
Identification of a Novel Gene MtbZIP60 as a Negative Regulator of Leaf Senescence through Transcriptome Analysis in Medicago truncatula


 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Changes in Physiological and Molecular Parameters during Age-Dependent Leaf Senescence
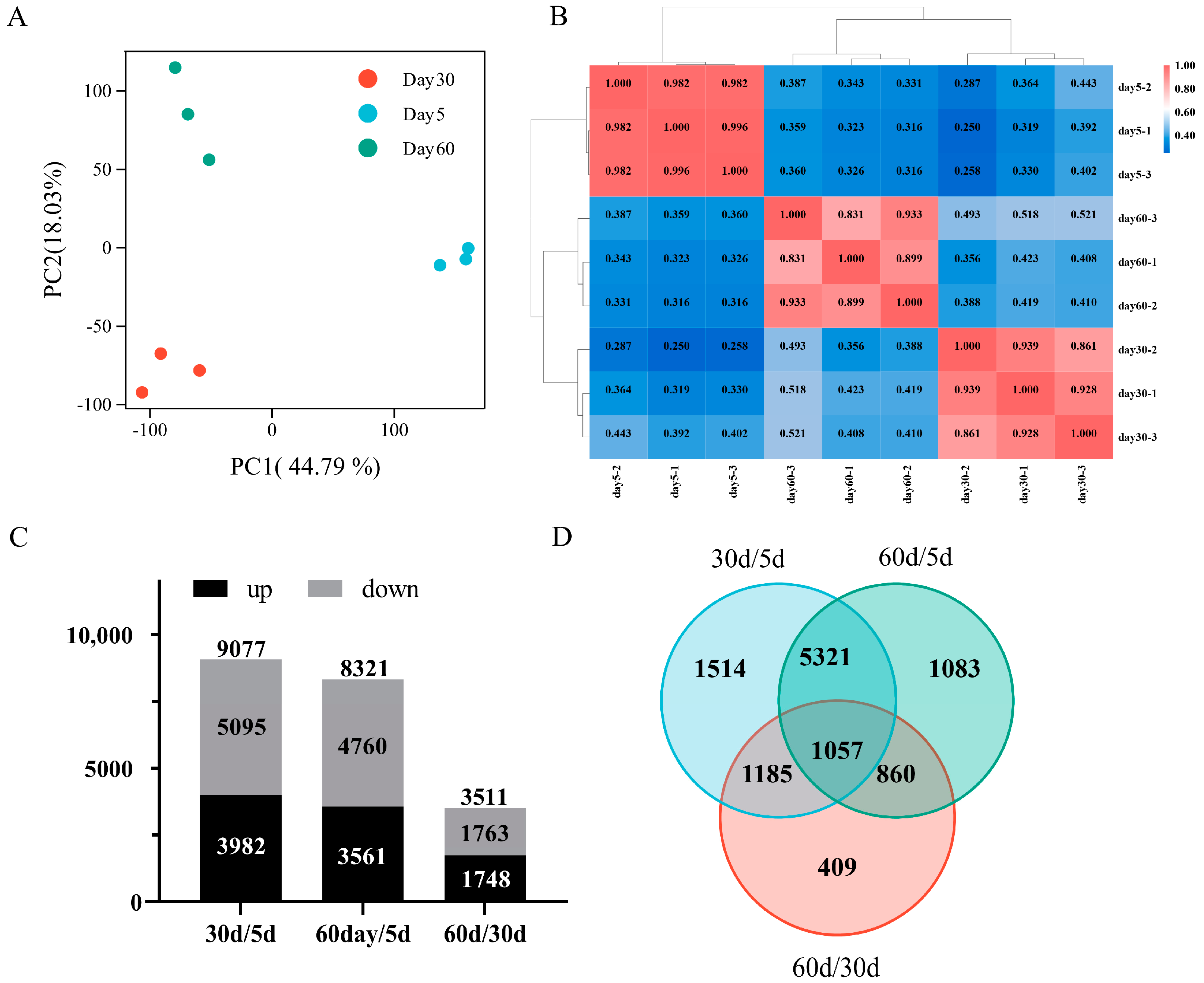
2.2. Transcriptome Sequencing
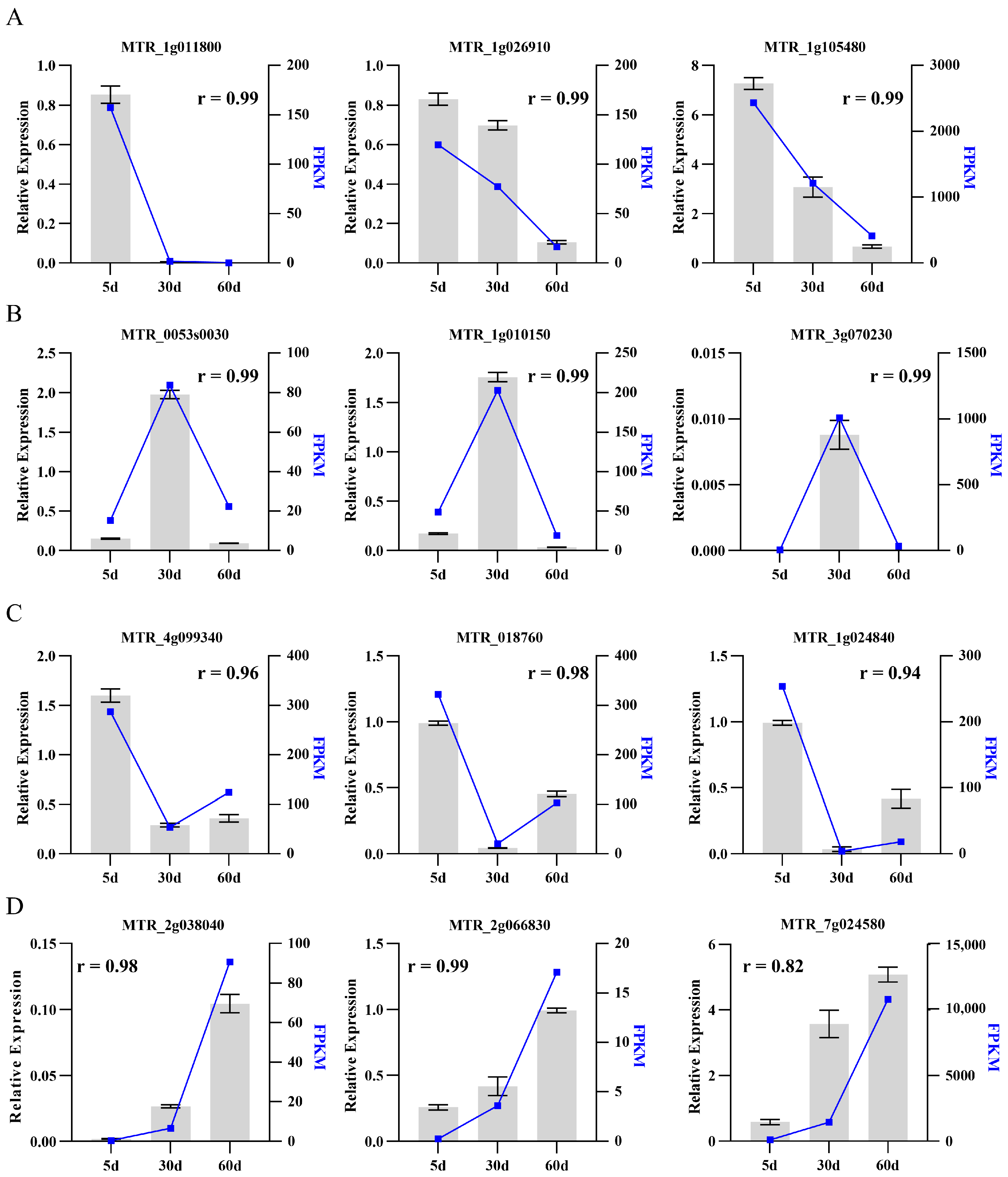
2.3. Validation of Transcriptome Data by qRT-PCR
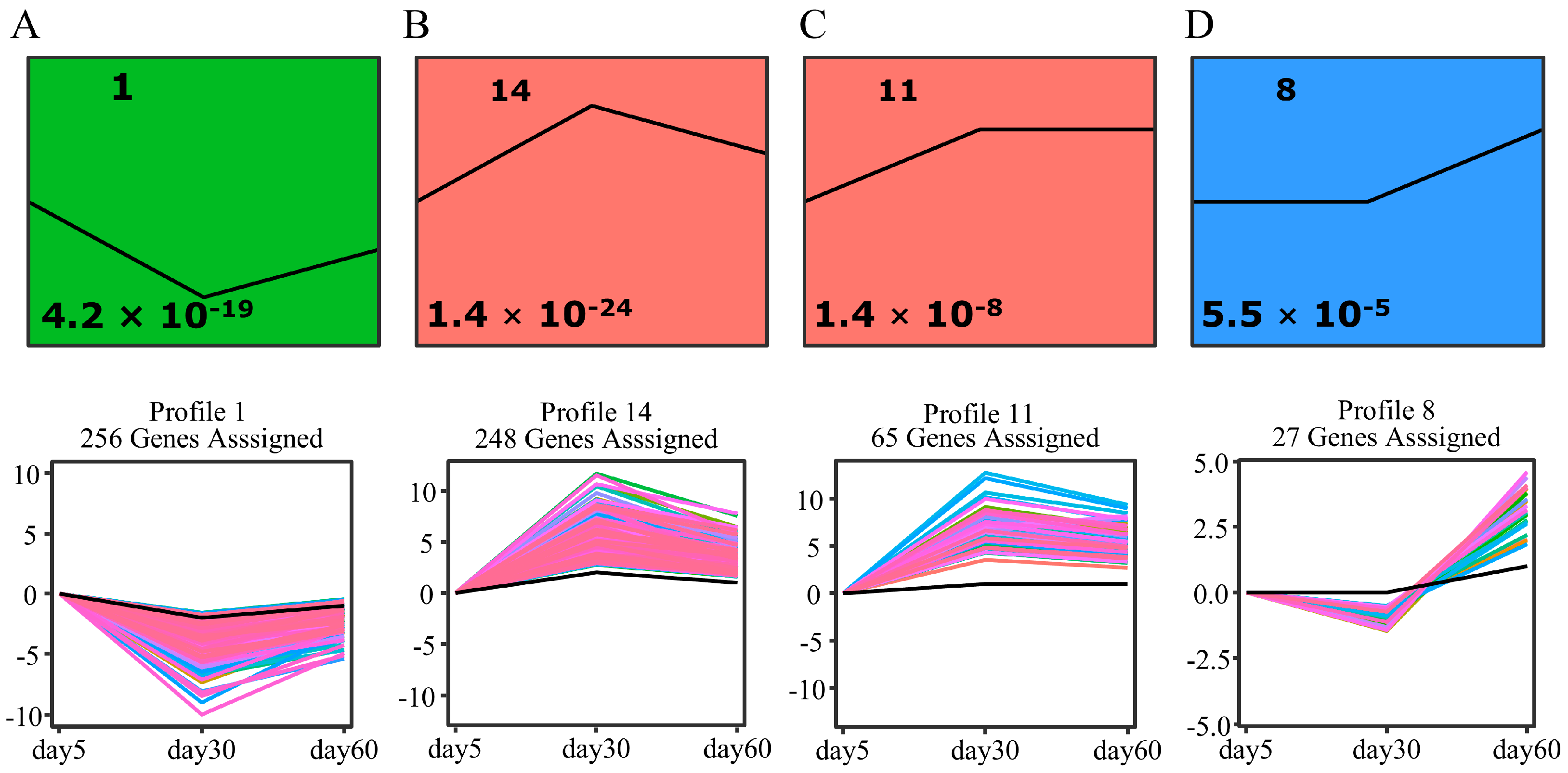
2.4. Short Time-Series Expression Miner (STEM) Clustering of DEGs during Leaf Growth and Senescence
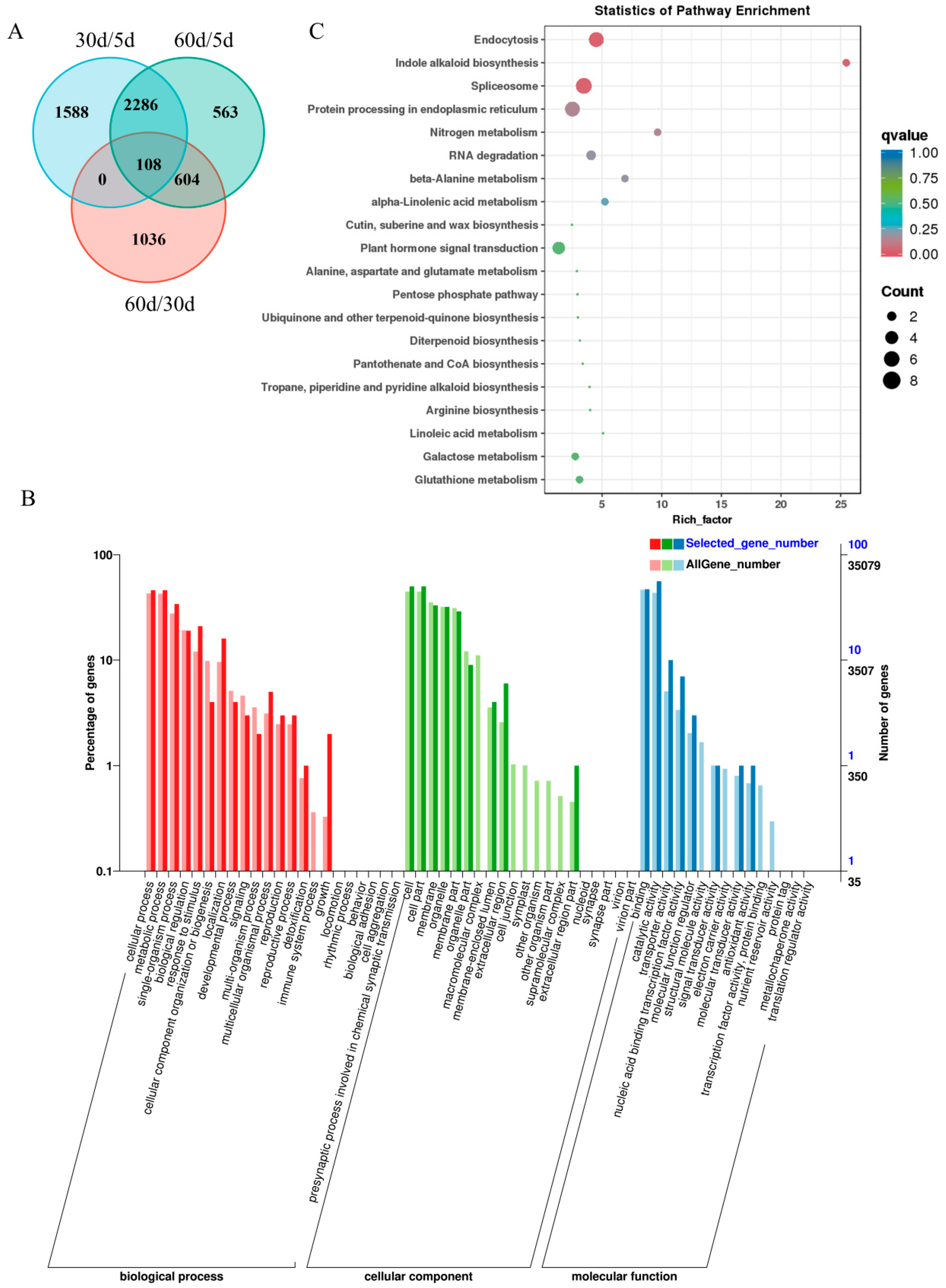
2.5. Gene Ontology Enrichment and Kyoto Encyclopedia of Genes and Genomes Pathway Enrichment Analysis of Senescence-Associated Genes
2.6. Comparison of Senescence-Associated Genes between Medicago and Arabidopsis
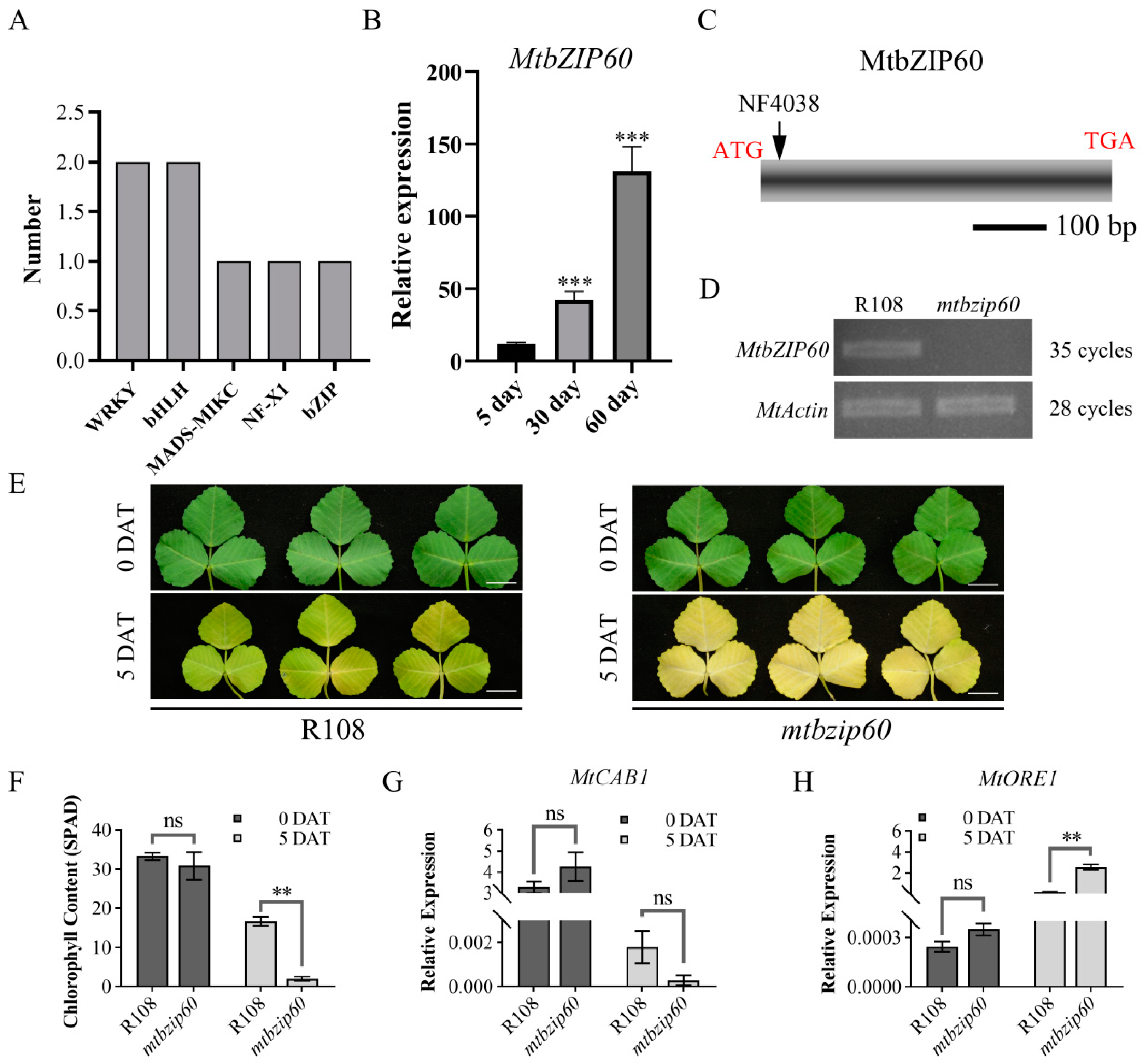
2.7. Transcription Factor Analysis
2.8. MtbZIP60 Functions as a Novel Regulator of Leaf Senescence
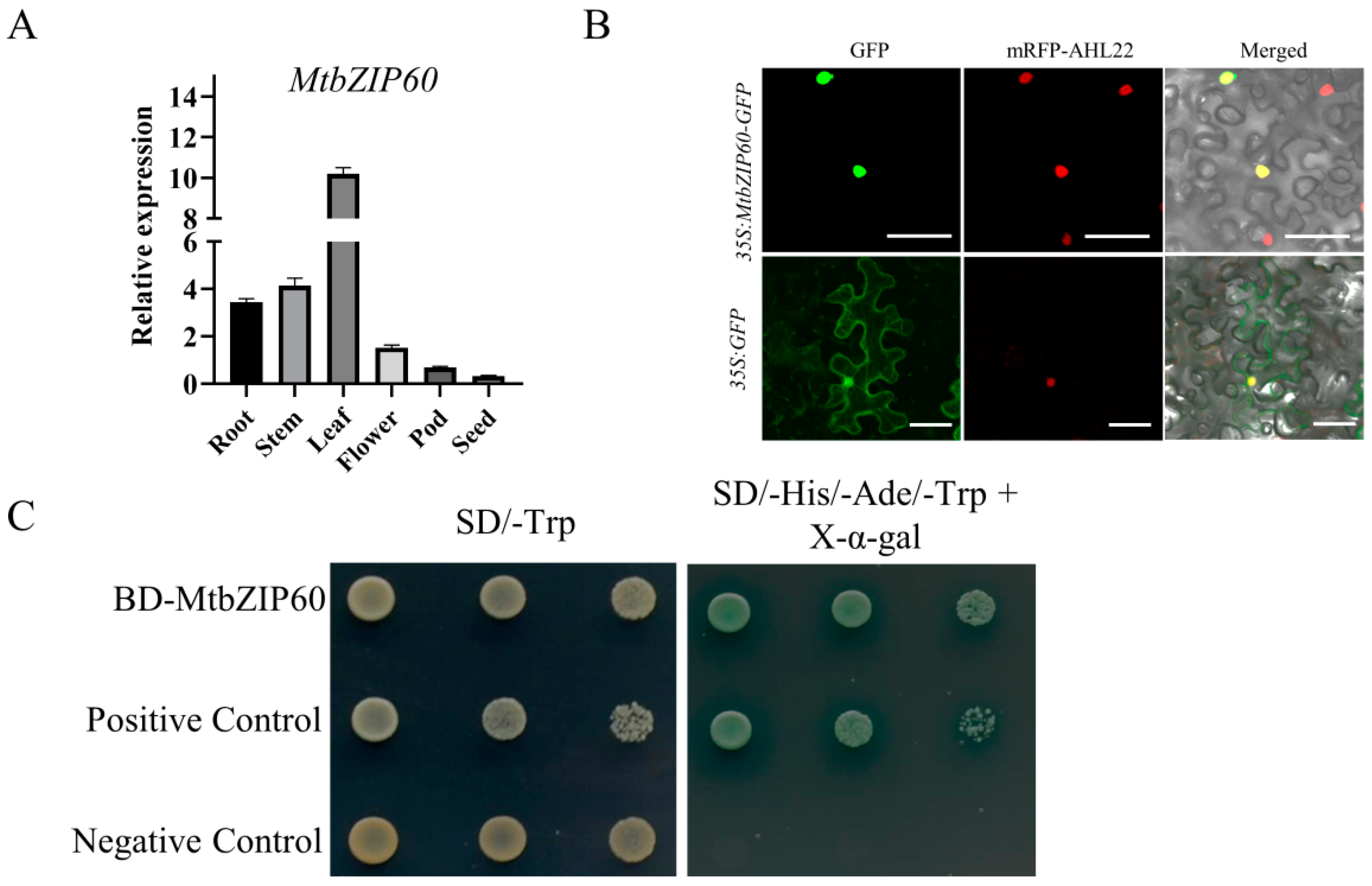
2.9. Expression Pattern, Subcellular Localization and Transcriptional Activity Analysis of MtbZIP60
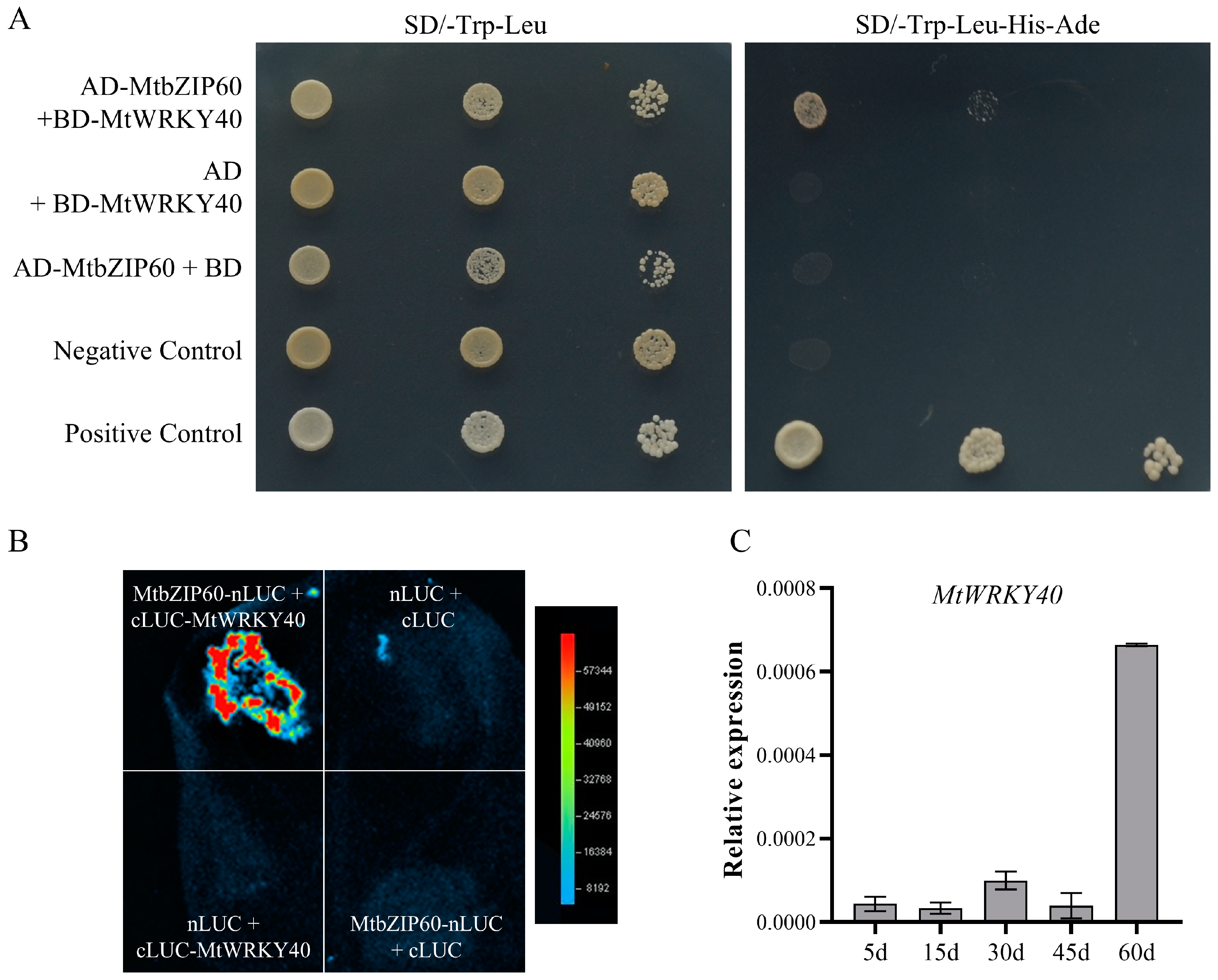
2.10. MtbZIP60 Interacts with MtWRKY40 in Regulation of Leaf Senescence
3. Discussion
4. Material and Methods
4.1. Plant Materials and Growth Condition
4.2. Determination of Chlorophylls and MDA Content
4.3. RNA Isolation and Gene Expression Analysis
4.4. Library Construction and Transcriptome Sequencing
4.5. Transcription Factor Analysis
4.6. Gene Ontology and Kyto Encyclopedia of Genes and Genomes Pathway Enrichment Analysis
4.7. Protein Subcellular Localization
4.8. Transactivation Activity Assay in Yeast
4.9. Yeast Two-Hybrid (Y2H) Interactions and Library Screening
4.10. Luciferase Complementation Imaging Assay (LCI)
4.11. Statistical Analysis
4.12. Accession Number
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Guo, Y.; Ren, G.; Zhang, K.; Li, Z.; Miao, Y.; Guo, H. Leaf senescence: Progression, regulation, and application. Mol. Hortic. 2021, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.R.; Kim, H.J.; Lim, P.O.; Nam, H.G. Leaf senescence: Systems and dynamics aspects. Annu. Rev. Plant Biol. 2019, 70, 347–376. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, P.L.; Holm, P.B.; Krupinska, K. Leaf senescence and nutrient remobilisation in barley and wheat. Plant Biol. 2008, 10 (Suppl. S1), 37–49. [Google Scholar] [CrossRef]
- Gregersen, P.L.; Culetic, A.; Boschian, L.; Krupinska, K. Plant senescence and crop productivity. Plant Mol. Biol. 2013, 82, 603–622. [Google Scholar] [CrossRef]
- Kim, T.; Kang, K.; Kim, S.H.; An, G.; Paek, N.C. OsWRKY5 promotes rice leaf senescence via senescence-associated NAC and abscisic acid biosynthesis pathway. Int. J. Mol. Sci. 2019, 20, 4437. [Google Scholar] [CrossRef]
- Joshi, S.; Choukimath, A.; Isenegger, D.; Panozzo, J.; Spangenberg, G.; Kant, S. Improved wheat growth and yield by delayed leaf senescence using developmentally regulated expression of a cytokinin biosynthesis gene. Front. Plant Sci. 2019, 10, 1285. [Google Scholar] [CrossRef]
- Dong, S.; Sang, L.; Xie, H.; Chai, M.; Wang, Z.Y. Comparative Transcriptome analysis of salt stress-induced leaf senescence in Medicago truncatula. Front. Plant Sci. 2021, 12, 666660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Xie, H.; Wen, J.; Zhang, J.; Wang, Z.-Y.; Xu, B.; Chai, M. Leaf Senescence in Forage and Turf Grass: Progress and Prospects; Maximum Academic Press: Fayetteville, GA, USA, 2024; Volume 4. [Google Scholar] [CrossRef]
- Li, Z.; Oelmüller, R.; Guo, H.; Miao, Y. Editorial: Signal transduction of plant organ senescence and cell death. Front. Plant Sci. 2023, 14, 1172373. [Google Scholar] [CrossRef]
- Ostrowska-Mazurek, A.; Kasprzak, P.; Kubala, S.; Zaborowska, M.; Sobieszczuk-Nowicka, E. Epigenetic landmarks of leaf senescence and crop improvement. Int. J. Mol. Sci. 2020, 21, 5125. [Google Scholar] [CrossRef]
- Guo, Y.; Gan, S. Leaf senescence: Signals, execution, and regulation. Curr. Top. Dev. Biol. 2005, 71, 83–112. [Google Scholar] [CrossRef]
- Lim, P.O.; Kim, H.J.; Nam, H.G. Leaf senescence. Annu. Rev. Plant Biol. 2007, 58, 115–136. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Liu, H.; Tan, S.; Li, Z. Transcription factors-regulated leaf senescence: Current knowledge, challenges and approaches. Int. J. Mol. Sci. 2023, 24, 9245. [Google Scholar] [CrossRef] [PubMed]
- Bengoa Luoni, S.; Astigueta, F.H.; Nicosia, S.; Moschen, S.; Fernandez, P.; Heinz, R. Transcription factors associated with leaf senescence in crops. Plants 2019, 8, 411. [Google Scholar] [CrossRef]
- Li, J.; Qiao, H.; Yin, P.; Liu, M.; Yang, Y.; Li, K.; Yang, L.; Yang, C.; Zhao, L.; Zhou, S.; et al. Increasingly amplified stimulation mediated by TaNAC69-B is crucial for the leaf senescence in wheat. Plant J. 2023, 114, 570–590. [Google Scholar] [CrossRef]
- Miao, Y.; Laun, T.; Zimmermann, P.; Zentgraf, U. Targets of the WRKY53 transcription factor and its role during leaf senescence in Arabidopsis. Plant Mol. Biol. 2004, 55, 853–867. [Google Scholar] [CrossRef]
- Lu, M.; Fu, B.; Meng, X.; Jia, T.; Lu, X.; Yang, C.; Li, K.; Yin, P.; Guo, Y.; Li, W.; et al. NtNAC028 and NtNAC080 form heterodimers to regulate jasmonic acid biosynthesis during leaf senescence in Nicotiana tabacum. J. Exp. Bot. 2024, 75, 2351–2371. [Google Scholar] [CrossRef]
- Chen, Q.; Yan, J.; Tong, T.; Zhao, P.; Wang, S.; Zhou, N.; Cui, X.; Dai, M.; Jiang, Y.Q.; Yang, B. ANAC087 transcription factor positively regulates age-dependent leaf senescence through modulating the expression of multiple target genes in Arabidopsis. J. Integr. Plant Biol. 2023, 65, 967–984. [Google Scholar] [CrossRef] [PubMed]
- Garapati, P.; Xue, G.P.; Munné-Bosch, S.; Balazadeh, S. Transcription factor ATAF1 in Arabidopsis promotes senescence by direct regulation of key chloroplast maintenance and senescence transcriptional cascades. Plant Physiol. 2015, 168, 1122–1139. [Google Scholar] [CrossRef]
- Kim, Y.S.; Sakuraba, Y.; Han, S.H.; Yoo, S.C.; Paek, N.C. Mutation of the Arabidopsis NAC016 transcription factor delays leaf senescence. Plant Cell Physiol. 2013, 54, 1660–1672. [Google Scholar] [CrossRef]
- Takasaki, H.; Maruyama, K.; Takahashi, F.; Fujita, M.; Yoshida, T.; Nakashima, K.; Myouga, F.; Toyooka, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K. SNAC-As, stress-responsive NAC transcription factors, mediate ABA-inducible leaf senescence. Plant J. 2015, 84, 1114–1123. [Google Scholar] [CrossRef]
- Guo, Y.; Gan, S. AtNAP, a NAC family transcription factor, has an important role in leaf senescence. Plant J. 2006, 46, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, K.; El-Kereamy, A.; Kim, S.H.; Nambara, E.; Rothstein, S.J. ANAC032 positively regulates age-dependent and stress-induced senescence in Arabidopsis thaliana. Plant Cell Physiol. 2016, 57, 2029–2046. [Google Scholar] [CrossRef] [PubMed]
- Oda-Yamamizo, C.; Mitsuda, N.; Sakamoto, S.; Ogawa, D.; Ohme-Takagi, M.; Ohmiya, A. The NAC transcription factor ANAC046 is a positive regulator of chlorophyll degradation and senescence in Arabidopsis leaves. Sci. Rep. 2016, 6, 23609. [Google Scholar] [CrossRef]
- Balazadeh, S.; Kwasniewski, M.; Caldana, C.; Mehrnia, M.; Zanor, M.I.; Xue, G.P.; Mueller-Roeber, B. ORS1, an H2O2-responsive NAC transcription factor, controls senescence in Arabidopsis thaliana. Mol. Plant 2011, 4, 346–360. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Gao, J.; Yao, L.; Ren, G.; Zhu, X.; Gao, S.; Qiu, K.; Zhou, X.; Kuai, B. The role of ANAC072 in the regulation of chlorophyll degradation during age- and dark-induced leaf senescence. Plant Cell Rep. 2016, 35, 1729–1741. [Google Scholar] [CrossRef]
- Balazadeh, S.; Siddiqui, H.; Allu, A.D.; Matallana-Ramirez, L.P.; Caldana, C.; Mehrnia, M.; Zanor, M.I.; Kohler, B.; Mueller-Roeber, B. A gene regulatory network controlled by the NAC transcription factor ANAC092/AtNAC2/ORE1 during salt-promoted senescence. Plant J. 2010, 62, 250–264. [Google Scholar] [CrossRef]
- Kim, H.J.; Park, J.H.; Kim, J.; Kim, J.J.; Hong, S.; Kim, J.; Kim, J.H.; Woo, H.R.; Hyeon, C.; Lim, P.O.; et al. Time-evolving genetic networks reveal a NAC troika that negatively regulates leaf senescence in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, E4930–E4939. [Google Scholar] [CrossRef]
- Wu, A.; Allu, A.D.; Garapati, P.; Siddiqui, H.; Dortay, H.; Zanor, M.I.; Asensi-Fabado, M.A.; Munné-Bosch, S.; Antonio, C.; Tohge, T.; et al. JUNGBRUNNEN1, a reactive oxygen species-responsive NAC transcription factor, regulates longevity in Arabidopsis. Plant Cell 2012, 24, 482–506. [Google Scholar] [CrossRef]
- Yang, S.D.; Seo, P.J.; Yoon, H.K.; Park, C.M. The Arabidopsis NAC transcription factor VNI2 integrates abscisic acid signals into leaf senescence via the COR/RD genes. Plant Cell 2011, 23, 2155–2168. [Google Scholar] [CrossRef]
- Kan, C.; Zhang, Y.; Wang, H.L.; Shen, Y.; Xia, X.; Guo, H.; Li, Z. Transcription factor NAC075 delays leaf senescence by deterring reactive oxygen species accumulation in Arabidopsis. Front. Plant Sci. 2021, 12, 634040. [Google Scholar] [CrossRef]
- Jiang, J.; Ma, S.; Ye, N.; Jiang, M.; Cao, J.; Zhang, J. WRKY transcription factors in plant responses to stresses. J. Integr. Plant Biol. 2017, 59, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Robatzek, S.; Somssich, I.E. Targets of AtWRKY6 regulation during plant senescence and pathogen defense. Genes Dev. 2002, 16, 1139–1149. [Google Scholar] [CrossRef]
- Zhou, X.; Jiang, Y.; Yu, D. WRKY22 transcription factor mediates dark-induced leaf senescence in Arabidopsis. Mol. Cells 2011, 31, 303–313. [Google Scholar] [CrossRef]
- Niu, F.; Cui, X.; Zhao, P.; Sun, M.; Yang, B.; Deyholos, M.K.; Li, Y.; Zhao, X.; Jiang, Y.Q. WRKY42 transcription factor positively regulates leaf senescence through modulating SA and ROS synthesis in Arabidopsis thaliana. Plant J. 2020, 104, 171–184. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, L.; Wu, S.; Chen, Y.; Yu, D.; Chen, L. AtWRKY75 positively regulates age-triggered leaf senescence through gibberellin pathway. Plant Divers. 2021, 43, 331–340. [Google Scholar] [CrossRef]
- Yu, Y.; Qi, Y.; Xu, J.; Dai, X.; Chen, J.; Dong, C.H.; Xiang, F. Arabidopsis WRKY71 regulates ethylene-mediated leaf senescence by directly activating EIN2, ORE1 and ACS2 genes. Plant J. 2021, 107, 1819–1836. [Google Scholar] [CrossRef]
- Chen, L.; Xiang, S.; Chen, Y.; Li, D.; Yu, D. Arabidopsis WRKY45 Interacts with the DELLA protein RGL1 to positively regulate age-triggered leaf senescence. Mol. Plant 2017, 10, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Besseau, S.; Li, J.; Palva, E.T. WRKY54 and WRKY70 co-operate as negative regulators of leaf senescence in Arabidopsis thaliana. J. Exp. Bot. 2012, 63, 2667–2679. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cui, X.; Yang, B.; Xu, S.; Wei, X.; Zhao, P.; Niu, F.; Sun, M.; Wang, C.; Cheng, H.; et al. WRKY55 transcription factor positively regulates leaf senescence and the defense response by modulating the transcription of genes implicated in the biosynthesis of reactive oxygen species and salicylic acid in Arabidopsis. Development 2020, 147, dev189647. [Google Scholar] [CrossRef]
- Smykowski, A.; Fischer, S.M.; Zentgraf, U. Phosphorylation affects dna-binding of the senescence-regulating bZIP transcription factor GBF1. Plants 2015, 4, 691–709. [Google Scholar] [CrossRef]
- Gao, S.; Gao, J.; Zhu, X.; Song, Y.; Li, Z.; Ren, G.; Zhou, X.; Kuai, B. ABF2, ABF3, and ABF4 promote ABA-mediated chlorophyll degradation and leaf senescence by transcriptional activation of chlorophyll catabolic genes and senescence-associated genes in Arabidopsis. Mol. Plant 2016, 9, 1272–1285. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Cheng, Q.; Zhao, Y.; Wu, F.; Mu, B.; Gao, J.; Yang, L.; Yan, J.; Zhang, H.; Cui, X.; et al. The abscisic acid-responsive element binding factors MAPKKK18 module regulates abscisic acid-induced leaf senescence in Arabidopsis. J. Biol. Chem. 2023, 299, 103060. [Google Scholar] [CrossRef] [PubMed]
- Hickman, R.; Hill, C.; Penfold, C.A.; Breeze, E.; Bowden, L.; Moore, J.D.; Zhang, P.; Jackson, A.; Cooke, E.; Bewicke-Copley, F.; et al. A local regulatory network around three NAC transcription factors in stress responses and senescence in Arabidopsis leaves. Plant J. 2013, 75, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, X.; Li, M.; Yan, Y.; Liu, X.; Li, L. Dual function of NAC072 in ABF3-mediated ABA-responsive gene regulation in Arabidopsis. Front. Plant Sci. 2016, 7, 1075. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, C.; Li, K.; Li, X.; Xu, M.; Guo, Y. CLE14 functions as a “brake signal” to suppress age-dependent and stress-induced leaf senescence by promoting JUB1-mediated ROS scavenging in Arabidopsis. Mol. Plant 2022, 15, 179–188. [Google Scholar] [CrossRef]
- Chen, D.; Shi, Y.; Zhang, P.; Xie, W.; Li, S.; Xiao, J.; Yuan, M. Deletion of the sugar importer gene OsSWEET1b accelerates sugar starvation-promoted leaf senescence in rice. Plant Physiol. 2024, 195, 2176–2194. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Wang, X.; Wei, Z.; Jin, B. Medicago truncatula (model legume), Medicago sativa (alfalfa), Medicago polymorpha (bur clover), and Medicago ruthenica. Trends Genet. 2022, 38, 782–783. [Google Scholar] [CrossRef]
- Chen, H.; Zeng, Y.; Yang, Y.; Huang, L.; Tang, B.; Zhang, H.; Hao, F.; Liu, W.; Li, Y.; Liu, Y.; et al. Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa. Nat. Commun. 2020, 11, 2494. [Google Scholar] [CrossRef]
- Shen, C.; Du, H.; Chen, Z.; Lu, H.; Zhu, F.; Chen, H.; Meng, X.; Liu, Q.; Liu, P.; Zheng, L.; et al. The chromosome-level genome sequence of the autotetraploid alfalfa and resequencing of core germplasms provide genomic resources for alfalfa research. Mol. Plant 2020, 13, 1250–1261. [Google Scholar] [CrossRef]
- Calderini, O.; Bovone, T.; Scotti, C.; Pupilli, F.; Piano, E.; Arcioni, S. Delay of leaf senescence in Medicago sativa transformed with the ipt gene controlled by the senescence-specific promoter SAG12. Plant Cell Rep. 2007, 26, 611–615. [Google Scholar] [CrossRef]
- Jiang, J.; Jia, H.; Feng, G.; Wang, Z.; Li, J.; Gao, H.; Wang, X. Overexpression of Medicago sativa TMT elevates the α-tocopherol content in Arabidopsis seeds, alfalfa leaves, and delays dark-induced leaf senescence. Plant Sci. 2016, 249, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Han, L.; Pislariu, C.; Nakashima, J.; Fu, C.; Jiang, Q.; Quan, L.; Blancaflor, E.B.; Tang, Y.; Bouton, J.H.; et al. From model to crop: Functional analysis of a STAY-GREEN gene in the model legume Medicago truncatula and effective use of the gene for alfalfa improvement. Plant Physiol. 2011, 157, 1483–1496. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xie, H.; Zhou, L.; Dong, D.; Liu, Y.; Jia, C.; Han, L.; Chao, Y.; Chen, Y. Overexpression of MsSAG113 gene promotes leaf senescence in alfalfa via participating in the hormone regulatory network. Front. Plant Sci. 2022, 13, 1085497. [Google Scholar] [CrossRef]
- Mahmood, K.; Torres-Jerez, I.; Krom, N.; Liu, W.; Udvardi, M.K. Transcriptional programs and regulators underlying age-dependent and dark-induced senescence in Medicago truncatula. Cells 2022, 11, 1570. [Google Scholar] [CrossRef]
- Dong, S.; Pang, W.; Liu, Z.; Li, H.; Zhang, K.; Cong, L.; Yang, G.; Wang, Z.Y.; Xie, H. Transcriptome analysis of leaf senescence regulation under alkaline stress in Medicago truncatula. Front. Plant Sci. 2022, 13, 881456. [Google Scholar] [CrossRef]
- Wen, Z.; Mei, Y.; Zhou, J.; Cui, Y.; Wang, D.; Wang, N.N. SAUR49 can positively regulate leaf senescence by suppressing SSPP in Arabidopsis. Plant Cell Physiol. 2020, 61, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Melzer, S.; Lens, F.; Gennen, J.; Vanneste, S.; Rohde, A.; Beeckman, T. Flowering-time genes modulate meristem determinacy and growth form in Arabidopsis thaliana. Nat. Genet. 2008, 40, 1489–1492. [Google Scholar] [CrossRef]
- Breeze, E.; Harrison, E.; McHattie, S.; Hughes, L.; Hickman, R.; Hill, C.; Kiddle, S.; Kim, Y.S.; Penfold, C.A.; Jenkins, D.; et al. High-resolution temporal profiling of transcripts during Arabidopsis leaf senescence reveals a distinct chronology of processes and regulation. Plant Cell 2011, 23, 873–894. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Z.; Gao, J.; Wang, P.; Hu, T.; Wang, Z.; Hou, Y.J.; Wan, Y.; Liu, W.; Xie, S.; et al. Arabidopsis duodecuple mutant of PYL ABA receptors reveals PYL repression of ABA-independent SnRK2 activity. Cell Rep. 2018, 23, 3340–3351.e5. [Google Scholar] [CrossRef]
- He, Y.; Fukushige, H.; Hildebrand, D.F.; Gan, S. Evidence supporting a role of jasmonic acid in Arabidopsis leaf senescence. Plant Physiol. 2002, 128, 876–884. [Google Scholar] [CrossRef]
- Yoshida, S.; Ito, M.; Nishida, I.; Watanabe, A. Isolation and RNA gel blot analysis of genes that could serve as potential molecular markers for leaf senescence in Arabidopsis thaliana. Plant Cell Physiol. 2001, 42, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Zhen, X.; Liu, C.; Guo, Y.; Yu, Z.; Han, Y.; Zhang, B.; Liang, Y. Leaf Senescence regulation mechanism based on comparative transcriptome analysis in Foxtail millet. Int. J. Mol. Sci. 2024, 25, 3905. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Pang, C.; Fan, S.; Song, M.; Wei, H.; Yu, S. Global analysis of the Gossypium hirsutum L. transcriptome during leaf senescence by RNA-Seq. BMC Plant Biol. 2015, 15, 43. [Google Scholar] [CrossRef]
- Wu, X.Y.; Hu, W.J.; Luo, H.; Xia, Y.; Zhao, Y.; Wang, L.D.; Zhang, L.M.; Luo, J.C.; Jing, H.C. Transcriptome profiling of developmental leaf senescence in sorghum (Sorghum bicolor). Plant Mol. Biol. 2016, 92, 555–580. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, R.S.; Saski, C.; Kumar, R.; Flinn, B.S.; Luo, F.; Beissinger, T.M.; Ackerman, A.J.; Breitzman, M.W.; Bridges, W.C.; de Leon, N.; et al. Integrated genome-scale analysis identifies novel genes and networks underlying senescence in maize. Plant Cell 2019, 31, 1968–1989. [Google Scholar] [CrossRef]
- Zhu, F.; Alseekh, S.; Koper, K.; Tong, H.; Nikoloski, Z.; Naake, T.; Liu, H.; Yan, J.; Brotman, Y.; Wen, W.; et al. Genome-wide association of the metabolic shifts underpinning dark-induced senescence in Arabidopsis. Plant Cell 2022, 34, 557–578. [Google Scholar] [CrossRef]
- Zhou, L.; Chang, G.; Shen, C.; Teng, W.; He, X.; Zhao, X.; Jing, Y.; Huang, Z.; Tong, Y. Functional divergences of natural variations of TaNAM-A1 in controlling leaf senescence during wheat grain filling. J. Integr. Plant Biol. 2024, 66, 1242–1260. [Google Scholar] [CrossRef]
- Saher, S.; Piqueras, A.; Hellin, E.; Olmos, E. Hyperhydricity in micropropagated carnation shoots: The role of oxidative stress. Physiol. Plant. 2004, 120, 152–161. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Yin, P.; Ma, Q.; Wang, H.; Feng, D.; Wang, X.; Pei, Y.; Wen, J.; Tadege, M.; Niu, L.; Lin, H. SMALL LEAF AND BUSHY1 controls organ size and lateral branching by modulating the stability of BIG SEEDS1 in Medicago truncatula. New Phytol. 2020, 226, 1399–1412. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zou, Y.; Shang, Y.; Lin, H.; Wang, Y.; Cai, R.; Tang, X.; Zhou, J.M. Firefly luciferase complementation imaging assay for protein-protein interactions in plants. Plant Physiol. 2008, 146, 368–376. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Medicago truncatula Gene Id | Homologous Gene Id in Arabidopsis thaliana | Arabidopsis thaliana Gene Name | Effect for Senescence | Reference |
|---|---|---|---|---|---|
| 1 | Medtr2g066830 | AT3G12830 | SAUR72 | Promote | [57] |
| 2 | Medtr1g100623 | AT5G52300 | RD29B | Delay | [30] |
| 3 | Medtr4g109830 | AT5G60910 | AGL8 | Promote | [58,59] |
| 4 | Medtr5g083270 | AT5G05440 | PYL5 | Promote | [60] |
| 5 | Medtr5g091390 | AT1G29860 | WRKY71 | Promote | [37] |
| 6 | Medtr8g018730 | AT1G55020 | LOX1 | Promote | [61] |
| 7 | Medtr8g074270 | AT3G51430 | YLS2 | Marker gene | [62] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, J.; Wang, J.; Cao, J.; Li, K.; Meng, X.; Wen, J.; Mysore, K.S.; Wang, G.; Zhou, C.; Yin, P. Identification of a Novel Gene MtbZIP60 as a Negative Regulator of Leaf Senescence through Transcriptome Analysis in Medicago truncatula. Int. J. Mol. Sci. 2024, 25, 10410. https://doi.org/10.3390/ijms251910410
Xing J, Wang J, Cao J, Li K, Meng X, Wen J, Mysore KS, Wang G, Zhou C, Yin P. Identification of a Novel Gene MtbZIP60 as a Negative Regulator of Leaf Senescence through Transcriptome Analysis in Medicago truncatula. International Journal of Molecular Sciences. 2024; 25(19):10410. https://doi.org/10.3390/ijms251910410
Chicago/Turabian StyleXing, Jiayu, Jialan Wang, Jianuo Cao, Ke Li, Xiao Meng, Jiangqi Wen, Kirankumar S. Mysore, Geng Wang, Chunjiang Zhou, and Pengcheng Yin. 2024. "Identification of a Novel Gene MtbZIP60 as a Negative Regulator of Leaf Senescence through Transcriptome Analysis in Medicago truncatula" International Journal of Molecular Sciences 25, no. 19: 10410. https://doi.org/10.3390/ijms251910410