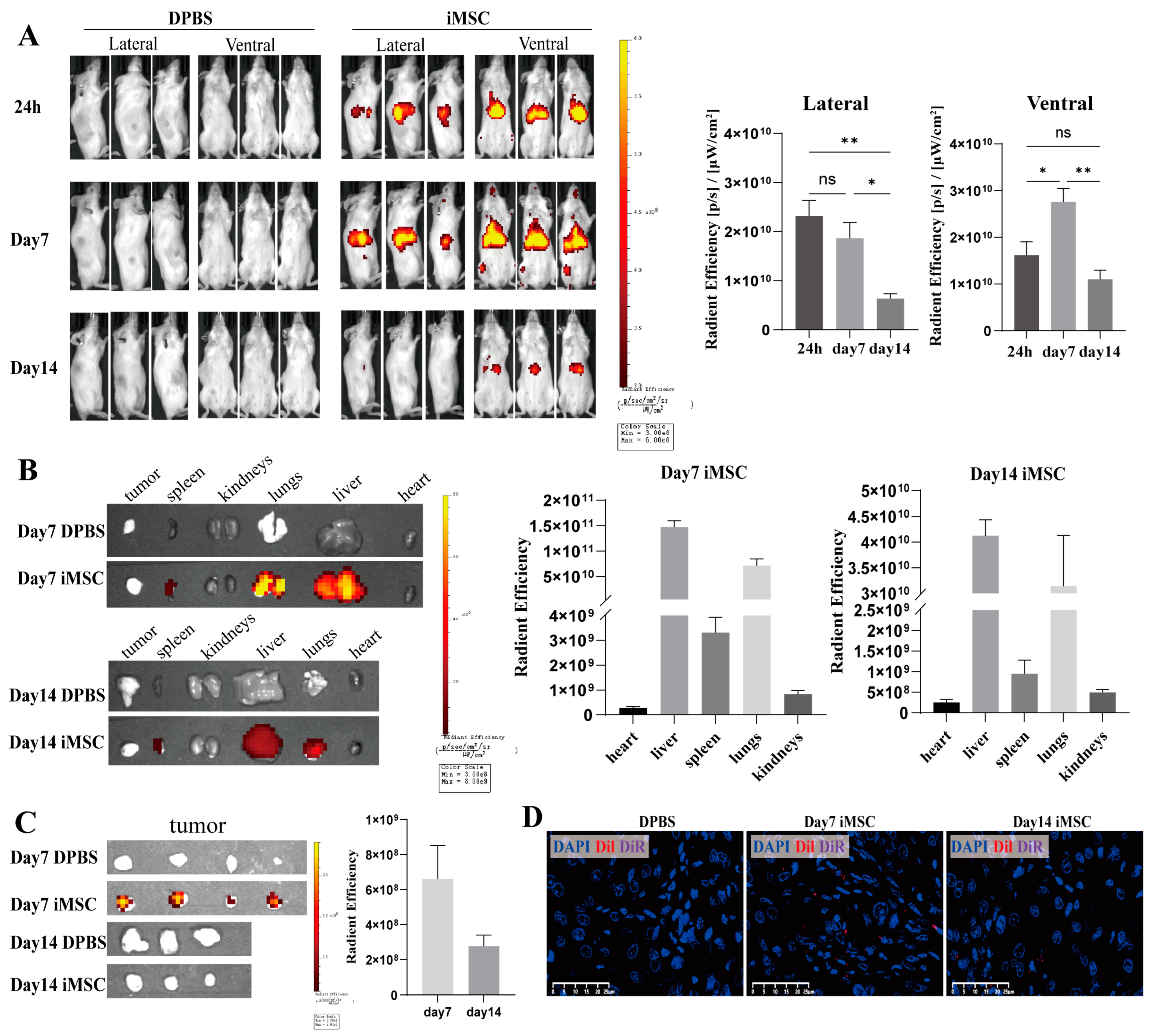
2.5. Biodistribution of iMSCs in the NCG Mouse Model
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Human iPSC Culture and Differentiation
4.3. Characterization of MSC Surface Markers Using Flow Cytometry
4.4. Osteogenic, Chondrogenic and Adipogenic Differentiation of MSCs
4.5. Alkaline Phosphatase Staining
4.6. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR) Analysis
- MIXL1-Forward: 5′GGCGTCAGAGTGGGAAATCC3′;
- MIXL1-Reverse: 5′GGCAGGCAGTTCACATCTACC3′;
- TBXT-Forward:5′CTATTCTGACAACTCACCTGCAT3′;
- TBXT-Reverse:5′ACAGGCTGGGGTACTGACT3′;
- PDGFRA-Forward:5′TGGCAGTACCCCATGTCTGAA3′;
- PDGFRA-Reverse: 5′CCAAGACCGTCACAAAAAGGC3′;
- EPCAM-Forward:5′AATCGTCAATGCCAGTGTACTT3′;
- EPCAM-Reverse:5′TCTCATCGCAGTCAGGATCATAA3′;
- IDO-Forward:5′TCTCATTTCGTGATGGAGACTGC3′;
- IDO-Reverse: 5′GTGTCCCGTTCTTGCATTTGC3′;
- ACTB-Forward:5′TGGTGGGTATGGGTCAGAAGGACTC 3′;
- ACTB-Reverse: 5′ CATGGCTGGGGTGTTGAAGGTCTCA 3′.
4.7. Cell Cycle Analysis
4.8. MSC Apoptosis Analysis Using Flow Cytometry
4.9. Carboxyfluorescein Succinimidyl Ester (CFSE) Proliferation Assay
4.10. Assessment of PBMC Subsets and Activation
4.11. MSC Flow Cytometry
4.12. Animal Experiments
4.13. mRNA Sequencing Analysis
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vodyanik, M.A.; Yu, J.; Zhang, X.; Tian, S.; Stewart, R.; Thomson, J.A.; Slukvin, I.I. A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell 2010, 7, 718–729. [Google Scholar] [CrossRef]
- Cheng, S.; Nethi, S.K.; Rathi, S.; Layek, B.; Prabha, S. Engineered Mesenchymal Stem Cells for Targeting Solid Tumors: Therapeutic Potential beyond Regenerative Therapy. J. Pharmacol. Exp. Ther. 2019, 370, 231–241. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Wong, K.L.; Lee, K.B.; Tai, B.C.; Law, P.; Lee, E.H.; Hui, J.H. Injectable cultured bone marrow-derived mesenchymal stem cells in varus knees with cartilage defects undergoing high tibial osteotomy: A prospective, randomized controlled clinical trial with 2 years’ follow-up. Arthroscopy 2013, 29, 2020–2028. [Google Scholar] [CrossRef]
- Orozco, L.; Munar, A.; Soler, R.; Alberca, M.; Soler, F.; Huguet, M.; Sentis, J.; Sanchez, A.; Garcia-Sancho, J. Treatment of knee osteoarthritis with autologous mesenchymal stem cells: A pilot study. Transplantation 2013, 95, 1535–1541. [Google Scholar] [CrossRef]
- Ohta, H.; Liu, X.; Maeda, M. Autologous adipose mesenchymal stem cell administration in arteriosclerosis and potential for anti-aging application: A retrospective cohort study. Stem Cell Res. Ther. 2020, 11, 538. [Google Scholar] [CrossRef]
- Ciccocioppo, R.; Bernardo, M.E.; Sgarella, A.; Maccario, R.; Avanzini, M.A.; Ubezio, C.; Minelli, A.; Alvisi, C.; Vanoli, A.; Calliada, F.; et al. Autologous bone marrow-derived mesenchymal stromal cells in the treatment of fistulising Crohn’s disease. Gut 2011, 60, 788–798. [Google Scholar] [CrossRef]
- Takayama, Y.; Kusamori, K.; Nishikawa, M. Mesenchymal stem/stromal cells as next-generation drug delivery vehicles for cancer therapeutics. Expert Opin. Drug Deliv. 2021, 18, 1627–1642. [Google Scholar] [CrossRef]
- Meng, M.; Zhang, W.W.; Chen, S.F.; Wang, D.R.; Zhou, C.H. Therapeutic utility of human umbilical cord-derived mesenchymal stem cells-based approaches in pulmonary diseases: Recent advancements and prospects. World J. Stem Cells 2024, 16, 70–88. [Google Scholar] [CrossRef]
- Le Thi Bich, P.; Nguyen Thi, H.; Dang Ngo Chau, H.; Phan Van, T.; Do, Q.; Dong Khac, H.; Le Van, D.; Nguyen Huy, L.; Mai Cong, K.; Ta Ba, T.; et al. Allogeneic umbilical cord-derived mesenchymal stem cell transplantation for treating chronic obstructive pulmonary disease: A pilot clinical study. Stem Cell Res. Ther. 2020, 11, 60. [Google Scholar] [CrossRef]
- Ridzuan, N.; Zakaria, N.; Widera, D.; Sheard, J.; Morimoto, M.; Kiyokawa, H.; Mohd Isa, S.A.; Chatar Singh, G.K.; Then, K.Y.; Ooi, G.C.; et al. Human umbilical cord mesenchymal stem cell-derived extracellular vesicles ameliorate airway inflammation in a rat model of chronic obstructive pulmonary disease (COPD). Stem Cell Res. Ther. 2021, 12, 54. [Google Scholar] [CrossRef]
- Wagner, W.; Horn, P.; Castoldi, M.; Diehlmann, A.; Bork, S.; Saffrich, R.; Benes, V.; Blake, J.; Pfister, S.; Eckstein, V.; et al. Replicative senescence of mesenchymal stem cells: A continuous and organized process. PLoS ONE 2008, 3, e2213. [Google Scholar] [CrossRef]
- Liu, J.; Ding, Y.; Liu, Z.; Liang, X. Senescence in Mesenchymal Stem Cells: Functional Alterations, Molecular Mechanisms, and Rejuvenation Strategies. Front. Cell Dev. Biol. 2020, 8, 258. [Google Scholar] [CrossRef]
- Costa, L.A.; Eiro, N.; Fraile, M.; Gonzalez, L.O.; Saa, J.; Garcia-Portabella, P.; Vega, B.; Schneider, J.; Vizoso, F.J. Functional heterogeneity of mesenchymal stem cells from natural niches to culture conditions: Implications for further clinical uses. Cell. Mol. Life Sci. 2021, 78, 447–467. [Google Scholar] [CrossRef]
- Wagner, W.; Bork, S.; Horn, P.; Krunic, D.; Walenda, T.; Diehlmann, A.; Benes, V.; Blake, J.; Huber, F.X.; Eckstein, V.; et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS ONE 2009, 4, e5846. [Google Scholar] [CrossRef]
- Bloor, A.J.C.; Patel, A.; Griffin, J.E.; Gilleece, M.H.; Radia, R.; Yeung, D.T.; Drier, D.; Larson, L.S.; Uenishi, G.I.; Hei, D.; et al. Production, safety and efficacy of iPSC-derived mesenchymal stromal cells in acute steroid-resistant graft versus host disease: A phase I, multicenter, open-label, dose-escalation study. Nat. Med. 2020, 26, 1720–1725. [Google Scholar] [CrossRef]
- Lian, Q.; Zhang, Y.; Zhang, J.; Zhang, H.K.; Wu, X.; Zhang, Y.; Lam, F.F.; Kang, S.; Xia, J.C.; Lai, W.H.; et al. Functional mesenchymal stem cells derived from human induced pluripotent stem cells attenuate limb ischemia in mice. Circulation 2010, 121, 1113–1123. [Google Scholar] [CrossRef]
- Pavathuparambil Abdul Manaph, N.; Al-Hawwas, M.; Bobrovskaya, L.; Coates, P.T.; Zhou, X.F. Urine-derived cells for human cell therapy. Stem Cell Res. Ther. 2018, 9, 189. [Google Scholar] [CrossRef]
- Zhou, T.; Benda, C.; Dunzinger, S.; Huang, Y.; Ho, J.C.; Yang, J.; Wang, Y.; Zhang, Y.; Zhuang, Q.; Li, Y.; et al. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 2012, 7, 2080–2089. [Google Scholar] [CrossRef]
- Balina-Sanchez, C.; Aguilera, Y.; Adan, N.; Sierra-Parraga, J.M.; Olmedo-Moreno, L.; Panadero-Moron, C.; Cabello-Laureano, R.; Marquez-Vega, C.; Martin-Montalvo, A.; Capilla-Gonzalez, V. Generation of mesenchymal stromal cells from urine-derived iPSCs of pediatric brain tumor patients. Front. Immunol. 2023, 14, 1022676. [Google Scholar] [CrossRef]
- Rajasingh, S.; Sigamani, V.; Selvam, V.; Gurusamy, N.; Kirankumar, S.; Vasanthan, J.; Rajasingh, J. Comparative analysis of human induced pluripotent stem cell-derived mesenchymal stem cells and umbilical cord mesenchymal stem cells. J. Cell. Mol. Med. 2021, 25, 8904–8919. [Google Scholar] [CrossRef]
- Zhou, M.; Hu, Z.; Qiu, L.; Zhou, T.; Feng, M.; Hu, Q.; Zeng, B.; Li, Z.; Sun, Q.; Wu, Y.; et al. Seamless Genetic Conversion of SMN2 to SMN1 via CRISPR/Cpf1 and Single-Stranded Oligodeoxynucleotides in Spinal Muscular Atrophy Patient-Specific Induced Pluripotent Stem Cells. Hum. Gene Ther. 2018, 29, 1252–1263. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, B.; Jia, L.; Huang, W.; Xiang, A.P.; Fang, C.; Liang, X.; Li, W. Lateral Mesoderm-Derived Mesenchymal Stem Cells With Robust Osteochondrogenic Potential and Hematopoiesis-Supporting Ability. Front. Mol. Biosci. 2022, 9, 767536. [Google Scholar] [CrossRef]
- Pereira, L.A.; Wong, M.S.; Mossman, A.K.; Sourris, K.; Janes, M.E.; Knezevic, K.; Hirst, C.E.; Lim, S.M.; Pimanda, J.E.; Stanley, E.G.; et al. Pdgfralpha and Flk1 are direct target genes of Mixl1 in differentiating embryonic stem cells. Stem Cell Res. 2012, 8, 165–179. [Google Scholar] [CrossRef]
- Kuan, I.I.; Lee, C.C.; Chen, C.H.; Lu, J.; Kuo, Y.S.; Wu, H.C. The extracellular domain of epithelial cell adhesion molecule (EpCAM) enhances multipotency of mesenchymal stem cells through EGFR-LIN28-LET7 signaling. J. Biol. Chem. 2019, 294, 7769–7786. [Google Scholar] [CrossRef]
- Frobel, J.; Hemeda, H.; Lenz, M.; Abagnale, G.; Joussen, S.; Denecke, B.; Saric, T.; Zenke, M.; Wagner, W. Epigenetic rejuvenation of mesenchymal stromal cells derived from induced pluripotent stem cells. Stem Cell Rep. 2014, 3, 414–422. [Google Scholar] [CrossRef]
- Xu, M.; Shaw, G.; Murphy, M.; Barry, F. Induced Pluripotent Stem Cell-Derived Mesenchymal Stromal Cells Are Functionally and Genetically Different From Bone Marrow-Derived Mesenchymal Stromal Cells. Stem Cells 2019, 37, 754–765. [Google Scholar] [CrossRef]
- Christodoulou, I.; Goulielmaki, M.; Devetzi, M.; Panagiotidis, M.; Koliakos, G.; Zoumpourlis, V. Mesenchymal stem cells in preclinical cancer cytotherapy: A systematic review. Stem Cell Res. Ther. 2018, 9, 336. [Google Scholar] [CrossRef]
- Zangi, L.; Margalit, R.; Reich-Zeliger, S.; Bachar-Lustig, E.; Beilhack, A.; Negrin, R.; Reisner, Y. Direct imaging of immune rejection and memory induction by allogeneic mesenchymal stromal cells. Stem Cells 2009, 27, 2865–2874. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, X.; Wang, H.; Liu, X.; Zhang, T.; Wang, Y.; Hu, D. The challenges and promises of allogeneic mesenchymal stem cells for use as a cell-based therapy. Stem Cell Res. Ther. 2015, 6, 234. [Google Scholar] [CrossRef]
- Sun, Y.Q.; Zhang, Y.; Li, X.; Deng, M.X.; Gao, W.X.; Yao, Y.; Chiu, S.M.; Liang, X.; Gao, F.; Chan, C.W.; et al. Insensitivity of Human iPS Cells-Derived Mesenchymal Stem Cells to Interferon-gamma-induced HLA Expression Potentiates Repair Efficiency of Hind Limb Ischemia in Immune Humanized NOD Scid Gamma Mice. Stem Cells 2015, 33, 3452–3467. [Google Scholar] [CrossRef]
- Li, X.; Xu, Z.; Bai, J.; Yang, S.; Zhao, S.; Zhang, Y.; Chen, X.; Wang, Y. Umbilical Cord Tissue-Derived Mesenchymal Stem Cells Induce T Lymphocyte Apoptosis and Cell Cycle Arrest by Expression of Indoleamine 2, 3-Dioxygenase. Stem Cells Int. 2016, 2016, 7495135. [Google Scholar] [CrossRef]
- Najar, M.; Raicevic, G.; Boufker, H.I.; Fayyad Kazan, H.; De Bruyn, C.; Meuleman, N.; Bron, D.; Toungouz, M.; Lagneaux, L. Mesenchymal stromal cells use PGE2 to modulate activation and proliferation of lymphocyte subsets: Combined comparison of adipose tissue, Wharton’s Jelly and bone marrow sources. Cell. Immunol. 2010, 264, 171–179. [Google Scholar] [CrossRef]
- Fechter, K.; Dorronsoro, A.; Jakobsson, E.; Ferrin, I.; Lang, V.; Sepulveda, P.; Pennington, D.J.; Trigueros, C. IFNgamma Regulates Activated Vdelta2+ T Cells through a Feedback Mechanism Mediated by Mesenchymal Stem Cells. PLoS ONE 2017, 12, e0169362. [Google Scholar] [CrossRef]
- Ribot, J.C.; de Barros, A.; Pang, D.J.; Neves, J.F.; Peperzak, V.; Roberts, S.J.; Girardi, M.; Borst, J.; Hayday, A.C.; Pennington, D.J.; et al. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing gammadelta T cell subsets. Nat. Immunol. 2009, 10, 427–436. [Google Scholar] [CrossRef]
- Zhang, C.; Han, X.; Liu, J.; Chen, L.; Lei, Y.; Chen, K.; Si, J.; Wang, T.Y.; Zhou, H.; Zhao, X.; et al. Single-cell Transcriptomic Analysis Reveals the Cellular Heterogeneity of Mesenchymal Stem Cells. Genom. Proteom. Bioinform. 2022, 20, 70–86. [Google Scholar] [CrossRef]
- Spitzhorn, L.S.; Megges, M.; Wruck, W.; Rahman, M.S.; Otte, J.; Degistirici, O.; Meisel, R.; Sorg, R.V.; Oreffo, R.O.C.; Adjaye, J. Human iPSC-derived MSCs (iMSCs) from aged individuals acquire a rejuvenation signature. Stem Cell Res. Ther. 2019, 10, 100. [Google Scholar] [CrossRef]
- Hou, G.; Dong, C.; Dong, Z.; Liu, G.; Xu, H.; Chen, L.; Liu, L.; Wang, H.; Zhou, W. Upregulate KIF4A Enhances Proliferation, Invasion of Hepatocellular Carcinoma and Indicates poor prognosis Across Human Cancer Types. Sci. Rep. 2017, 7, 4148. [Google Scholar] [CrossRef]
- Lian, R.; Ma, H.; Wu, Z.; Zhang, G.; Jiao, L.; Miao, W.; Jin, Q.; Li, R.; Chen, P.; Shi, H.; et al. EZH2 promotes cell proliferation by regulating the expression of RUNX3 in laryngeal carcinoma. Mol. Cell. Biochem. 2018, 439, 35–43. [Google Scholar] [CrossRef]
- Zhou, C.; Gao, Y.; Ding, P.; Wu, T.; Ji, G. The role of CXCL family members in different diseases. Cell Death Discov. 2023, 9, 212. [Google Scholar] [CrossRef]
- Mebarki, M.; Abadie, C.; Larghero, J.; Cras, A. Human umbilical cord-derived mesenchymal stem/stromal cells: A promising candidate for the development of advanced therapy medicinal products. Stem Cell Res. Ther. 2021, 12, 152. [Google Scholar] [CrossRef]
- de Witte, S.F.H.; Merino, A.M.; Franquesa, M.; Strini, T.; van Zoggel, J.A.A.; Korevaar, S.S.; Luk, F.; Gargesha, M.; O’Flynn, L.; Roy, D.; et al. Cytokine treatment optimises the immunotherapeutic effects of umbilical cord-derived MSC for treatment of inflammatory liver disease. Stem Cell Res. Ther. 2017, 8, 140. [Google Scholar] [CrossRef]
- Lee, H.R.; Kim, S.; Shin, S.; Jeong, S.Y.; Lee, D.W.; Lim, S.U.; Kang, J.Y.; Son, M.Y.; Lee, C.; Yu, K.R.; et al. iPSC-Derived MSCs Are a Distinct Entity of MSCs with Higher Therapeutic Potential than Their Donor-Matched Parental MSCs. Int. J. Mol. Sci. 2023, 24, 881. [Google Scholar] [CrossRef]
- Sanchez-Diaz, M.; Quinones-Vico, M.I.; Sanabria de la Torre, R.; Montero-Vilchez, T.; Sierra-Sanchez, A.; Molina-Leyva, A.; Arias-Santiago, S. Biodistribution of Mesenchymal Stromal Cells after Administration in Animal Models and Humans: A Systematic Review. J. Clin. Med. 2021, 10, 2925. [Google Scholar] [CrossRef]
- Chatterjee, D.; Marquardt, N.; Tufa, D.M.; Hatlapatka, T.; Hass, R.; Kasper, C.; von Kaisenberg, C.; Schmidt, R.E.; Jacobs, R. Human Umbilical Cord-Derived Mesenchymal Stem Cells Utilize Activin-A to Suppress Interferon-gamma Production by Natural Killer Cells. Front. Immunol. 2014, 5, 662. [Google Scholar] [CrossRef]
- Xin, J.; Xu, R.; Lin, S.; Xin, M.; Cai, W.; Zhou, J.; Fu, C.; Zhen, G.; Lai, J.; Li, Y.; et al. Clinical potential of TCF21 methylation in the diagnosis of renal cell carcinoma. Oncol. Lett. 2016, 12, 1265–1270. [Google Scholar] [CrossRef]
- Shao, K.; Koch, C.; Gupta, M.K.; Lin, Q.; Lenz, M.; Laufs, S.; Denecke, B.; Schmidt, M.; Linke, M.; Hennies, H.C.; et al. Induced pluripotent mesenchymal stromal cell clones retain donor-derived differences in DNA methylation profiles. Mol. Ther. 2013, 21, 240–250. [Google Scholar] [CrossRef]
- Ankrum, J.A.; Ong, J.F.; Karp, J.M. Mesenchymal stem cells: Immune evasive, not immune privileged. Nat. Biotechnol. 2014, 32, 252–260. [Google Scholar] [CrossRef]
- Kidd, S.; Spaeth, E.; Dembinski, J.L.; Dietrich, M.; Watson, K.; Klopp, A.; Battula, V.L.; Weil, M.; Andreeff, M.; Marini, F.C. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells 2009, 27, 2614–2623. [Google Scholar] [CrossRef]
- Wu, H.H.; Zhou, Y.; Tabata, Y.; Gao, J.Q. Mesenchymal stem cell-based drug delivery strategy: From cells to biomimetic. J. Control. Release 2019, 294, 102–113. [Google Scholar] [CrossRef]
- Soontararak, S.; Chow, L.; Johnson, V.; Coy, J.; Wheat, W.; Regan, D.; Dow, S. Mesenchymal Stem Cells (MSC) Derived from Induced Pluripotent Stem Cells (iPSC) Equivalent to Adipose-Derived MSC in Promoting Intestinal Healing and Microbiome Normalization in Mouse Inflammatory Bowel Disease Model. Stem Cells Transl. Med. 2018, 7, 456–467. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Zhang, Y.; Li, Z.; Zhou, S.; Tang, Q.; Wang, Z.; Xiao, R.; Feng, M.; Wu, L.; Liang, D. Mesenchymal Stem Cells Derived from Human Urine-Derived iPSCs Exhibit Low Immunogenicity and Reduced Immunomodulatory Profile. Int. J. Mol. Sci. 2024, 25, 10394. https://doi.org/10.3390/ijms251910394
Wang P, Zhang Y, Li Z, Zhou S, Tang Q, Wang Z, Xiao R, Feng M, Wu L, Liang D. Mesenchymal Stem Cells Derived from Human Urine-Derived iPSCs Exhibit Low Immunogenicity and Reduced Immunomodulatory Profile. International Journal of Molecular Sciences. 2024; 25(19):10394. https://doi.org/10.3390/ijms251910394
Chicago/Turabian StyleWang, Peiyun, Ying Zhang, Zhixing Li, Shenglan Zhou, Qiyu Tang, Zujia Wang, Rou Xiao, Mai Feng, Lingqian Wu, and Desheng Liang. 2024. "Mesenchymal Stem Cells Derived from Human Urine-Derived iPSCs Exhibit Low Immunogenicity and Reduced Immunomodulatory Profile" International Journal of Molecular Sciences 25, no. 19: 10394. https://doi.org/10.3390/ijms251910394