
Crosstalk between BER and NHEJ in XRCC4-Deficient Cells Depending on hTERT Overexpression

 ,
,
Abstract
:1. Introduction
2. Results
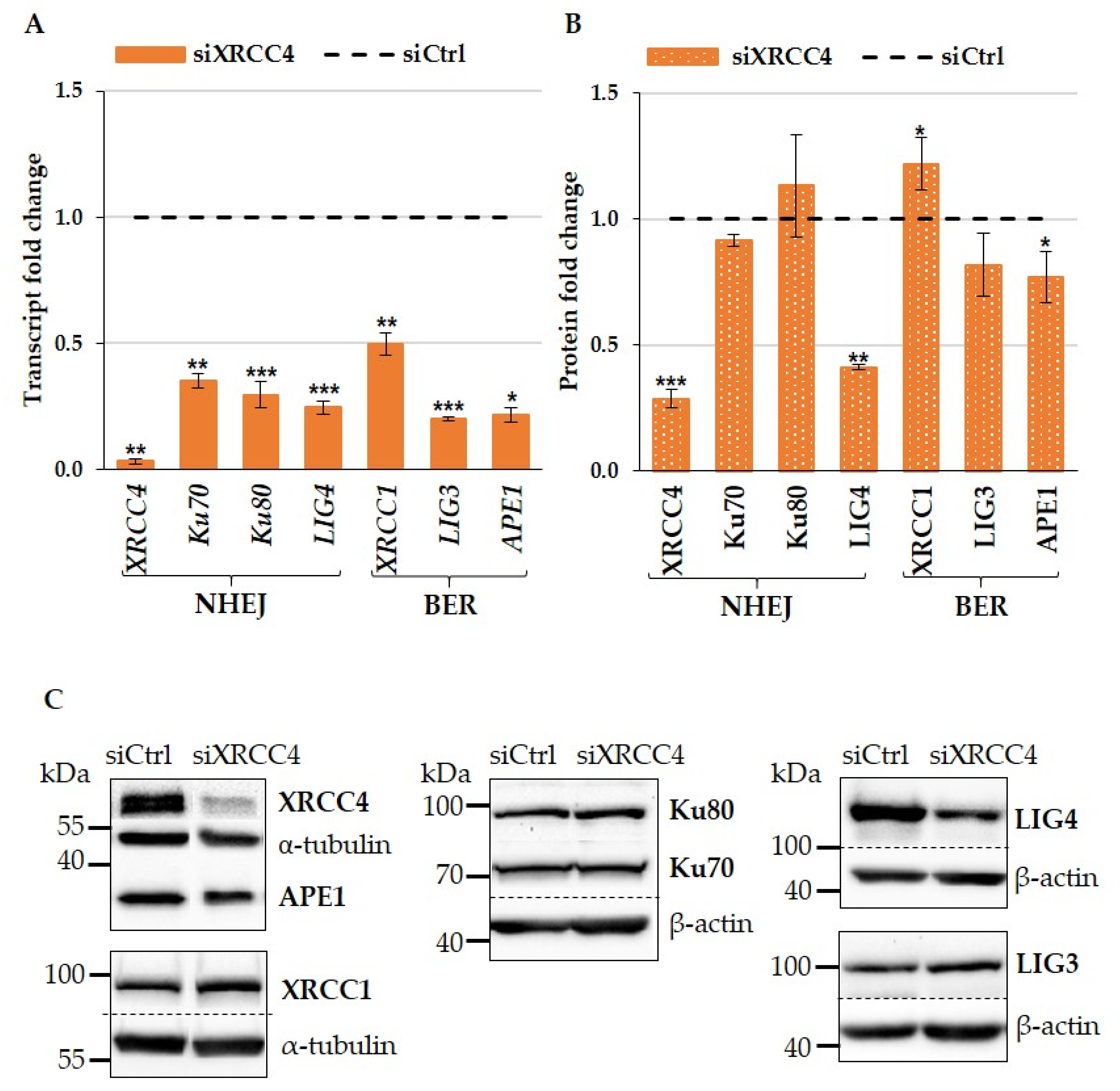
2.1. XRCC4 Knockdown Caused NHEJ Deficiency in Normal TIG-1 Cells
2.2. Effect of XRCC4 Knockdown on Expression of BER Components in Normal TIG-1 Cells
2.3. Effects of XRCC4 Knockdown on BER and NHEJ Expression in hTERT Modified NBE1 Cells
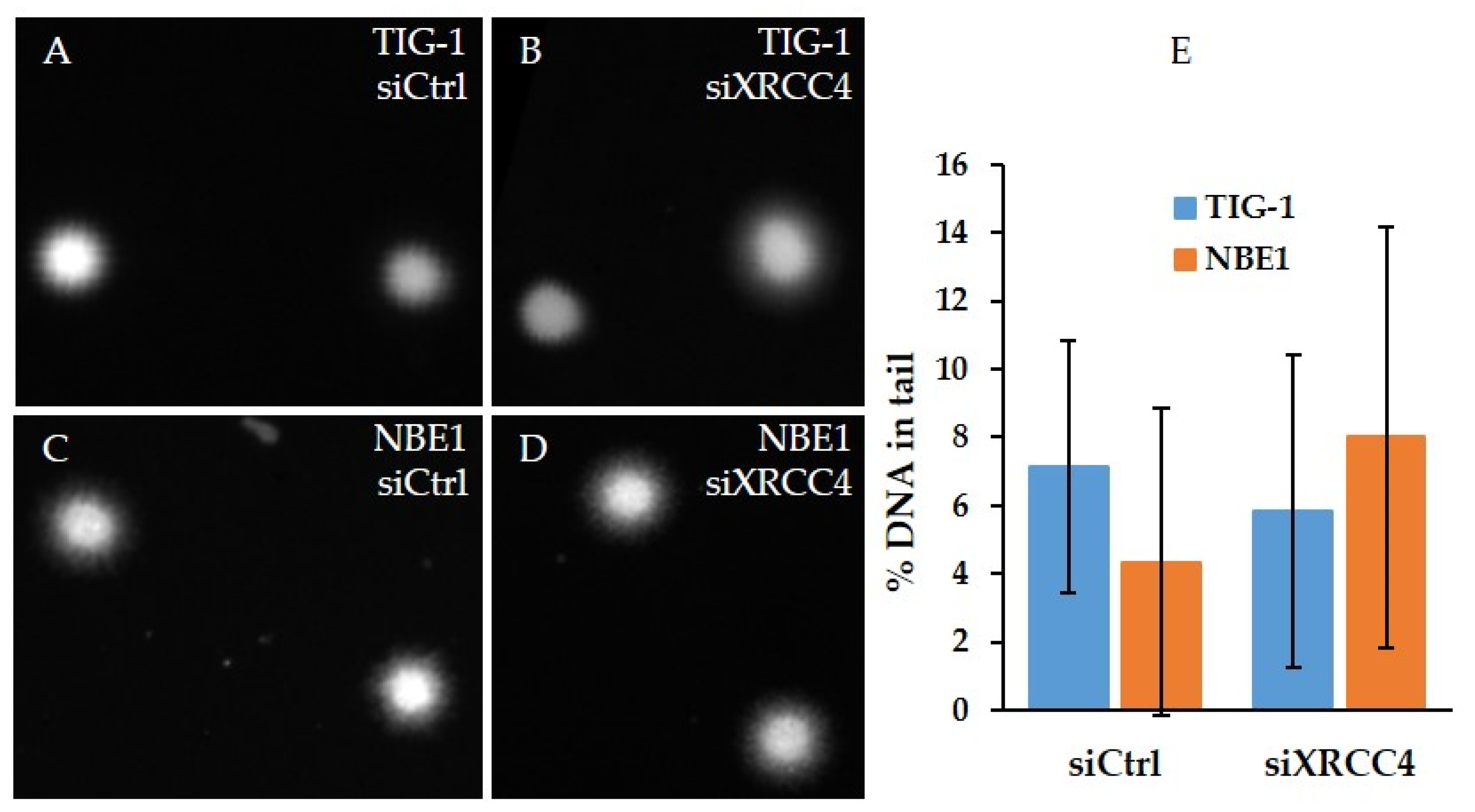
2.4. Analysis of DNA Damage after XRCC4 Knockdown in Normal and hTERT-Modified Cells
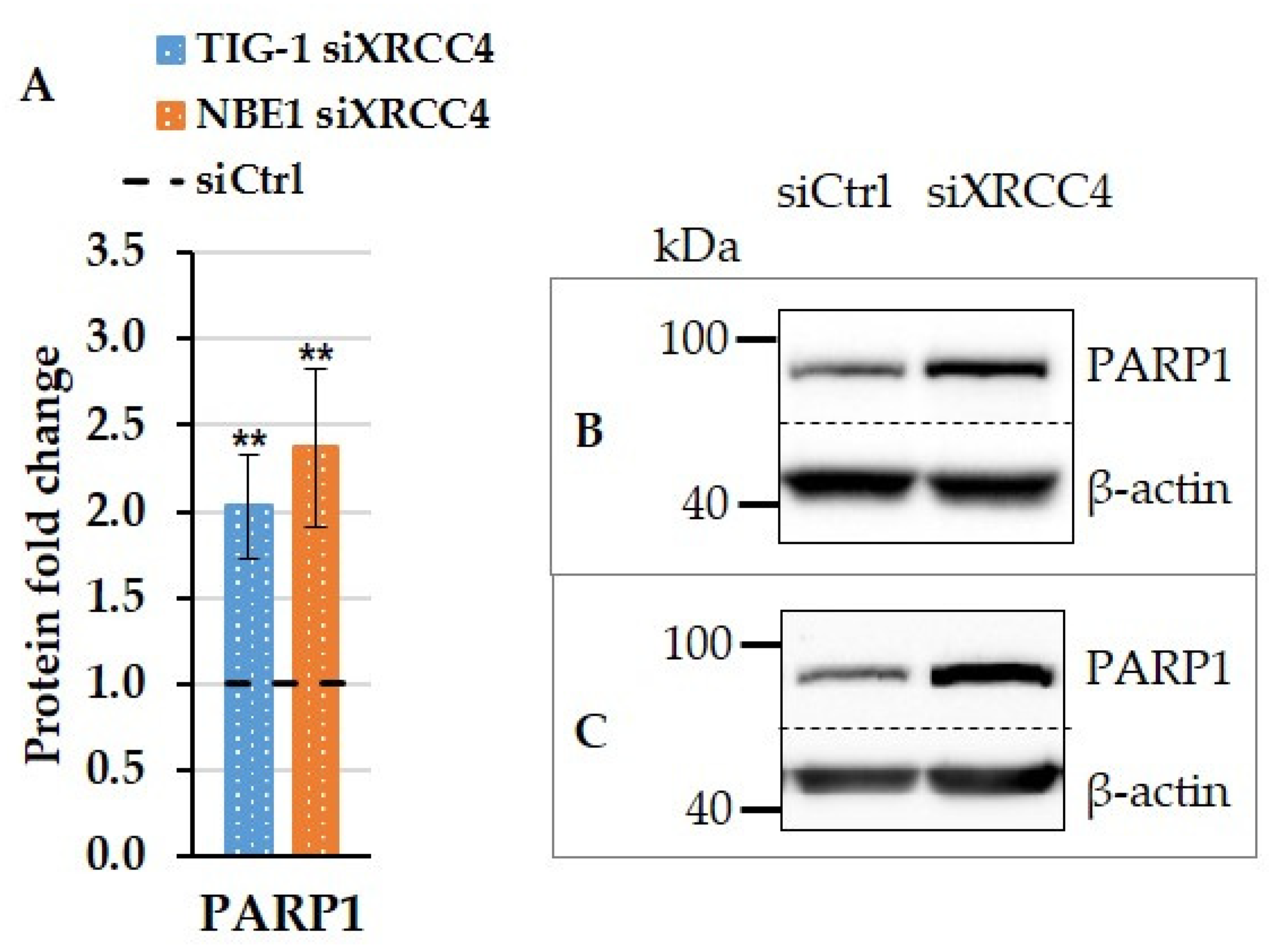
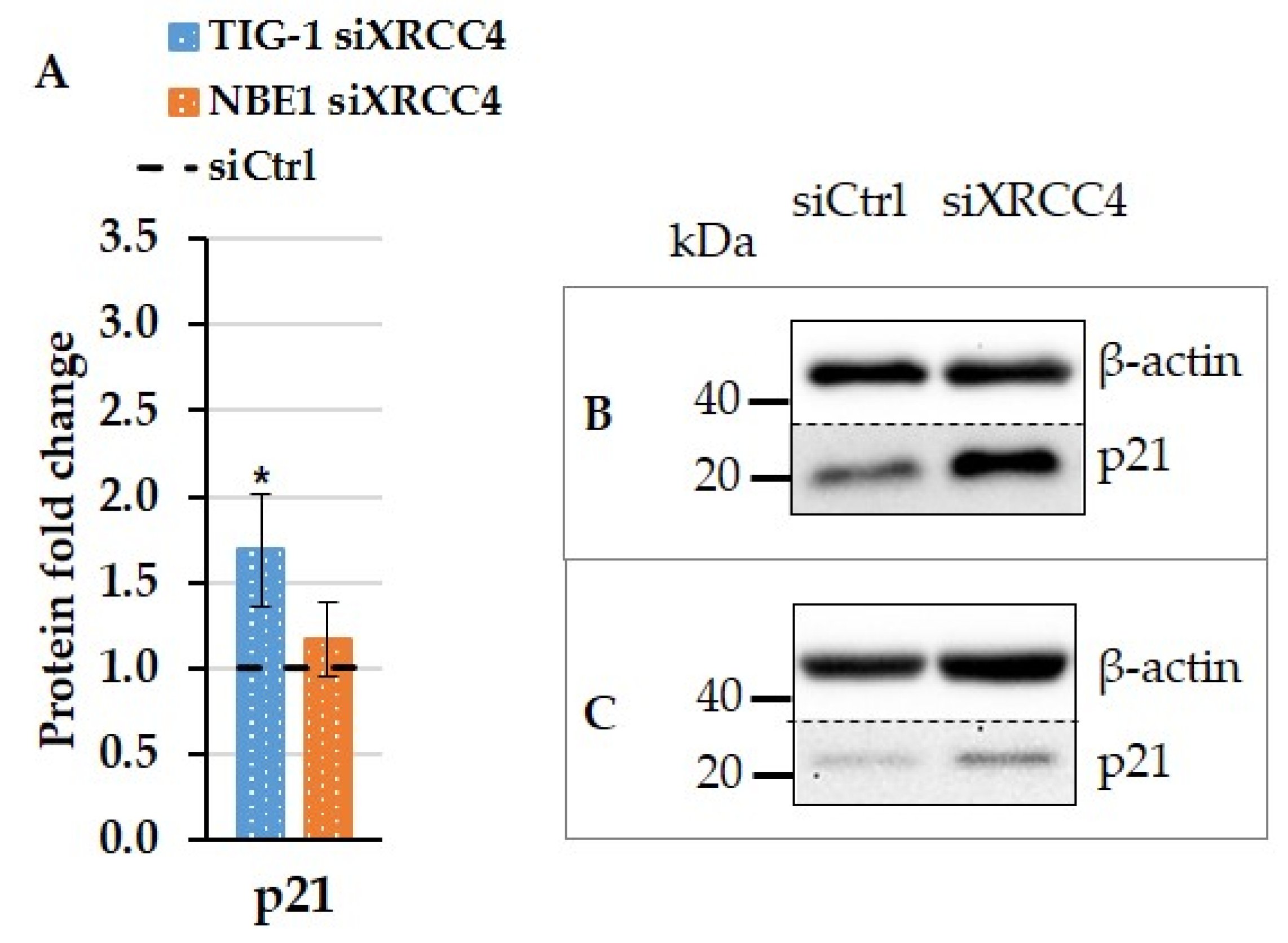
2.5. Effect of XRCC4 Knockdown on PARP1 Expression in Normal and hTERT-Modified Cells
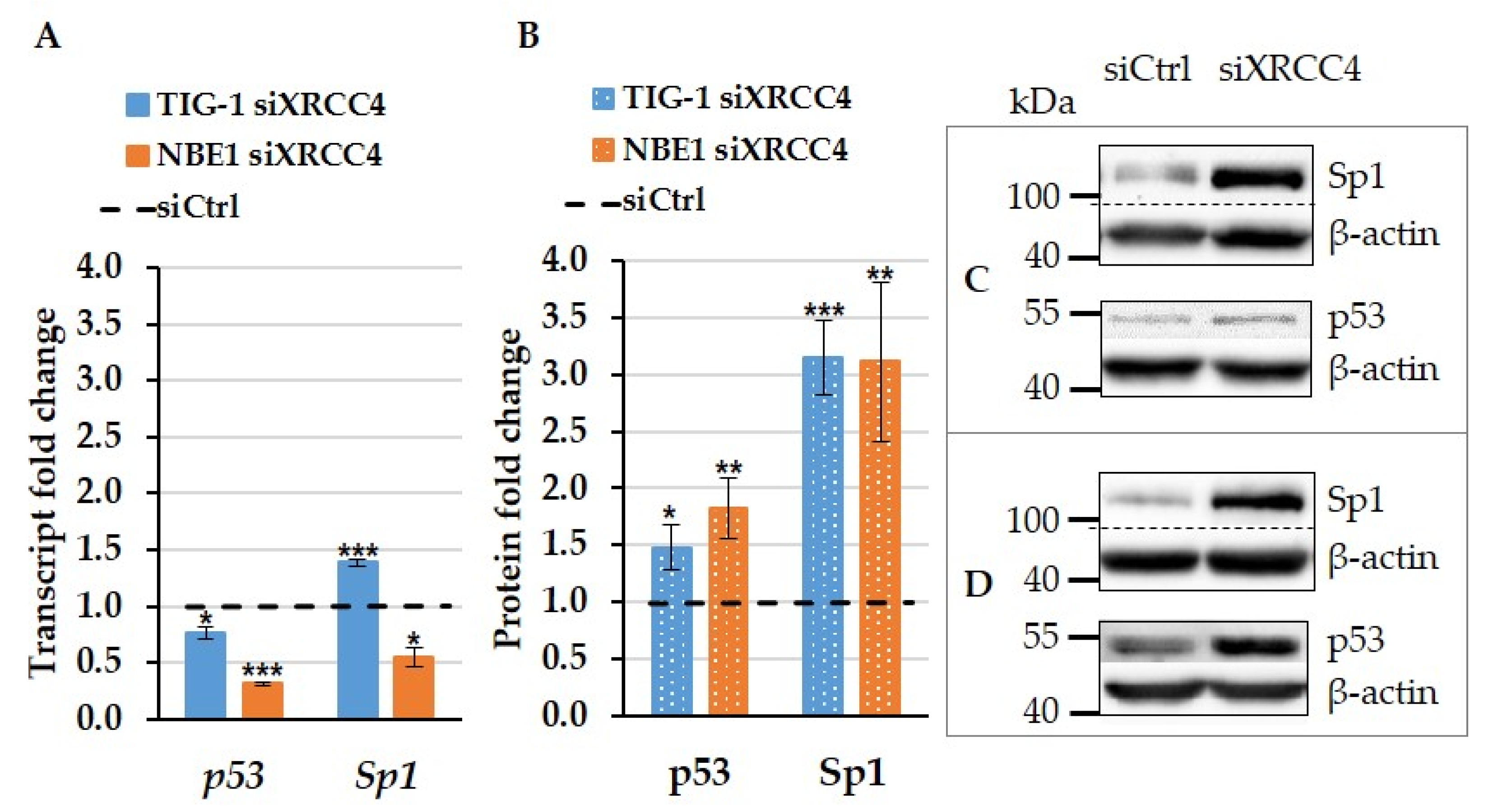
2.6. The Effect of XRCC4 Knockdown on Transcription Factors p53 and Sp1 in Normal and hTERT Modified Cells
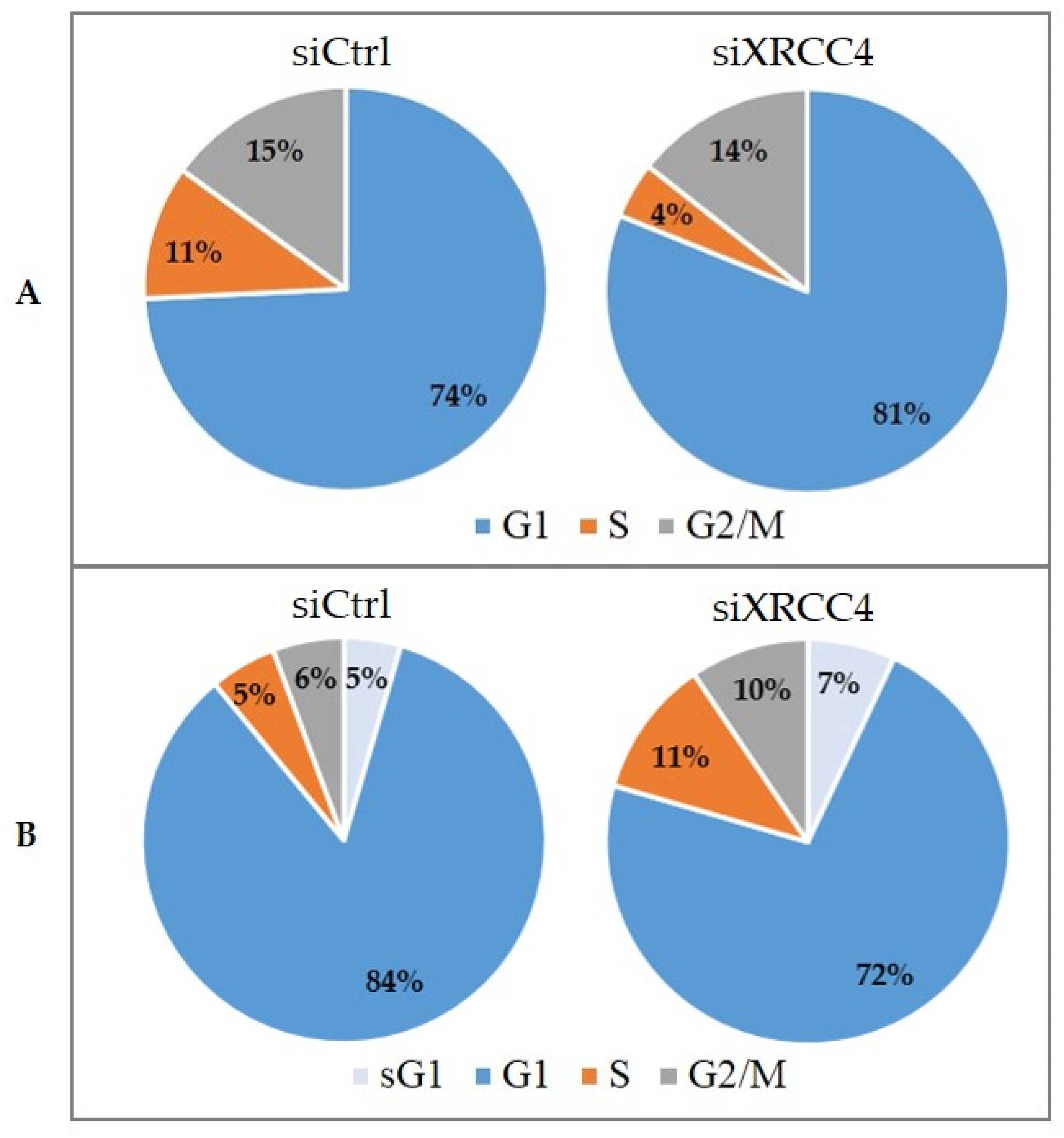
2.7. Cell Cycle Alterations after XRCC4 Knockdown in Normal and hTERT-Modified Cells
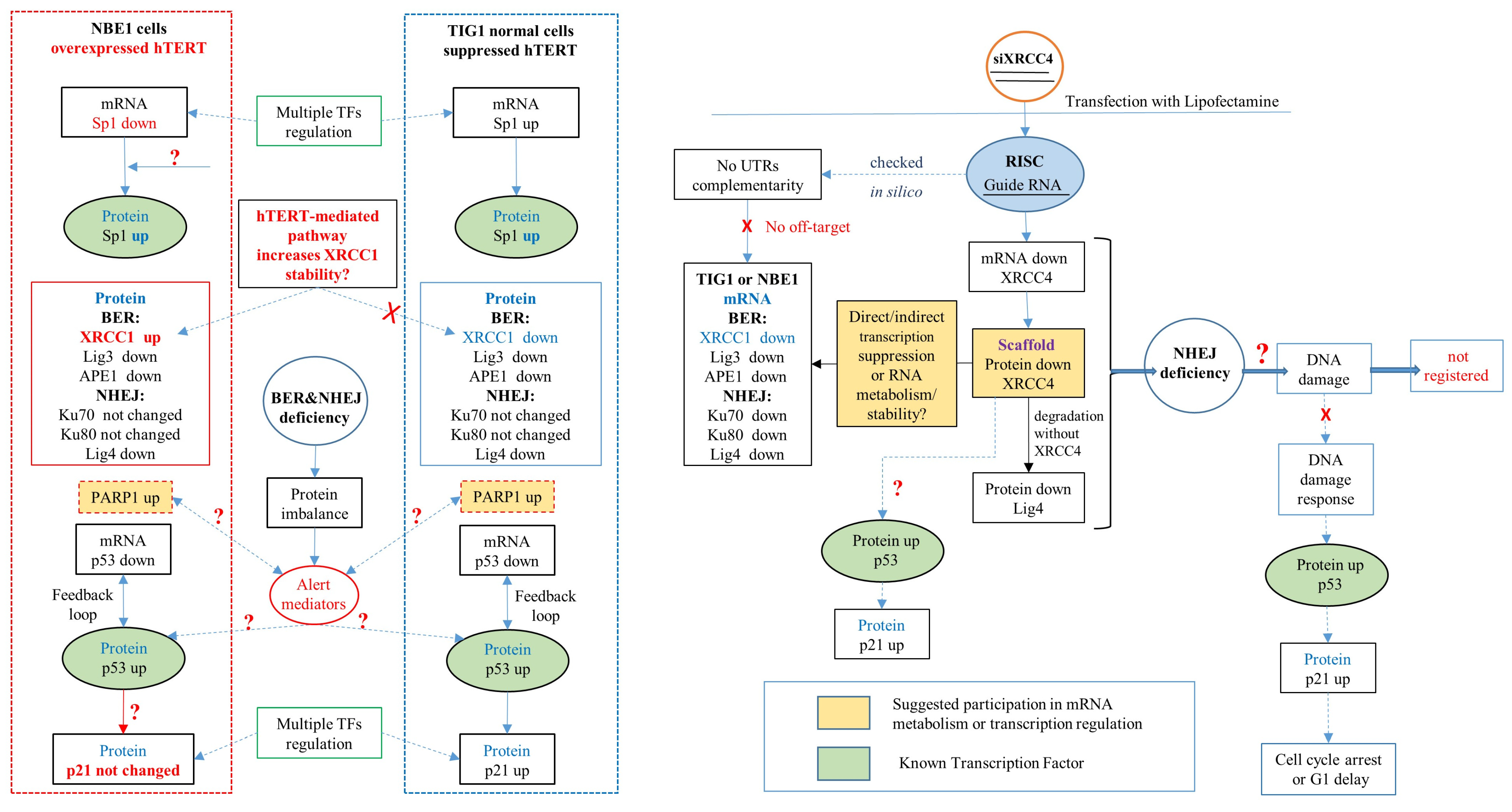
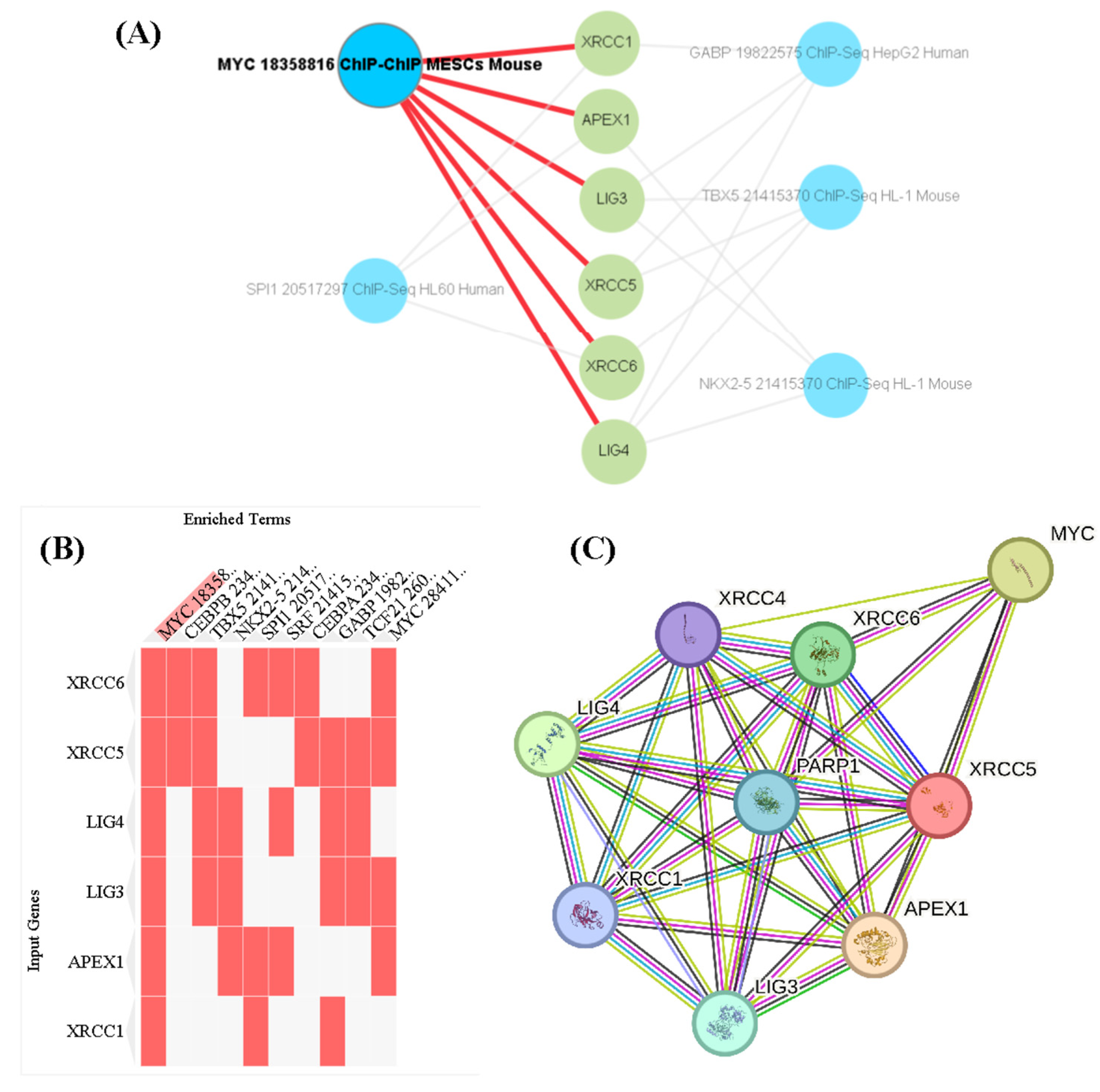
3. Discussion
4. Materials and Methods
4.1. Cells and Cell Culture
4.2. siRNA Transfections
4.3. Real-Time PCR (qPCR)
4.4. Western Blot
4.5. Alkaline Comet Assay
4.6. Image Analysis
4.7. Cell Cycle Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic Instability—An Evolving Hallmark of Cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.-K. DNA Damage Repair: Historical Perspectives, Mechanistic Pathways and Clinical Translation for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-Art Strategies for Targeting the DNA Damage Response in Cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Curtin, N.J. DNA Repair Dysregulation from Cancer Driver to Therapeutic Target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.O.; Sekiguchi, J.M.; Chang, S.; Frank, K.M.; Gao, Y.; DePinho, R.A.; Alt, F.W. The Nonhomologous End-Joining Pathway of DNA Repair Is Required for Genomic Stability and the Suppression of Translocations. Proc. Natl. Acad. Sci. USA 2000, 97, 6630–6633. [Google Scholar] [CrossRef]
- Horikawa, I.; Barrett, J.C. Transcriptional Regulation of the Telomerase hTERT Gene as a Target for Cellular and Viral Oncogenic Mechanisms. Carcinogenesis 2003, 24, 1167–1176. [Google Scholar] [CrossRef]
- Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor P21cip1/Waf1 in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef]
- Sorteberg, A.L.; Halipi, V.; Wickström, M.; Shirazi Fard, S. The Cyclin Dependent Kinase Inhibitor p21Cip1/Waf1 Is a Therapeutic Target in High-Risk Neuroblastoma. Front. Oncol. 2022, 12, 906194. [Google Scholar] [CrossRef]
- Li, L.; Guan, Y.; Chen, X.; Yang, J.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11, 629266. [Google Scholar] [CrossRef]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms Underlying the Activation of TERT Transcription and Telomerase Activity in Human Cancer: Old Actors and New Players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef]
- Yang, R.; Han, Y.; Guan, X.; Hong, Y.; Meng, J.; Ding, S.; Long, Q.; Yi, W. Regulation and Clinical Potential of Telomerase Reverse Transcriptase (TERT/hTERT) in Breast Cancer. Cell Commun. Signal 2023, 21, 218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Cheng, D.; Wang, S.; Zhu, J. Human Specific Regulation of the Telomerase Reverse Transcriptase Gene. Genes 2016, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Leão, R.; Apolónio, J.D.; Lee, D.; Figueiredo, A.; Tabori, U.; Castelo-Branco, P. Mechanisms of Human Telomerase Reverse Transcriptase (hTERT) Regulation: Clinical Impacts in Cancer. J. Biomed. Sci. 2018, 25, 22. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.-S.; Wright, W.E.; Shay, J.W. Human Telomerase and Its Regulation. Microbiol. Mol. Biol. Rev. 2002, 66, 407–425. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, R.-L.; Liu, J.-J.; Zhou, J.; Li, X.; Hu, W.-W.; Jiang, W.-J.; Hao, N.-B. The Prognostic Significance of hTERT Overexpression in Cancers: A Systematic Review and Meta-Analysis. Medicine 2018, 97, e11794. [Google Scholar] [CrossRef]
- Tomlinson, R.L.; Ziegler, T.D.; Supakorndej, T.; Terns, R.M.; Terns, M.P. Cell Cycle-Regulated Trafficking of Human Telomerase to Telomeres. MBoC 2006, 17, 955–965. [Google Scholar] [CrossRef]
- Maida, Y.; Yasukawa, M.; Furuuchi, M.; Lassmann, T.; Possemato, R.; Okamoto, N.; Kasim, V.; Hayashizaki, Y.; Hahn, W.C.; Masutomi, K. An RNA-Dependent RNA Polymerase Formed by TERT and the RMRP RNA. Nature 2009, 461, 230–235. [Google Scholar] [CrossRef]
- Chiodi, I.; Mondello, C. Telomere-Independent Functions of Telomerase in Nuclei, Cytoplasm, and Mitochondria. Front. Oncol. 2012, 2, 133. [Google Scholar] [CrossRef]
- Ghareghomi, S.; Ahmadian, S.; Zarghami, N.; Kahroba, H. Fundamental Insights into the Interaction between Telomerase/TERT and Intracellular Signaling Pathways. Biochimie 2021, 181, 12–24. [Google Scholar] [CrossRef]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. The Cell Cycle: A Review of Regulation, Deregulation and Therapeutic Targets in Cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef]
- Hume, S.; Dianov, G.L.; Ramadan, K. A Unified Model for the G1/S Cell Cycle Transition. Nucleic Acids Res. 2020, 48, 12483–12501. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.M.; Carr, A.M. Integrating DNA Damage Repair with the Cell Cycle. Curr. Opin. Cell Biol. 2018, 52, 120–125. [Google Scholar] [CrossRef]
- Clay, D.E.; Fox, D.T. DNA Damage Responses during the Cell Cycle: Insights from Model Organisms and Beyond. Genes 2021, 12, 1882. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.; Clemente-Blanco, A. Cell Cycle and DNA Repair Regulation in the Damage Response: Protein Phosphatases Take Over the Reins. Int. J. Mol. Sci. 2020, 21, 446. [Google Scholar] [CrossRef]
- Barnum, K.J.; O’Connell, M.J. Cell Cycle Regulation by Checkpoints. In Cell Cycle Control; Noguchi, E., Gadaleta, M.C., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2014; Volume 1170, pp. 29–40. ISBN 978-1-4939-0887-5. [Google Scholar]
- Reisman, D.; Takahashi, P.; Polson, A.; Boggs, K. Transcriptional Regulation of the p53 Tumor Suppressor Gene in S-Phase of the Cell-Cycle and the Cellular Response to DNA Damage. Biochem. Res. Int. 2012, 2012, 808934. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, P.; Polson, A.; Reisman, D. Elevated Transcription of the P53 Gene in Early S-Phase Leads to a Rapid DNA-Damage Response during S-Phase of the Cell Cycle. Apoptosis 2011, 16, 950–958. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. Pathways Governing G1/S Transition and Their Response to DNA Damage. FEBS Lett. 2001, 490, 117–122. [Google Scholar] [CrossRef]
- Xia, W.; Ci, S.; Li, M.; Wang, M.; Dianov, G.L.; Ma, Z.; Li, L.; Hua, K.; Alagamuthu, K.K.; Qing, L.; et al. Two-way Crosstalk between BER and c-NHEJ Repair Pathway Is Mediated by Pol-β and Ku70. FASEB J. 2019, 33, 11668–11681. [Google Scholar] [CrossRef]
- Yu, Y.; Sun, Y.; Li, Z.; Li, J.; Tian, D. Systematic Analysis Identifies XRCC4 as a Potential Immunological and Prognostic Biomarker Associated with Pan-Cancer. BMC Bioinform. 2023, 24, 44. [Google Scholar] [CrossRef]
- Wu, C.-N.; Liang, S.-Y.; Tsai, C.-W.; Bau, D.-T. The Role of XRCC4 in Carcinogenesis and Anticancer Drug Discovery. Recent Pat. Anti-Cancer Drug Discov. 2008, 3, 209–219. [Google Scholar] [CrossRef]
- Lu, J.; Wang, X.-Z.; Zhang, T.-Q.; Huang, X.-Y.; Yao, J.-G.; Wang, C.; Wei, Z.-H.; Ma, Y.; Wu, X.-M.; Luo, C.-Y.; et al. Prognostic Significance of XRCC4 Expression in Hepatocellular Carcinoma. Oncotarget 2017, 8, 87955–87970. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA Repair by Nonhomologous End Joining and Homologous Recombination during Cell Cycle in Human Cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.L.; Dianov, G.L. Co-Ordination of Base Excision Repair and Genome Stability. DNA Repair. 2013, 12, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Khoronenkova, S.V.; Dianov, G.L. ATM Prevents DSB Formation by Coordinating SSB Repair and Cell Cycle Progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3997–4002. [Google Scholar] [CrossRef] [PubMed]
- Stinson, B.M.; Loparo, J.J. Repair of DNA Double-Strand Breaks by the Nonhomologous End Joining Pathway. Annu. Rev. Biochem. 2021, 90, 137–164. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA End-Joining for Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef]
- Stadler, J.; Richly, H. Regulation of DNA Repair Mechanisms: How the Chromatin Environment Regulates the DNA Damage Response. Int. J. Mol. Sci. 2017, 18, 1715. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Ho, T.L.F.; Hariharan, A.; Goh, H.C.; Wong, Y.L.; Verkaik, N.S.; Lee, M.Y.; Tam, W.L.; Van Gent, D.C.; Venkitaraman, A.R.; et al. Rapid Recruitment of P53 to DNA Damage Sites Directs DNA Repair Choice and Integrity. Proc. Natl. Acad. Sci. USA 2022, 119, e2113233119. [Google Scholar] [CrossRef]
- Beishline, K.; Kelly, C.M.; Olofsson, B.A.; Koduri, S.; Emrich, J.; Greenberg, R.A.; Azizkhan-Clifford, J. Sp1 Facilitates DNA Double-Strand Break Repair through a Nontranscriptional Mechanism. Mol. Cell. Biol. 2012, 32, 3790–3799. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How Does P53 Induce Apoptosis and How Does This Relate to P53-Mediated Tumour Suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, L.; Gilmour, J.; Bonifer, C. The Role of the Ubiquitously Expressed Transcription Factor Sp1 in Tissue-Specific Transcriptional Regulation and in Disease. Yale J. Biol. Med. 2016, 89, 513–525. [Google Scholar]
- Helton, E.S.; Chen, X. P53 Modulation of the DNA Damage Response. J. Cell. Biochem. 2007, 100, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Wierstra, I. Sp1: Emerging Roles--beyond Constitutive Activation of TATA-Less Housekeeping Genes. Biochem. Biophys. Res. Commun. 2008, 372, 1–13. [Google Scholar] [CrossRef]
- Poletto, M.; Legrand, A.J.; Fletcher, S.C.; Dianov, G.L. P53 Coordinates Base Excision Repair to Prevent Genomic Instability. Nucleic Acids Res. 2016, 44, 3165–3175. [Google Scholar] [CrossRef]
- Loshchenova, P.S.; Sergeeva, S.V.; Limonov, D.V.; Guo, Z.; Dianov, G.L. Sp1-Independent Downregulation of NHEJ in Response to BER Deficiency. DNA Repair. 2020, 86, 102740. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Aizawa, S.; Ooka, H.; Ohsawa, T.; Kaji, K.; Kondo, H.; Kobayashi, T.; Noumura, T.; Matsuo, M.; Mitsui, Y.; et al. A New Human Diploid Cell Strain, TIG-1, for the Research on Cellular Aging. Exp. Gerontol. 1980, 15, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Ouellette, M.M.; McDaniel, L.D.; Wright, W.E.; Shay, J.W.; Schultz, R.A. The Establishment of Telomerase-Immortalized Cell Lines Representing Human Chromosome Instability Syndromes. Hum. Mol. Genet. 2000, 9, 403–411. [Google Scholar] [CrossRef]
- Stead, L.F.; Berri, S.; Wood, H.M.; Egan, P.; Conway, C.; Daly, C.; Papagiannopoulos, K.; Rabbitts, P. The Transcriptional Consequences of Somatic Amplifications, Deletions, and Rearrangements in a Human Lung Squamous Cell Carcinoma. Neoplasia 2012, 14, 1075–1086. [Google Scholar] [CrossRef]
- Chen, J.; Peng, Y.; Zhang, H.; Wang, K.; Zhao, C.; Zhu, G.; Reddy Palli, S.; Han, Z. Off-target effects of RNAi correlate with the mismatch rate between dsRNA and non-target mRNA. RNA Biol. 2021, 18, 1747–1759. [Google Scholar] [CrossRef]
- Critchlow, S.E.; Bowater, R.P.; Jackson, S.P. Mammalian DNA Double-Strand Break Repair Protein XRCC4 Interacts with DNA Ligase IV. Curr. Biol. 1997, 7, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Bowden, R.D.; Buckwalter, M.R.; McBride, J.F.; Johnson, D.A.; Murray, B.K.; O’Neill, K.L. Tail Profile: A More Accurate System for Analyzing DNA Damage Using the Comet Assay. Mutat. Res. 2003, 537, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.L.; Ren, E.C. Functional Aspects of PARP1 in DNA Repair and Transcription. Biomolecules 2012, 2, 524–548. [Google Scholar] [CrossRef]
- Wei, H.; Yu, X. Functions of PARylation in DNA Damage Repair Pathways. Genom. Proteom. Bioinform. 2016, 14, 131–139. [Google Scholar] [CrossRef]
- Tufan, A.B.; Lazarow, K.; Kolesnichenko, M.; Sporbert, A.; von Kries, J.P.; Scheidereit, C. TSG101 Associates with PARP1 and Is Essential for PARylation and DNA Damage-Induced NF-κB Activation. EMBO J. 2022, 41, e110372. [Google Scholar] [CrossRef]
- Orlando, G.; Khoronenkova, S.V.; Dianova, I.I.; Parsons, J.L.; Dianov, G.L. ARF Induction in Response to DNA Strand Breaks Is Regulated by PARP1. Nucleic Acids Res. 2014, 42, 2320–2329. [Google Scholar] [CrossRef] [PubMed]
- Süsse, S.; Scholz, C.-J.; Bürkle, A.; Wiesmüller, L. Poly(ADP-Ribose) Polymerase (PARP-1) and P53 Independently Function in Regulating Double-Strand Break Repair in Primate Cells. Nucleic Acids Res. 2004, 32, 669–680. [Google Scholar] [CrossRef]
- Wei, L.; Nakajima, S.; Hsieh, C.-L.; Kanno, S.; Masutani, M.; Levine, A.S.; Yasui, A.; Lan, L. Damage Response of XRCC1 at Sites of DNA Single Strand Breaks Is Regulated by Phosphorylation and Ubiquitylation after Degradation of Poly(ADP-Ribose). J. Cell Sci. 2013, 126, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Zaniolo, K.; Desnoyers, S.; Leclerc, S.; Guérin, S.L. Regulation of Poly(ADP-Ribose) Polymerase-1 (PARP-1) Gene Expression through the Post-Translational Modification of Sp1: A Nuclear Target Protein of PARP-1. BMC Mol. Biol. 2007, 8, 96. [Google Scholar] [CrossRef]
- Kumar, V.; Kumar, A.; Mir, K.U.I.; Yadav, V.; Chauhan, S.S. Pleiotropic Role of PARP1: An Overview. 3 Biotech 2022, 12, 3. [Google Scholar] [CrossRef]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-Ribosyl)Ation by PARP1: Reaction Mechanism and Regulatory Proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.A.; Kim, N.S.; Yu, S.-W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(ADP-Ribose) (PAR) Polymer Is a Death Signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef] [PubMed]
- Boehi, F.; Manetsch, P.; Hottiger, M.O. Interplay between ADP-Ribosyltransferases and Essential Cell Signaling Pathways Controls Cellular Responses. Cell Discov. 2021, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Terasawa, M.; Shinohara, A.; Shinohara, M. Canonical Non-Homologous End Joining in Mitosis Induces Genome Instability and Is Suppressed by M-Phase-Specific Phosphorylation of XRCC4. PLoS Genet. 2014, 10, e1004563. [Google Scholar] [CrossRef]
- Drouet, J.; Delteil, C.; Lefrançois, J.; Concannon, P.; Salles, B.; Calsou, P. DNA-Dependent Protein Kinase and XRCC4-DNA Ligase IV Mobilization in the Cell in Response to DNA Double Strand Breaks. J. Biol. Chem. 2005, 280, 7060–7069. [Google Scholar] [CrossRef]
- Berg, E.; Christensen, M.O.; Dalla Rosa, I.; Wannagat, E.; Jänicke, R.U.; Rösner, L.M.; Dirks, W.G.; Boege, F.; Mielke, C. XRCC4 Controls Nuclear Import and Distribution of Ligase IV and Exchanges Faster at Damaged DNA in Complex with Ligase IV. DNA Repair 2011, 10, 1232–1242. [Google Scholar] [CrossRef]
- Mahaney, B.L.; Hammel, M.; Meek, K.; Tainer, J.A.; Lees-Miller, S.P. XRCC4 and XLF Form Long Helical Protein Filaments Suitable for DNA End Protection and Alignment to Facilitate DNA Double Strand Break Repair. Biochem. Cell Biol. 2013, 91, 31–41. [Google Scholar] [CrossRef]
- Kumari, N.; Antil, H.; Kumari, S.; Raghavan, S.C. Deficiency of Ligase IV Leads to Reduced NHEJ, Accumulation of DNA Damage, and Can Sensitize Cells to Cancer Therapeutics. Genomics 2023, 115, 110731. [Google Scholar] [CrossRef]
- Bryans, M.; Valenzano, M.C.; Stamato, T.D. Absence of DNA Ligase IV Protein in XR-1 Cells: Evidence for Stabilization by XRCC4. Mutat. Res. 1999, 433, 53–58. [Google Scholar] [CrossRef]
- Zahid, S.; Seif El Dahan, M.; Iehl, F.; Fernandez-Varela, P.; Le Du, M.-H.; Ropars, V.; Charbonnier, J.B. The Multifaceted Roles of Ku70/80. Int. J. Mol. Sci. 2021, 22, 4134. [Google Scholar] [CrossRef]
- Calsou, P.; Delteil, C.; Frit, P.; Drouet, J.; Salles, B. Coordinated Assembly of Ku and P460 Subunits of the DNA-Dependent Protein Kinase on DNA Ends Is Necessary for XRCC4-Ligase IV Recruitment. J. Mol. Biol. 2003, 326, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Shyu, A.-B.; Wilkinson, M.F.; van Hoof, A. Messenger RNA Regulation: To Translate or to Degrade. EMBO J. 2008, 27, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Schwalb, B.; Pirkl, N.; Maier, K.C.; Schenk, A.; Failmezger, H.; Tresch, A.; Cramer, P. Global Analysis of Eukaryotic mRNA Degradation Reveals Xrn1-Dependent Buffering of Transcript Levels. Mol. Cell 2013, 52, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Haimovich, G.; Medina, D.A.; Causse, S.Z.; Garber, M.; Millán-Zambrano, G.; Barkai, O.; Chávez, S.; Pérez-Ortín, J.E.; Darzacq, X.; Choder, M. Gene Expression Is Circular: Factors for mRNA Degradation Also Foster mRNA Synthesis. Cell 2013, 153, 1000–1011. [Google Scholar] [CrossRef]
- Hata, Y.; Iida, J. Scaffold Protein. In Encyclopedia of Neuroscience; Binder, M.D., Hirokawa, N., Windhorst, U., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 3613–3616. ISBN 978-3-540-23735-8. [Google Scholar]
- DiRusso, C.J.; Dashtiahangar, M.; Gilmore, T.D. Scaffold Proteins as Dynamic Integrators of Biological Processes. J. Biol. Chem. 2022, 298, 102628. [Google Scholar] [CrossRef]
- Su, Z.; Dhusia, K.; Wu, Y. Understand the Functions of Scaffold Proteins in Cell Signaling by a Mesoscopic Simulation Method. Biophys. J. 2020, 119, 2116–2126. [Google Scholar] [CrossRef]
- Buday, L.; Tompa, P. Functional Classification of Scaffold Proteins and Related Molecules. FEBS J. 2010, 277, 4348–4355. [Google Scholar] [CrossRef]
- Hazegh Nikroo, A.; Lemmens, L.J.M.; Wezeman, T.; Ottmann, C.; Merkx, M.; Brunsveld, L. Switchable Control of Scaffold Protein Activity via Engineered Phosphoregulated Autoinhibition. ACS Synth. Biol. 2022, 11, 2464–2472. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Intrinsic Disorder in Scaffold Proteins: Getting More from Less. Prog. Biophys. Mol. Biol. 2008, 98, 85–106. [Google Scholar] [CrossRef]
- Balázs, A.; Csizmok, V.; Buday, L.; Rakács, M.; Kiss, R.; Bokor, M.; Udupa, R.; Tompa, K.; Tompa, P. High Levels of Structural Disorder in Scaffold Proteins as Exemplified by a Novel Neuronal Protein, CASK-Interactive Protein1. FEBS J. 2009, 276, 3744–3756. [Google Scholar] [CrossRef]
- Hammel, M.; Rey, M.; Yu, Y.; Mani, R.S.; Classen, S.; Liu, M.; Pique, M.E.; Fang, S.; Mahaney, B.L.; Weinfeld, M.; et al. XRCC4 Protein Interactions with XRCC4-like Factor (XLF) Create an Extended Grooved Scaffold for DNA Ligation and Double Strand Break Repair. J. Biol. Chem. 2011, 286, 32638–32650. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The Harmonizome: A Collection of Processed Datasets Gathered to Serve and Mine Knowledge about Genes and Proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Y.; Li, B.; Chen, F.; Shen, W.; Lu, Y.; Zhong, C.; Zhang, C.; Xie, H.; Katanaev, V.L.; et al. WDR74 Modulates Melanoma Tumorigenesis and Metastasis through the RPL5-MDM2-P53 Pathway. Oncogene 2020, 39, 2741–2755. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, J.E.; Xie, Z.; Marino, G.B.; Nguyen, N.; Clarke, D.J.B.; Ma’ayan, A. Enrichr-KG: Bridging enrichment analysis across multiple libraries. Nucleic Acids Res 2023, 51, W168–W179. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Dataset—CHEA Transcription Factor Targets 2022. Available online: https://maayanlab.cloud/Harmonizome/dataset/CHEA+Transcription+Factor+Targets+2022 (accessed on 20 September 2020).
- Ahmadi, S.E.; Rahimi, S.; Zarandi, B.; Chegeni, R.; Safa, M. MYC: A multipurpose oncogene with prognostic and therapeutic implications in blood malignancies. J. Hematol. Oncol. 2021, 14, 121. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res 2023, 6, D638–D646. [Google Scholar] [CrossRef]
- Ratnaparkhe, M.; Wong, J.K.L.; Wei, P.C.; Hlevnjak, M.; Kolb, T.; Simovic, M.; Haag, D.; Paul, Y.; Devens, F.; Northcott, P.; et al. Defective DNA damage repair leads to frequent catastrophic genomic events in murine and human tumors. Nat. Commun. 2018, 9, 4760. [Google Scholar] [CrossRef]
- Kraus, W.L.; Lis, J.T. PARP Goes Transcription. Cell 2003, 113, 677–683. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Kraus, W.L. The PARP Side of the Nucleus: Molecular Actions, Physiological Outcomes, and Clinical Targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Stilmann, M.; Hinz, M.; Arslan, S.C.; Zimmer, A.; Schreiber, V.; Scheidereit, C. A Nuclear Poly(ADP-Ribose)-Dependent Signalosome Confers DNA Damage-Induced IkappaB Kinase Activation. Mol. Cell 2009, 36, 365–378. [Google Scholar] [CrossRef]
- David, K.K.; Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Parthanatos, a Messenger of Death. Front. Biosci. (Landmark Ed.) 2009, 14, 1116–1128. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Huang, K.; Li, X.; Du, M.; Kang, X.; Luo, X.; Gao, L.; Wang, C.; Zhang, Y.; Zhang, C.; et al. Identification of Poly(ADP-Ribose) Polymerase-1 as a Cell Cycle Regulator through Modulating Sp1 Mediated Transcription in Human Hepatoma Cells. PLoS ONE 2013, 8, e82872. [Google Scholar] [CrossRef]
- Oppenheim, A.; Lahav, G. The Puzzling Interplay between P53 and Sp1. Aging 2017, 9, 1355–1356. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Chen, J. Mechanism of P53 Stabilization by ATM after DNA Damage. Cell Cycle 2010, 9, 472–478. [Google Scholar] [CrossRef]
- Mendoza, M.; Mandani, G.; Momand, J. The MDM2 Gene Family. Biomol. Concepts 2014, 5, 9–19. [Google Scholar] [CrossRef]
- Chen, J.; Kastan, M.B. 5′-3′-UTR Interactions Regulate P53 mRNA Translation and Provide a Target for Modulating P53 Induction after DNA Damage. Genes. Dev. 2010, 24, 2146–2156. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; Ströse, A.; Tedesco, D.; Gurova, K.; Selivanova, G. Integrated High-Throughput Analysis Identifies Sp1 as a Crucial Determinant of P53-Mediated Apoptosis. Cell Death Differ. 2014, 21, 1493–1502. [Google Scholar] [CrossRef]
- Tapias, A.; Ciudad, C.J.; Roninson, I.B.; Noé, V. Regulation of Sp1 by Cell Cycle Related Proteins. Cell Cycle 2008, 7, 2856–2867. [Google Scholar] [CrossRef]
- Deniaud, E.; Baguet, J.; Chalard, R.; Blanquier, B.; Brinza, L.; Meunier, J.; Michallet, M.-C.; Laugraud, A.; Ah-Soon, C.; Wierinckx, A.; et al. Overexpression of Transcription Factor Sp1 Leads to Gene Expression Perturbations and Cell Cycle Inhibition. PLoS ONE 2009, 4, e7035. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Struhl, K. Different SP1 Binding Dynamics at Individual Genomic Loci in Human Cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2113579118. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-A.; Suh, D.-C.; Kang, J.-E.; Kim, M.-H.; Park, H.; Lee, M.-N.; Kim, J.-M.; Jeon, B.-N.; Roh, H.-E.; Yu, M.-Y.; et al. Transcriptional Activity of Sp1 Is Regulated by Molecular Interactions between the Zinc Finger DNA Binding Domain and the Inhibitory Domain with Corepressors, and This Interaction Is Modulated by MEK. J. Biol. Chem. 2005, 280, 28061–28071. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Philipsen, S. Regulation of the Activity of Sp1-Related Transcription Factors. Mol. Cell Endocrinol. 2002, 195, 27–38. [Google Scholar] [CrossRef]
- Tanno, Y.; Hosoi, Y.; Li, Z.; Cai, K.; Matsumoto, Y.; Enomoto, A.; Morita, A.; Takakura, K.; Sakai, K.; Tomita, M.; et al. Transcriptional Regulation of DNA Double-Strand Break Repair Genes by Sp1. The Japan Radiation Research Society Annual Meeting Abstracts, 2005, Volume 2005, The 48th Annual Meeting of The Japan Radiation Research Society, Session ID P-A-006, Pages 143, Released on J-STAGE May 25, 2006. Available online: https://www.jstage.jst.go.jp/article/jrrsabst/2005/0/2005_0_143/_article/-char/en (accessed on 21 September 2024).
- Zaky, A.; Busso, C.; Izumi, T.; Chattopadhyay, R.; Bassiouny, A.; Mitra, S.; Bhakat, K.K. Regulation of the Human AP-Endonuclease (APE1/Ref-1) Expression by the Tumor Suppressor P53 in Response to DNA Damage. Nucleic Acids Res. 2008, 36, 1555–1566. [Google Scholar] [CrossRef]
- Engeland, K. Cell Cycle Regulation: P53-P21-RB Signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Giono, L.E.; Manfredi, J.J. The P53 Tumor Suppressor Participates in Multiple Cell Cycle Checkpoints. J. Cell Physiol. 2006, 209, 13–20. [Google Scholar] [CrossRef]
- Lazo, P.A. Reverting P53 Activation after Recovery of Cellular Stress to Resume with Cell Cycle Progression. Cell Signal 2017, 33, 49–58. [Google Scholar] [CrossRef]
- Lai, L.; Shin, G.Y.; Qiu, H. The Role of Cell Cycle Regulators in Cell Survival-Dual Functions of Cyclin-Dependent Kinase 20 and p21Cip1/Waf1. Int. J. Mol. Sci. 2020, 21, 8504. [Google Scholar] [CrossRef]
- Manu, K.A.; Cao, P.H.A.; Chai, T.F.; Casey, P.J.; Wang, M. P21cip1/Waf1 Coordinate Autophagy, Proliferation and Apoptosis in Response to Metabolic Stress. Cancers 2019, 11, 1112. [Google Scholar] [CrossRef]
- Saramäki, A.; Banwell, C.M.; Campbell, M.J.; Carlberg, C. Regulation of the Human P21(Waf1/Cip1) Gene Promoter via Multiple Binding Sites for P53 and the Vitamin D3 Receptor. Nucleic Acids Res. 2006, 34, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Shats, I.; Milyavsky, M.; Tang, X.; Stambolsky, P.; Erez, N.; Brosh, R.; Kogan, I.; Braunstein, I.; Tzukerman, M.; Ginsberg, D.; et al. P53-Dependent down-Regulation of Telomerase Is Mediated by P21waf1. J. Biol. Chem. 2004, 279, 50976–50985. [Google Scholar] [CrossRef] [PubMed]
- Ramlee, M.K.; Wang, J.; Toh, W.X.; Li, S. Transcription Regulation of the Human Telomerase Reverse Transcriptase (hTERT) Gene. Genes 2016, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Won, J.; Yim, J.; Kim, T.K. Sp1 and Sp3 Recruit Histone Deacetylase to Repress Transcription of Human Telomerase Reverse Transcriptase (hTERT) Promoter in Normal Human Somatic Cells. J. Biol. Chem. 2002, 277, 38230–38238. [Google Scholar] [CrossRef]
- Cheng, D.; Zhao, Y.; Wang, S.; Jia, W.; Kang, J.; Zhu, J. Human Telomerase Reverse Transcriptase (hTERT) Transcription Requires Sp1/Sp3 Binding to the Promoter and a Permissive Chromatin Environment. J. Biol. Chem. 2015, 290, 30193–30203. [Google Scholar] [CrossRef]
- Chai, W.; Ford, L.P.; Lenertz, L.; Wright, W.E.; Shay, J.W. Human Ku70/80 Associates Physically with Telomerase through Interaction with hTERT. J. Biol. Chem. 2002, 277, 47242–47247. [Google Scholar] [CrossRef]
- Park, J.-I.; Venteicher, A.S.; Hong, J.Y.; Choi, J.; Jun, S.; Shkreli, M.; Chang, W.; Meng, Z.; Cheung, P.; Ji, H.; et al. Telomerase Modulates Wnt Signalling by Association with Target Gene Chromatin. Nature 2009, 460, 66–72. [Google Scholar] [CrossRef]
- Zhou, J.; Ding, D.; Wang, M.; Cong, Y.-S. Telomerase Reverse Transcriptase in the Regulation of Gene Expression. BMB Rep. 2014, 47, 8–14. [Google Scholar] [CrossRef]
- Rahman, R.; Latonen, L.; Wiman, K.G. hTERT Antagonizes P53-Induced Apoptosis Independently of Telomerase Activity. Oncogene 2005, 24, 1320–1327. [Google Scholar] [CrossRef]
- Wu, L.; Wang, S.; Tang, B.; Tang, L.; Lei, Y.; Liu, Y.; Yang, M.; Yang, G.; Zhang, D.; Liu, E. Human Telomerase Reverse Transcriptase (hTERT) Synergistic with Sp1 Upregulate Gli1 Expression and Increase Gastric Cancer Invasion and Metastasis. J. Mol. Histol. 2021, 52, 1165–1175. [Google Scholar] [CrossRef]
- Liu, N.; Ding, D.; Hao, W.; Yang, F.; Wu, X.; Wang, M.; Xu, X.; Ju, Z.; Liu, J.-P.; Song, Z.; et al. hTERT Promotes Tumor Angiogenesis by Activating VEGF via Interactions with the Sp1 Transcription Factor. Nucleic Acids Res. 2016, 44, 8693–8703. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, S.; Banerjee, P.P. Telomerase Reverse Transcriptase Regulates the Expression of a Key Cell Cycle Regulator, Cyclin D1. Biochem. Biophys. Res. Commun. 2006, 347, 774–780. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Sequence (5′ to 3′) |
|---|---|
| XRCC4 | AAUCUUGGGACAGAACCUAAA |
| Target | Forward Primer (5′→3′) | Reverse Primer (5′→3′) |
|---|---|---|
| B2M | ATGTCTCGCTCCGTGGCCTTA | ATCTTGGGCTGTGACAAAGTC |
| XRCC1 | CACGGATGAGAACACGGACA | TCCCCATTGAAGGCTGTGAC |
| LIG4 | GAAGGCATCTGGTAAGCTCG | TTTCTTCATCTTTGGGGCAG |
| XRCC4 | TGCAAAGAAATCTTGGGACAG | TGCTCCTTTTTAGACGTCTC |
| Ku70 | GTTGATGCCTCCAAGGCTATG | CCCCTTAAACTGGTCAAGCTCTA |
| Ku80 | TGGAAGTGTGAATCCTGCTG | TCCAAAAACTGTTCGATGTGA |
| Sp1 | GCCTCCAGACCATTAACCTCAG | TCATGTATTCCATCACCACCAG |
| p53 | GACATAGTGTGGTGGTGCCC | CAAAGCTGTTCCGTCCCAGT |
| APE1 | GGGCGTTCGTAACGGGAATG | TGCGGCCGTCTTACTCTTCT |
| LIG3 | TGTGTTAGACGCCCTTGACC | TACTCAACGGACTTGCAGGC |
| Target | Antibodies |
|---|---|
| XRCC1 | MS-1993-P0 (Thermo Scientific, Wilmington, DE, USA) |
| DNA Ligase 3 | GTX70143 (GeneTex, Irvine, CA, USA) |
| APE1 | NB100-101 (Novus Biologicals, Toronto, ON, Canada) |
| XRCC4 | 66621-1-Ig (Proteintech, Solana Beach, CA, USA) |
| DNA Ligase 4 | ab193353 (Abcam, Waltham, MA, USA) |
| Ku80 | ab119935 Abcam (Abcam, Waltham, MA, USA) |
| Ku70 | NB100-1915 (Novus Biologicals, Toronto, ON, Canada) |
| Sp1 | 07-645 (Merck Millipore, Carrigtwohill, Ireland) |
| p53 | sc-126 (SANTA CRUZ, Dallas, TX, USA) |
| p21 Waf1/Cip1 (12D1) | #2947 (Cell signaling technology, Danvers, MA, USA) |
| PARP1 | ab227244 Abcam (Abcam, Waltham, MA, USA) |
| β-actin | ab6276 Abcam (Abcam, Waltham, MA, USA) |
| α-tubulin | T6199 (Merck KGaA, Darmstadt, Germany) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sergeeva, S.V.; Loshchenova, P.S.; Oshchepkov, D.Y.; Orishchenko, K.E. Crosstalk between BER and NHEJ in XRCC4-Deficient Cells Depending on hTERT Overexpression. Int. J. Mol. Sci. 2024, 25, 10405. https://doi.org/10.3390/ijms251910405
Sergeeva SV, Loshchenova PS, Oshchepkov DY, Orishchenko KE. Crosstalk between BER and NHEJ in XRCC4-Deficient Cells Depending on hTERT Overexpression. International Journal of Molecular Sciences. 2024; 25(19):10405. https://doi.org/10.3390/ijms251910405
Chicago/Turabian StyleSergeeva, Svetlana V., Polina S. Loshchenova, Dmitry Yu. Oshchepkov, and Konstantin E. Orishchenko. 2024. "Crosstalk between BER and NHEJ in XRCC4-Deficient Cells Depending on hTERT Overexpression" International Journal of Molecular Sciences 25, no. 19: 10405. https://doi.org/10.3390/ijms251910405