
Hypoxia Pathways in Parkinson’s Disease: From Pathogenesis to Therapeutic Targets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Hypoxia Pathways
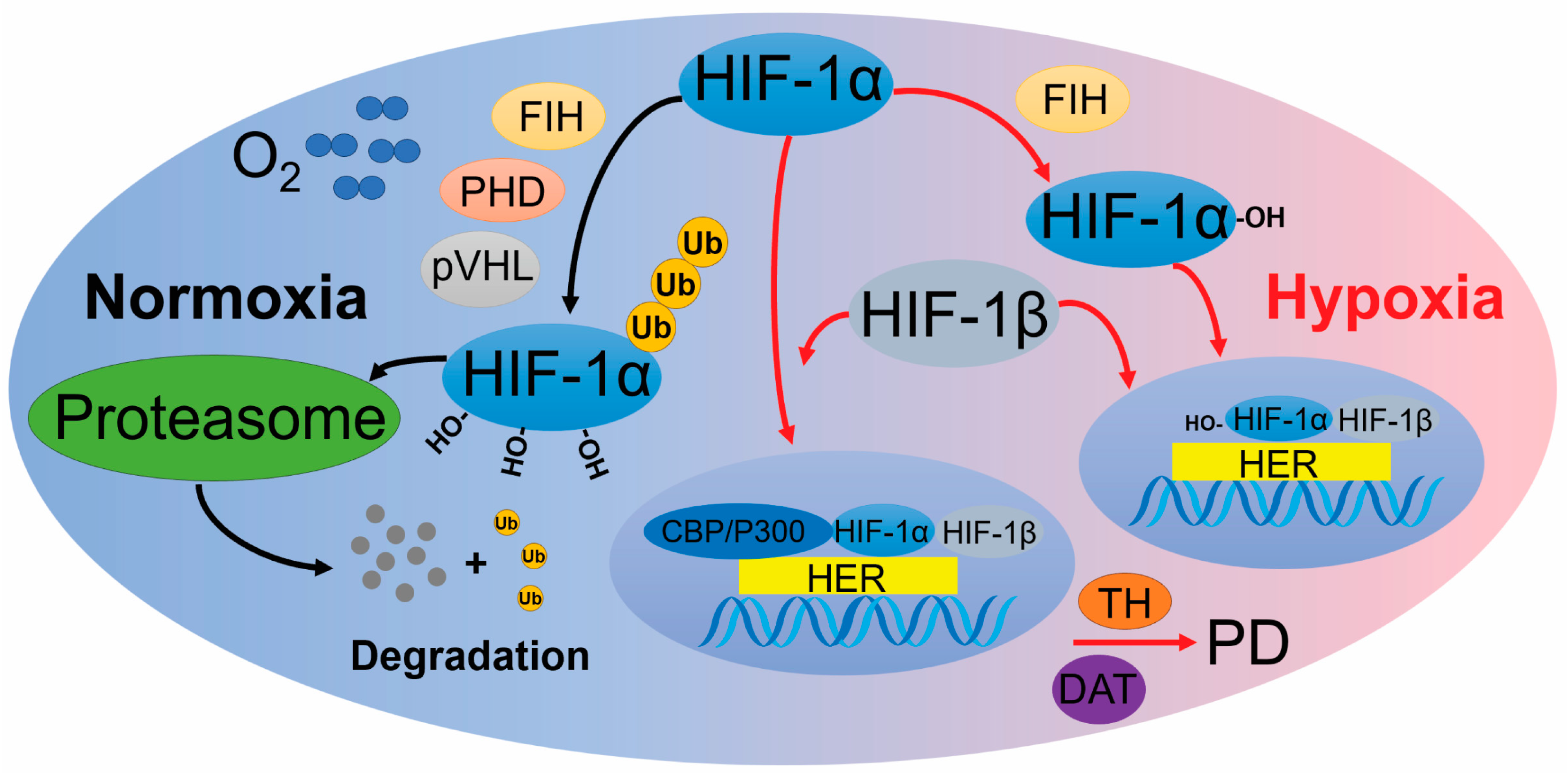
2.1. HIF Pathway
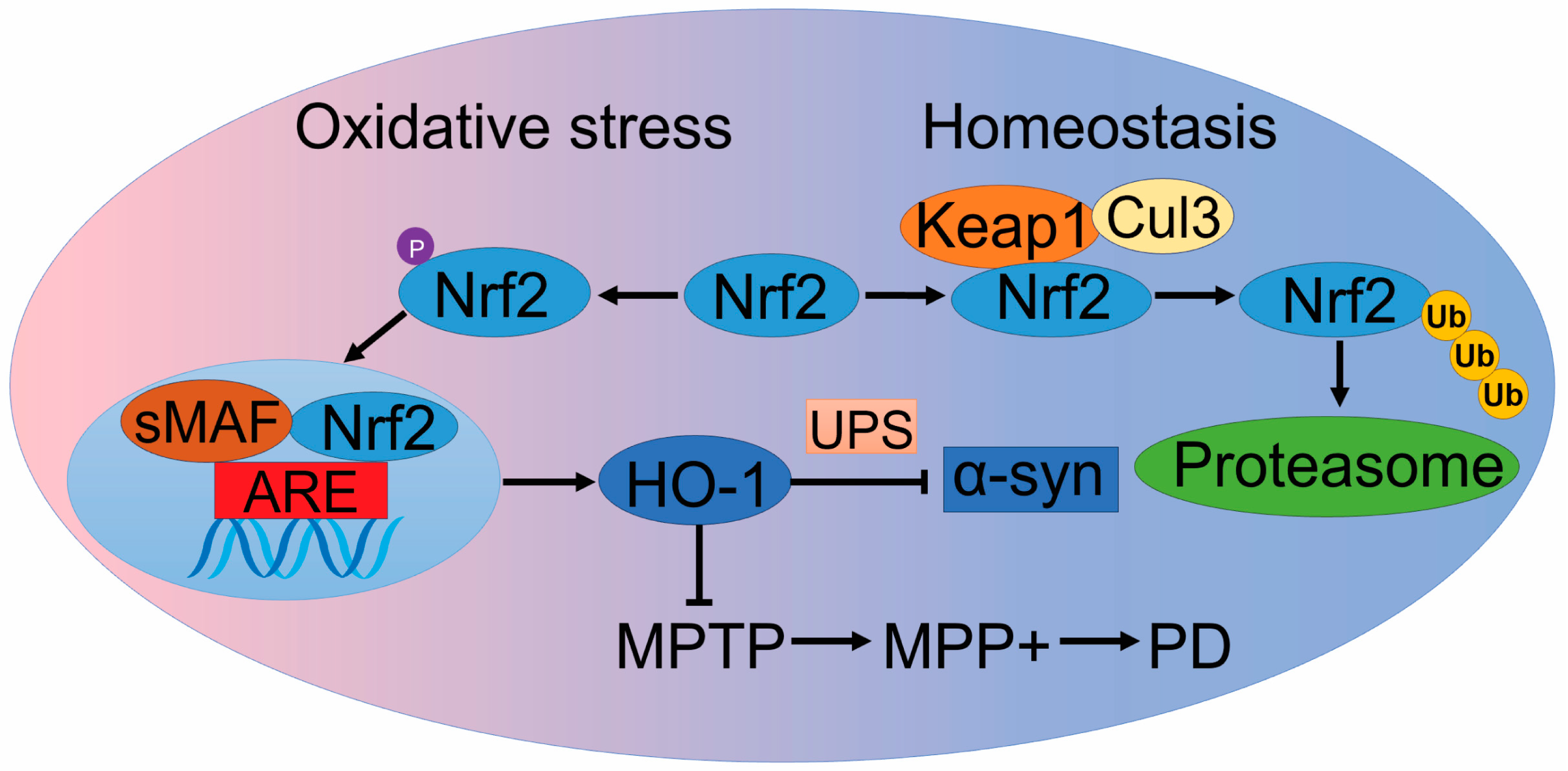
2.2. Nrf2/HO-1 Pathway
3. The Correlation between Hypoxia and PD Pathogenesis
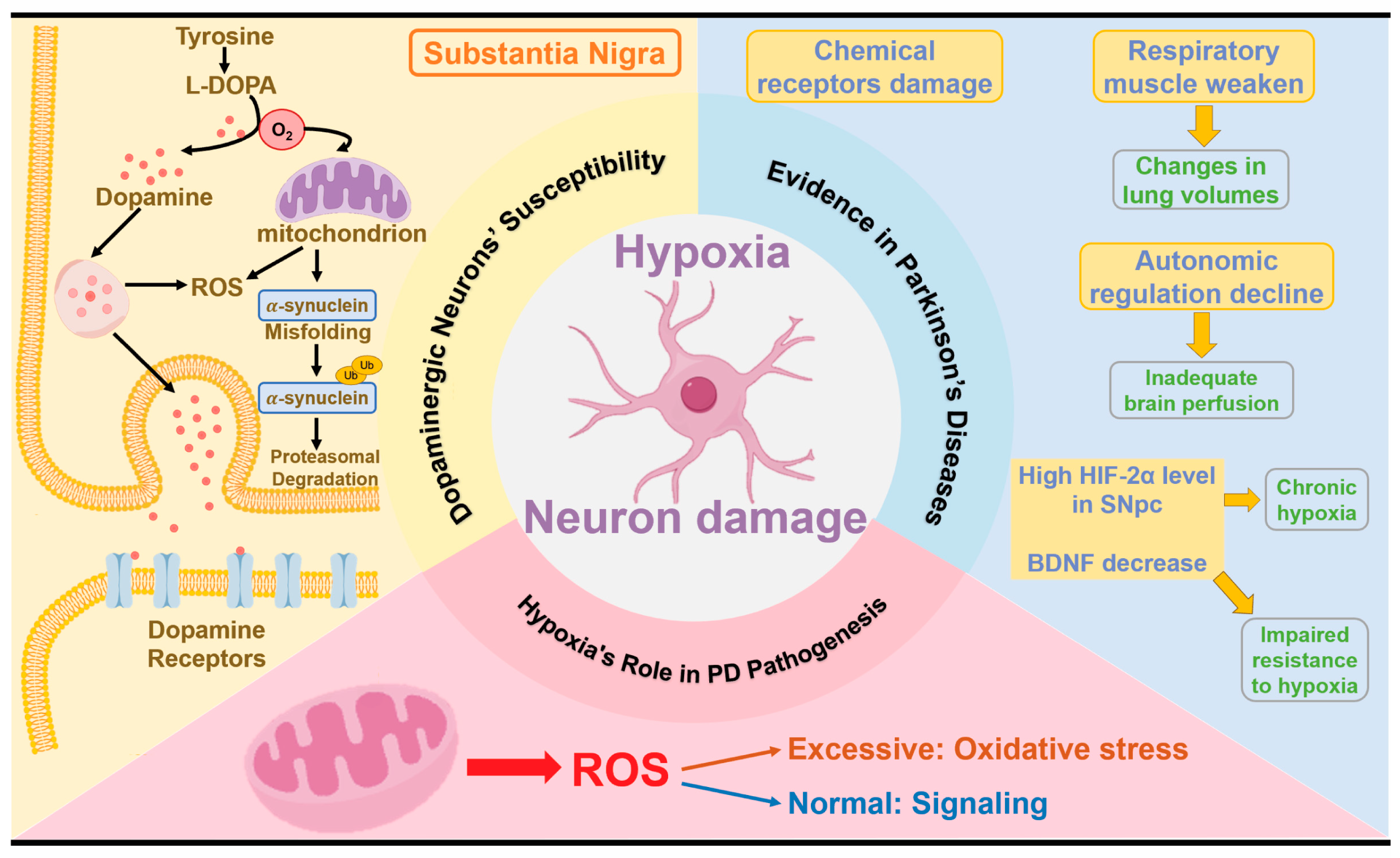
3.1. Dopaminergic Neurons’ Susceptibility to Hypoxia
3.2. Evidence of Hypoxia in PD
3.3. The Role of Hypoxia in PD Pathogenesis
3.4. The Role of Hypoxia in Other Neurodegenerative Diseases
4. The Mechanism of Hypoxia in Causing PD
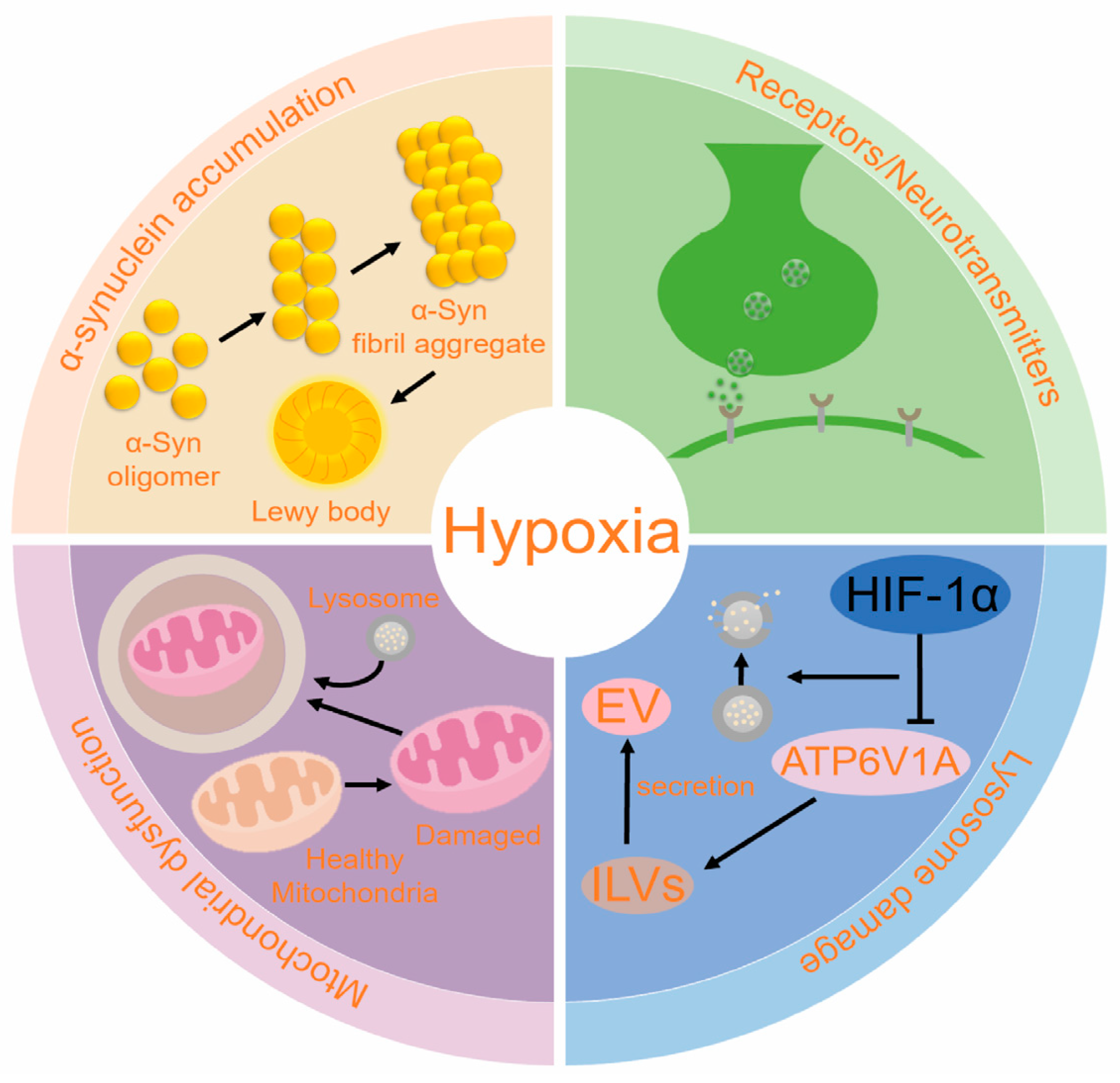
4.1. α-Syn Accumulation
4.2. Mitochondrion Dysfunction
4.3. Lysosome Damage
4.4. PD and Receptors/Neurotransmitters
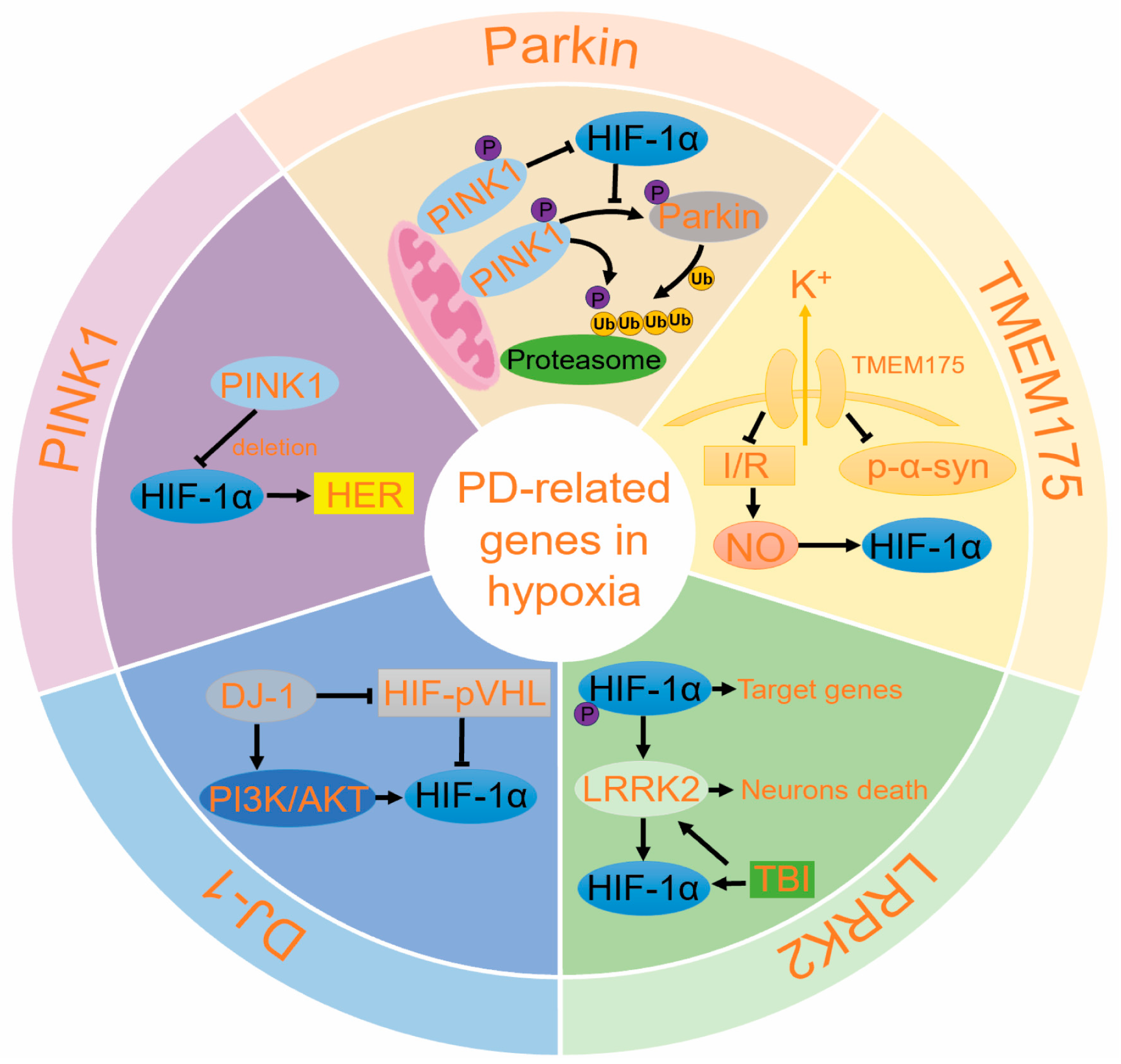
5. The Role of PD-Related Genes in Hypoxia
5.1. Hypoxia Promotes Parkin/PINK1 Pathway
5.2. DJ-1 Reduces Oxidative Stress
5.3. LRRK2 Can Be Activated by HIF-1α
5.4. TMEM175 May Influence HIF-1α
6. Hypoxia-Based Therapeutic Strategies for PD
6.1. Drugs Targeting Hypoxia Pathways
6.2. Behavioral Modulation Improving Hypoxia
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lestón Pinilla, L.; Ugun-Klusek, A.; Rutella, S.; De Girolamo, L.A. Hypoxia signaling in parkinson’s disease: There is use in asking “What HIF?”. Biology 2021, 10, 723. [Google Scholar] [CrossRef] [PubMed]
- Mitroshina, E.V.; Vedunova, M.V. The Role of Oxygen Homeostasis and the HIF-1 Factor in the Development of Neurodegeneration. Int. J. Mol. Sci. 2024, 25, 4581. [Google Scholar] [CrossRef] [PubMed]
- Pinky; Neha; Salman, M.; Kumar, P.; Khan, M.A.; Jamal, A.; Parvez, S. Age-related pathophysiological alterations in molecular stress markers and key modulators of hypoxia. Ageing Res. Rev. 2023, 90, 102022. [Google Scholar] [CrossRef] [PubMed]
- Auti, A.; Alessio, N.; Ballini, A.; Dioguardi, M.; Cantore, S.; Scacco, S.; Vitiello, A.; Quagliuolo, L.; Rinaldi, B.; Santacroce, L. Protective effect of resveratrol against hypoxia-induced neural oxidative stress. J. Pers. Med. 2022, 12, 1202. [Google Scholar] [CrossRef] [PubMed]
- Fiskum, V.; Sandvig, A.; Sandvig, I. Silencing of activity during hypoxia improves functional outcomes in motor neuron networks in vitro. Front. Integr. Neurosci. 2021, 15, 792863. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.C.; Carvalho, C.; Cardoso, S.; Santos, R.X.; Plácido, A.I.; Candeias, E.; Duarte, A.I.; Moreira, P.I. Defective HIF signaling pathway and brain response to hypoxia in neurodegenerative diseases: Not an “iffy” question! Curr. Pharm. Des. 2013, 19, 6809–6822. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.L.; Sun, B.L.; Chen, D.W.; Chen, Y.; Li, W.W.; Xu, M.Y.; Shen, Y.Y.; Xu, Z.Q.; Wang, Y.J.; Bu, X.L. Plasma α-synuclein levels are increased in patients with obstructive sleep apnea syndrome. Ann. Clin. Transl. Neurol. 2019, 6, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Mirelman, A.; Bonato, P.; Camicioli, R.; Ellis, T.D.; Giladi, N.; Hamilton, J.L.; Hass, C.J.; Hausdorff, J.M.; Pelosin, E.; Almeida, Q.J. Gait impairments in Parkinson’s disease. Lancet Neurol. 2019, 18, 697–708. [Google Scholar] [CrossRef]
- Sanchez-Luengos, I.; Lucas-Jiménez, O.; Ojeda, N.; Peña, J.; Gómez-Esteban, J.C.; Gómez-Beldarrain, M.Á.; Vázquez-Picón, R.; Foncea-Beti, N.; Ibarretxe-Bilbao, N. Predictors of health-related quality of life in Parkinson’s disease: The impact of overlap between health-related quality of life and clinical measures. Qual. Life Res. 2022, 31, 3241–3252. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Robak, L.A.; Yu, M.; Cykowski, M.; Shulman, J.M. Genetics and pathogenesis of Parkinson’s syndrome. Annu. Rev. Pathol. Mech. Dis. 2023, 18, 95–121. [Google Scholar] [CrossRef] [PubMed]
- Soares, N.M.; da Silva, P.H.R.; Pereira, G.M.; Leoni, R.F.; Rieder, C.R.d.M.; Alva, T.A.P. Diffusion tensor metrics, motor and non-motor symptoms in de novo Parkinson’s disease. Neuroradiology 2024. online ahead of print. [Google Scholar]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Alster, P.; Madetko-Alster, N. Significance of dysautonomia in Parkinson’s Disease and atypical parkinsonisms. Neurol. Neurochir. Pol. 2024, 58, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Brimson, J.M.; Tencomnao, T. Rhinacanthus nasutus protects cultured neuronal cells against hypoxia induced cell death. Molecules 2011, 16, 6322–6338. [Google Scholar] [CrossRef]
- Burtscher, J.; Syed, M.M.K.; Lashuel, H.A.; Millet, G.P. Hypoxia conditioning as a promising therapeutic target in Parkinson’s disease? Mov. Disord. 2021, 36, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Opazo, A.; Mitchell, G.S. Therapeutic potential of intermittent hypoxia: A matter of dose. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2014, 307, R1181–R1197. [Google Scholar] [CrossRef] [PubMed]
- Janssen Daalen, J.M.; Koopman, W.J.; Saris, C.G.; Meinders, M.J.; Thijssen, D.H.; Bloem, B.R. The Hypoxia Response Pathway: A Potential Intervention Target in Parkinson’s Disease? Mov. Disord. 2024, 39, 273–293. [Google Scholar] [CrossRef]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: Preclinical and clinical outcomes. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2014, 1842, 1282–1294. [Google Scholar] [CrossRef]
- Jha, N.K.; Jha, S.K.; Sharma, R.; Kumar, D.; Ambasta, R.K.; Kumar, P. Hypoxia-induced signaling activation in neurodegenerative diseases: Targets for new therapeutic strategies. J. Alzheimer’s Dis. 2018, 62, 15–38. [Google Scholar] [CrossRef]
- Jagečić, D.; Petrović, D.J.; Šimunić, I.; Isaković, J.; Mitrečić, D. The Oxygen and Glucose Deprivation of Immature Cells of the Nervous System Exerts Distinct Effects on Mitochondria, Mitophagy, and Autophagy, Depending on the Cells’ Differentiation Stage. Brain Sci. 2023, 13, 910. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.-Y.; Huang, B.-Y.; Nie, H.-F.; Chen, X.-Y.; Zhou, Y.; Yang, T.; Cheng, S.-W.; Mei, Z.-G.; Ge, J.-W. The interplay between mitochondrial dysfunction and ferroptosis during ischemia-associated central nervous system diseases. Brain Sci. 2023, 13, 1367. [Google Scholar] [CrossRef] [PubMed]
- Diptendu, S.; Dev, M.G.; Basha, K.A.; Kumar, R.T.; Jahirhussain, G.; Sarasa, D. Reactive oxygen species effects on mitochondrial dynamicity that may lead to Parkinson’s disease: A review. Intern. J. Zool. Invest. 2022, 8, 237–244. [Google Scholar] [CrossRef]
- Baranich, T.; Voronkova, A.; Anufriev, P.; Brydun, A.; Turygina, S.; Glinkina, V.; Sukhorukov, V. Hypoxia-inducible Factors—Their Regulation and Function in Neural Tissue. Hum. Physiol. 2020, 46, 895–899. [Google Scholar] [CrossRef]
- Lewczuk, A.; Zablocka, B.; Beresewicz-Haller, M. Is Nrf2 Behind Endogenous Neuroprotection of the Hippocampal CA2-4, DG Region? Mol. Neurobiol. 2023, 60, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
- Potteti, H.R.; Noone, P.M.; Tamatam, C.R.; Ankireddy, A.; Noel, S.; Rabb, H.; Reddy, S.P. Nrf2 mediates hypoxia-inducible HIF1α activation in kidney tubular epithelial cells. Am. J. Physiol.-Ren. Physiol. 2021, 320, F464–F474. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, J.; Chao, D.; Sandhu, H.K.; Liao, X.; Zhao, J.; Wen, G.; Xia, Y. δ-Opioid receptor activation reduces α-synuclein overexpression and oligomer formation induced by MPP+ and/or hypoxia. Exp. Neurol. 2014, 255, 127–136. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, Z.; Fan, Y.; Fang, Y. An overview of biological research on hypoxia-inducible factors (HIFs). Endokrynol. Pol. 2020, 71, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef]
- Wenger, R.H. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002, 16, 1151–1162. [Google Scholar] [CrossRef]
- Cowman, S.J.; Koh, M.Y. Revisiting the HIF switch in the tumor and its immune microenvironment. Trends Cancer 2022, 8, 28–42. [Google Scholar] [CrossRef]
- Wu, D.; Potluri, N.; Lu, J.; Kim, Y.; Rastinejad, F. Structural integration in hypoxia-inducible factors. Nature 2015, 524, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, A.; Heikkilä, M.; Rautavuoma, K.; Hirsilä, M.; Kivirikko, K.I.; Myllyharju, J. Hypoxia-inducible factor (HIF)-3α is subject to extensive alternative splicing in human tissues and cancer cells and is regulated by HIF-1 but not HIF-2. Int. J. Biochem. Cell Biol. 2010, 42, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Kriegsheim, A.V.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Paltoglou, S.; Roberts, B. HIF-1α and EPAS ubiquitination mediated by the VHL tumour suppressor involves flexibility in the ubiquitination mechanism, similar to other RING E3 ligases. Oncogene 2007, 26, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Camagni, G.F.; Minervini, G.; Tosatto, S.C. Structural Characterization of Hypoxia Inducible Factor α—Prolyl Hydroxylase Domain 2 Interaction through MD Simulations. Int. J. Mol. Sci. 2023, 24, 4710. [Google Scholar] [CrossRef]
- Yamashita, K.; Discher, D.J.; Hu, J.; Bishopric, N.H.; Webster, K.A. Molecular regulation of the endothelin-1 gene by hypoxia: Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, and p300/CBP. J. Biol. Chem. 2001, 276, 12645–12653. [Google Scholar] [CrossRef]
- Lyubimov, A.V.; Cherkashin, D.V.; Efimov, S.V.; Alanichev, A.E.; Ivanov, V.S.; Kutelev, G.G. Mechanisms and triggers of adaptation to hypoxia. Rev. Clin. Pharmacol. Drug Ther. 2021, 19, 269–280. [Google Scholar] [CrossRef]
- Ivy, C.M.; Velotta, J.P.; Cheviron, Z.A.; Scott, G.R. Genetic variation in HIF-2α attenuates ventilatory sensitivity and carotid body growth in chronic hypoxia in high-altitude deer mice. J. Physiol. 2022, 600, 4207–4225. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ma, D.; Yue, F.; Qi, Y.; Dou, M.; Cui, L.; Xing, Y. The potential role of hypoxia-inducible factor-1 in the progression and therapy of central nervous system diseases. Curr. Neuropharmacol. 2022, 20, 1651. [Google Scholar] [CrossRef] [PubMed]
- Mitroshina, E.V.; Savyuk, M.O.; Ponimaskin, E.; Vedunova, M.V. Hypoxia-inducible factor (HIF) in ischemic stroke and neurodegenerative disease. Front. Cell Dev. Biol. 2021, 9, 703084. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-Y.; Wu, J.; Wang, R.; Zhang, S.; Xu, H.; Kaznacheyeva, Е.; Lu, X.-J.; Ren, H.-G.; Wang, G.-H. Pazopanib alleviates neuroinflammation and protects dopaminergic neurons in LPS-stimulated mouse model by inhibiting MEK4-JNK-AP-1 pathway. Acta Pharmacol. Sin. 2023, 44, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, M.A.; Malik, Y.S.; Xing, Z.; Guo, Z.; Tian, H.; Zhu, X.; Chen, X. Polylysine-modified polyethylenimine (PEI-PLL) mediated VEGF gene delivery protects dopaminergic neurons in cell culture and in rat models of Parkinson’s Disease (PD). Acta Biomater. 2017, 54, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, T.; Shingo, T.; Muraoka, K.; Kameda, M.; Takeuchi, A.; Yano, A.; Nishio, S.; Matsui, T.; Miyoshi, Y.; Hamada, H. The differences between high and low-dose administration of VEGF to dopaminergic neurons of in vitro and in vivo Parkinson’s disease model. Brain Res. 2005, 1038, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef]
- Choi, Y.K. Detrimental Roles of Hypoxia-Inducible Factor-1α in Severe Hypoxic Brain Diseases. Int. J. Mol. Sci. 2024, 25, 4465. [Google Scholar] [CrossRef]
- Barteczek, P.; Li, L.; Ernst, A.-S.; Böhler, L.-I.; Marti, H.H.; Kunze, R. Neuronal HIF-1α and HIF-2α deficiency improves neuronal survival and sensorimotor function in the early acute phase after ischemic stroke. J. Cereb. Blood Flow Metab. 2017, 37, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef]
- Fei, P.; Wang, W.; Kim, S.-H.; Wang, S.; Burns, T.F.; Sax, J.K.; Buzzai, M.; Dicker, D.T.; McKenna, W.G.; Bernhard, E.J. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell 2004, 6, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Vatte, S.; Ugale, R. HIF-1, an important regulator in potential new therapeutic approaches to ischemic stroke. Neurochem. Int. 2023, 170, 105605. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Moszyńska, A.; Jaśkiewicz, M.; Serocki, M.; Cabaj, A.; Crossman, D.K.; Bartoszewska, S.; Gebert, M.; Dąbrowski, M.; Collawn, J.F.; Bartoszewski, R. The hypoxia induced changes in miRNA-mRNA in RNA-induced silencing complexes and HIF-2 induced miRNAs in human endothelial cells. FASEB J. 2022, 36, e22412. [Google Scholar] [CrossRef] [PubMed]
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin. Drug Discov. 2019, 14, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Formica, V.; Riondino, S.; Morelli, C.; Guerriero, S.; D’Amore, F.; Di Grazia, A.; Del Vecchio Blanco, G.; Sica, G.; Arkenau, H.-T.; Monteleone, G. HIF2α, Hepcidin and their crosstalk as tumour-promoting signalling. Br. J. Cancer 2023, 129, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Liu, J.; Guo, M.; Gu, Y.; Guan, Y.; Shao, Q.; Ma, W.; Ji, X. Chronic hypoxia leads to cognitive impairment by promoting HIF-2α-mediated ceramide catabolism and alpha-synuclein hyperphosphorylation. Cell Death Discov. 2022, 8, 473. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a transcription factor for stress response and beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- La Vitola, P.; Balducci, C.; Baroni, M.; Artioli, L.; Santamaria, G.; Castiglioni, M.; Cerovic, M.; Colombo, L.; Caldinelli, L.; Pollegioni, L. Peripheral inflammation exacerbates α-synuclein toxicity and neuropathology in Parkinson’s models. Neuropathol. Appl. Neurobiol. 2021, 47, 43–60. [Google Scholar] [CrossRef]
- Yu, C.; Xiao, J.-H. The Keap1-Nrf2 system: A mediator between oxidative stress and aging. Oxidative Med. Cell. Longev. 2021, 2021, 6635460. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a therapeutic target in neurodegenerative diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, G.; Hwang, V.; Ando, D.M.; Daub, A.; Lee, A.K.; Ravisankar, A.; Modan, S.; Finucane, M.M.; Shaby, B.A.; Finkbeiner, S. Nrf2 mitigates LRRK2-and α-synuclein–induced neurodegeneration by modulating proteostasis. Proc. Natl. Acad. Sci. USA 2017, 114, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A. Brain-protective mechanisms of transcription factor NRF2: Toward a common strategy for neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 255–277. [Google Scholar] [CrossRef] [PubMed]
- Padda, I.; Sethi, Y.; Das, M.; Fabian, D.; Ralhan, T.; Aziz, D.; Sexton, J.; Johal, G. Heme Oxygenase-1, Cardiac Senescence, and Myocardial Infarction: A Critical Review of the Triptych. Cardiovasc. Drugs Ther. 2024, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.V.; Sapochnik, D.; Garcia Solá, M.; Coso, O. Regulation of the expression of heme oxygenase-1: Signal transduction, gene promoter activation, and beyond. Antioxid. Redox Signal. 2020, 32, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zheng, J.; Ma, J.; Wang, Z.; Shi, X.; Li, M.; Huang, S.; Hu, S.; Zhao, Z.; Li, D. Increased plasma heme oxygenase-1 levels in patients with early-stage Parkinson’s disease. Front. Aging Neurosci. 2021, 13, 621508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, J.; Duan, H.; Li, R.; Peng, W.; Wu, C. Activation of Nrf2/HO-1 signaling: An important molecular mechanism of herbal medicine in the treatment of atherosclerosis via the protection of vascular endothelial cells from oxidative stress. J. Adv. Res. 2021, 34, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-H.; Hsieh, H.-L. Roles of heme oxygenase-1 in neuroinflammation and brain disorders. Antioxidants 2022, 11, 923. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dou, S.; Zhu, J.; Shao, Z.; Wang, C.; Cheng, B. Ghrelin mitigates MPP+-induced cytotoxicity: Involvement of ERK1/2-mediated Nrf2/HO-1 and endoplasmic reticulum stress PERK signaling pathway. Peptides 2020, 133, 170374. [Google Scholar] [CrossRef]
- Jo, M.G.; Ikram, M.; Jo, M.H.; Yoo, L.; Chung, K.C.; Nah, S.-Y.; Hwang, H.; Rhim, H.; Kim, M.O. Gintonin mitigates MPTP-induced loss of nigrostriatal dopaminergic neurons and accumulation of α-synuclein via the Nrf2/HO-1 pathway. Mol. Neurobiol. 2019, 56, 39–55. [Google Scholar] [CrossRef]
- Mallet, R.T.; Burtscher, J.; Pialoux, V.; Pasha, Q.; Ahmad, Y.; Millet, G.P.; Burtscher, M. Molecular mechanisms of high-altitude acclimatization. Int. J. Mol. Sci. 2023, 24, 1698. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.Y.-Y.; Ho, P.W.-L.; Liu, H.-F.; Leung, C.-T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.-L. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Mallet, R.T.; Burtscher, M.; Millet, G.P. Hypoxia and brain aging: Neurodegeneration or neuroprotection? Ageing Res. Rev. 2021, 68, 101343. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Li, X.; Li, X.; Liu, Q.; Cheng, Y. Oxidative stress in Parkinson’s disease: A systematic review and meta-analysis. Front. Mol. Neurosci. 2018, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Rajagopalan, S.; Siddiq, A.; Gwiazda, R.; Yang, L.; Beal, M.F.; Ratan, R.R.; Andersen, J.K. Inhibition of prolyl hydroxylase protects against 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-induced neurotoxicity: Model for the potential involvement of the hypoxia-inducible factor pathway in Parkinson disease. J. Biol. Chem. 2009, 284, 29065–29076. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Rajagopalan, S.; Ganesan, A.; Andersen, J.K. A Possible Novel Anti-Inflammatory Mechanism for the Pharmacological Prolyl Hydroxylase Inhibitor 3, 4-Dihydroxybenzoate: Implications for Use as a Therapeutic for Parkinson’s Disease. Park. Dis. 2012, 2012, 364684. [Google Scholar] [CrossRef]
- Qin, L.; Shu, L.; Zhong, J.; Pan, H.; Guo, J.; Sun, Q.; Yan, X.; Tang, B.; Xu, Q. Association of HIF1A and Parkinson’s disease in a Han Chinese population demonstrated by molecular inversion probe analysis. Neurol. Sci. 2019, 40, 1927–1931. [Google Scholar] [CrossRef]
- Burtscher, J.; Duderstadt, Y.; Gatterer, H.; Burtscher, M.; Vozdek, R.; Millet, G.P.; Hicks, A.A.; Ehrenreich, H.; Kopp, M. Hypoxia Sensing and Responses in Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 1759. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, C.; Giguère, N.; Bourque, M.-J.; Lévesque, M.; Slack, R.S.; Trudeau, L.-É. Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Meiser, J.; Weindl, D.; Hiller, K. Complexity of dopamine metabolism. Cell Commun. Signal. 2013, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef] [PubMed]
- Karunasinghe, R.N.; Lipski, J. Oxygen and glucose deprivation (OGD)-induced spreading depression in the Substantia Nigra. Brain Res. 2013, 1527, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Onodera, H.; Okabe, S.; Kikuchi, Y.; Tsuda, T.; Itoyama, Y. Impaired chemosensitivity and perception of dyspnoea in Parkinson’s disease. Lancet 2000, 356, 739–740. [Google Scholar] [CrossRef]
- Baille, G.; Perez, T.; Devos, D.; Deken, V.; Defebvre, L.; Moreau, C. Early occurrence of inspiratory muscle weakness in Parkinson’s disease. PLoS ONE 2018, 13, e0190400. [Google Scholar] [CrossRef] [PubMed]
- Herer, B.; Arnulf, I.; Housset, B. Effects of levodopa on pulmonary function in Parkinson’s disease. Chest 2001, 119, 387–393. [Google Scholar] [CrossRef]
- Pal, P.K.; Sathyaprabha, T.N.; Tuhina, P.; Thennarasu, K. Pattern of subclinical pulmonary dysfunctions in Parkinson’s disease and the effect of levodopa. Mov. Disord. 2007, 22, 420–424. [Google Scholar] [CrossRef]
- Sabaté, M.; Gonzalez, I.; Ruperez, F.; Rodríguez, M. Obstructive and restrictive pulmonary dysfunctions in Parkinson’s disease. J. Neurol. Sci. 1996, 138, 114–119. [Google Scholar] [CrossRef]
- Fujii, Y. Olprinone/dopamine combination for improving diaphragmatic fatigue in pentobarbital-anesthetized dogs. Curr. Ther. Res. 2006, 67, 204–213. [Google Scholar] [CrossRef]
- Aubier, M.; Murciano, D.; Menu, Y.; Boczkowski, J.; Mal, H.; Pariente, R. Dopamine effects on diaphragmatic strength during acute respiratory failure in chronic obstructive pulmonary disease. Ann. Intern. Med. 1989, 110, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.K. Neurotrophins in the dentate gyrus. Prog. Brain Res. 2007, 163, 371–397. [Google Scholar] [PubMed]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Hartman, W.; Helan, M.; Smelter, D.; Sathish, V.; Thompson, M.; Pabelick, C.M.; Johnson, B.; Prakash, Y. Role of hypoxia-induced brain derived neurotrophic factor in human pulmonary artery smooth muscle. PLoS ONE 2015, 10, e0129489. [Google Scholar] [CrossRef]
- Burtscher, J.; Citherlet, T.; Camacho-Cardenosa, A.; Camacho-Cardenosa, M.; Raberin, A.; Krumm, B.; Hohenauer, E.; Egg, M.; Lichtblau, M.; Müller, J. Mechanisms underlying the health benefits of intermittent hypoxia conditioning. J. Physiol. 2023; in press. [Google Scholar] [CrossRef]
- Pena, E.; San Martin-Salamanca, R.; El Alam, S.; Flores, K.; Arriaza, K. Tau Protein Alterations Induced by Hypobaric Hypoxia Exposure. Int. J. Mol. Sci. 2024, 25, 889. [Google Scholar] [CrossRef] [PubMed]
- Kazim, S.F.; Sharma, A.; Saroja, S.R.; Seo, J.H.; Larson, C.S.; Ramakrishnan, A.; Wang, M.; Blitzer, R.D.; Shen, L.; Peña, C.J. Chronic intermittent hypoxia enhances pathological tau seeding, propagation, and accumulation and exacerbates Alzheimer-like memory and synaptic plasticity deficits and molecular signatures. Biol. Psychiatry 2022, 91, 346–358. [Google Scholar] [CrossRef]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxidative Med. Cell. Longev. 2015, 2015, 151979. [Google Scholar] [CrossRef] [PubMed]
- Heras-Garvin, A.; Danninger, C.; Eschlböck, S.; Holton, J.L.; Wenning, G.K.; Stefanova, N. Signs of Chronic Hypoxia Suggest a Novel Pathophysiological Event in α-Synucleinopathies. Mov. Disord. 2020, 35, 2333–2338. [Google Scholar] [CrossRef] [PubMed]
- Kalva-Filho, C.A.; Faria, M.H.; Papoti, M.; Barbieri, F.A. Acute and cumulative effects of hypoxia exposure in people with Parkinson’s disease: A scoping review and evidence map. Park. Relat. Disord. 2023, 118, 105885. [Google Scholar] [CrossRef]
- McDonald, C.; Newton, J.L.; Burn, D.J. Orthostatic hypotension and cognitive impairment in Parkinson’s disease: Causation or association? Mov. Disord. 2016, 31, 937–946. [Google Scholar] [CrossRef]
- Hatano, T.; Okuzumi, A.; Matsumoto, G.; Tsunemi, T.; Hattori, N. α-Synuclein: A Promising Biomarker for Parkinson’s Disease and Related Disorders. J. Mov. Disord. 2024, 17, 127. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Longhena, F.; Faustini, G.; Spillantini, M.G.; Bellucci, A. Living in promiscuity: The multiple partners of alpha-synuclein at the synapse in physiology and pathology. Int. J. Mol. Sci. 2019, 20, 141. [Google Scholar] [CrossRef] [PubMed]
- Salmina, A.B.; Kapkaeva, M.R.; Vetchinova, A.S.; Illarioshkin, S.N. Novel approaches used to examine and control neurogenesis in Parkinson′s disease. Int. J. Mol. Sci. 2021, 22, 9608. [Google Scholar] [CrossRef]
- Fusco, G.; Sanz-Hernandez, M.; De Simone, A. Order and disorder in the physiological membrane binding of α-synuclein. Curr. Opin. Struct. Biol. 2018, 48, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Booms, A.; Coetzee, G.A. Functions of intracellular alpha-synuclein in microglia: Implications for Parkinson’s disease risk. Front. Cell. Neurosci. 2021, 15, 759571. [Google Scholar] [CrossRef]
- Tripathi, T.; Chattopadhyay, K. Interaction of α-Synuclein with ATP Synthase: Switching Role from Physiological to Pathological. ACS Chem. Neurosci. 2018, 10, 16–17. [Google Scholar] [CrossRef]
- Han, D.; Zheng, W.; Wang, X.; Chen, Z. Proteostasis of α-synuclein and its role in the pathogenesis of Parkinson’s disease. Front. Cell. Neurosci. 2020, 14, 45. [Google Scholar] [CrossRef]
- Irwin, D.J.; Abrams, J.Y.; Schonberger, L.B.; Leschek, E.W.; Mills, J.L.; Lee, V.M.-Y.; Trojanowski, J.Q. Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol. 2013, 70, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Gao, J.; Wang, J.; Xie, A. Prion-like mechanisms in Parkinson’s disease. Front. Neurosci. 2019, 13, 552. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Lim, Y.-J.; Liu, Z.; Long, H.; Sun, Y.; Hu, J.-J.; Zhao, C.; Tao, Y.; Zhang, X.; Li, D. Parkinson’s disease-related phosphorylation at Tyr39 rearranges α-synuclein amyloid fibril structure revealed by cryo-EM. Proc. Natl. Acad. Sci. USA 2020, 117, 20305–20315. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; He, X.; Zhang, Q.; Zhang, N.; Li, L.; Hui, Y.; Li, W.; Wang, D.; Wu, Z. A-synuclein induces microglial cell migration through stimulating HIF-1α accumulation. J. Neurosci. Res. 2017, 95, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Zhang, H.; Zhang, J.; Huang, K.; Chen, Y.; Jing, X.; Tao, E. α-Synuclein induces neuroinflammation injury through the IL6ST-AS/STAT3/HIF-1α axis. Int. J. Mol. Sci. 2023, 24, 1436. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Mehta, S.L.; Kaimal, B.; Lyons, K.; Dempsey, R.J.; Vemuganti, R. Poststroke induction of α-synuclein mediates ischemic brain damage. J. Neurosci. 2016, 36, 7055–7065. [Google Scholar] [CrossRef] [PubMed]
- Suthar, S.K.; Lee, S.-Y. Truncation or proteolysis of α-synuclein in Parkinsonism. Ageing Res. Rev. 2023, 90, 101978. [Google Scholar] [CrossRef] [PubMed]
- Weimers, P.; Halfvarson, J.; Sachs, M.C.; Saunders-Pullman, R.; Ludvigsson, J.F.; Peter, I.; Burisch, J.; Olén, O. Inflammatory bowel disease and Parkinson’s disease: A nationwide Swedish cohort study. Inflamm. Bowel Dis. 2019, 25, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Goodwin, J.; Engelborghs, Y.; Pountney, D. Raised calcium promotes α-synuclein aggregate formation. Mol. Cell. Neurosci. 2011, 46, 516–526. [Google Scholar] [CrossRef]
- Martinez, J.; Moeller, I.; Erdjument-Bromage, H.; Tempst, P.; Lauring, B. Parkinson’s disease-associated α-synuclein is a calmodulin substrate. J. Biol. Chem. 2003, 278, 17379–17387. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Nanduri, J.; Bhasker, C.R.; Semenza, G.L.; Prabhakar, N.R. Ca2+/calmodulin kinase-dependent activation of hypoxia inducible factor 1 transcriptional activity in cells subjected to intermittent hypoxia. J. Biol. Chem. 2005, 280, 4321–4328. [Google Scholar] [CrossRef]
- Zhang, Z.; Yan, J.; Chang, Y.; ShiDu Yan, S.; Shi, H. Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Curr. Med. Chem. 2011, 18, 4335–4343. [Google Scholar] [CrossRef]
- Chen, B.; Wen, X.; Jiang, H.; Wang, J.; Song, N.; Xie, J. Interactions between iron and α-synuclein pathology in Parkinson’s disease. Free Radic. Biol. Med. 2019, 141, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, C.; Tsuchiya, K.; Maeda, K. Hypoxia-inducible factor prolyl hydroxylase inhibitors and iron metabolism. Int. J. Mol. Sci. 2023, 24, 3037. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, J.; Tresse, E.; Ejlerskov, P.; Hu, E.; Liu, Y.; Marin, A.; Montalant, A.; Satriano, L.; Rundsten, C.F.; Carlsen, E.M.M. PIAS2-mediated blockade of IFN-β signaling: A basis for sporadic Parkinson disease dementia. Mol. Psychiatry 2021, 26, 6083–6099. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J. Direct membrane association drives mitochondrial fission by the parkinson disease-associated protein α-Synuclein*♦. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Du, T.; Wang, X.; Duan, C.; Gao, G.; Zhang, J.; Lu, L.; Yang, H. α-Synuclein amino terminus regulates mitochondrial membrane permeability. Brain Res. 2014, 1591, 14–26. [Google Scholar] [CrossRef]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s disease: From pathogenesis to treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef]
- Kong, Q.; Lin, C.-l.G. Oxidative damage to RNA: Mechanisms, consequences, and diseases. Cell. Mol. Life Sci. 2010, 67, 1817–1829. [Google Scholar] [CrossRef]
- Burmann, B.M.; Gerez, J.A.; Matečko-Burmann, I.; Campioni, S.; Kumari, P.; Ghosh, D.; Mazur, A.; Aspholm, E.E.; Šulskis, D.; Wawrzyniuk, M. Regulation of α-synuclein by chaperones in mammalian cells. Nature 2020, 577, 127–132. [Google Scholar] [CrossRef]
- Weng, M.; Xie, X.; Liu, C.; Lim, K.-L.; Zhang, C.-w.; Li, L. The sources of reactive oxygen species and its possible role in the pathogenesis of Parkinson’s disease. Park. Dis. 2018, 2018, 9163040. [Google Scholar] [CrossRef]
- Peng, Y.; He, J.; Xiang, H.; Xie, L.; She, J.; Cheng, D.; Liu, B.; Hu, J.; Qian, H. Potential Impact of Hypoxic Astrocytes on the Aggravation of Depressive Symptoms in Parkinson’s Disease. J. Mol. Neurosci. 2024, 74, 28. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-N.; Xi, M.-M.; Guo, Y.; Hai, C.-X.; Yang, W.-L.; Qin, X.-J. NADPH oxidase-mitochondria axis-derived ROS mediate arsenite-induced HIF-1α stabilization by inhibiting prolyl hydroxylases activity. Toxicol. Lett. 2014, 224, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Köhl, R.; Zhou, J.; Brüne, B. Reactive oxygen species attenuate nitric-oxide-mediated hypoxia-inducible factor-1α stabilization. Free. Radic. Biol. Med. 2006, 40, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Lafleur, V.N.; Robitaille, G.A.; Chan, D.A.; Giaccia, A.J.; Richard, D.E. Hypoxia-inducible factor-1 activation in nonhypoxic conditions: The essential role of mitochondrial-derived reactive oxygen species. Mol. Biol. Cell 2010, 21, 3247–3257. [Google Scholar] [CrossRef]
- Hu, Z.; Dong, N.; Lu, D.; Jiang, X.; Xu, J.; Wu, Z.; Zheng, D.; Wechsler, D.S. A positive feedback loop between ROS and Mxi1-0 promotes hypoxia-induced VEGF expression in human hepatocellular carcinoma cells. Cell. Signal. 2017, 31, 79–86. [Google Scholar] [CrossRef]
- Chen, C.; Guo, Z.; Shi, X.; Guo, Y.; Ma, G.; Ma, J.; Yu, Q. H2O2-induced oxidative stress improves meat tenderness by accelerating glycolysis via hypoxia-inducible factor-1α signaling pathway in postmortem bovine muscle. Food Chem. X 2022, 16, 100466. [Google Scholar] [CrossRef]
- Gremmels, H.; De Jong, O.G.; Hazenbrink, D.H.; Fledderus, J.O.; Verhaar, M.C. The transcription factor Nrf2 protects angiogenic capacity of endothelial colony-forming cells in high-oxygen radical stress conditions. Stem Cells Int. 2017, 2017, 4680612. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta BBA-Gen. Subj. 2015, 1850, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Srinivas, V.; Leshchinsky, I.; Sang, N.; King, M.P.; Minchenko, A.; Caro, J. Oxygen sensing and HIF-1 activation does not require an active mitochondrial respiratory chain electron-transfer pathway. J. Biol. Chem. 2001, 276, 21995–21998. [Google Scholar] [CrossRef]
- Baixauli, F.; Acín-Pérez, R.; Villarroya-Beltrí, C.; Mazzeo, C.; Nuñez-Andrade, N.; Gabandé-Rodriguez, E.; Ledesma, M.D.; Blázquez, A.; Martin, M.A.; Falcón-Pérez, J.M. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Mosquera, L.; Yambire, K.F.; Couto, R.; Pereyra, L.; Pabis, K.; Ponsford, A.H.; Diogo, C.V.; Stagi, M.; Milosevic, I.; Raimundo, N. Mitochondrial respiratory chain deficiency inhibits lysosomal hydrolysis. Autophagy 2019, 15, 1572–1591. [Google Scholar] [CrossRef] [PubMed]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Bélanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of mitochondrial function impairs lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Coody, T.K.; Jeong, M.-Y.; Berg, J.A.; Winge, D.R.; Hughes, A.L. Cysteine toxicity drives age-related mitochondrial decline by altering iron homeostasis. Cell 2020, 180, 296–310.e18. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.A.; Yen, F.S.; Nicholson, S.P.; Alwaseem, H.; Bayraktar, E.C.; Alam, M.; Timson, R.C.; La, K.; Abu-Remaileh, M.; Molina, H. Maintaining iron homeostasis is the key role of lysosomal acidity for cell proliferation. Mol. Cell 2020, 77, 645–655.e7. [Google Scholar] [CrossRef] [PubMed]
- Deus, C.M.; Yambire, K.F.; Oliveira, P.J.; Raimundo, N. Mitochondria–lysosome crosstalk: From physiology to neurodegeneration. Trends Mol. Med. 2020, 26, 71–88. [Google Scholar] [CrossRef]
- Hao, T.; Yu, J.; Wu, Z.; Jiang, J.; Gong, L.; Wang, B.; Guo, H.; Zhao, H.; Lu, B.; Engelender, S. Hypoxia-reprogramed megamitochondrion contacts and engulfs lysosome to mediate mitochondrial self-digestion. Nat. Commun. 2023, 14, 4105. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef]
- Wallings, R.L.; Humble, S.W.; Ward, M.E.; Wade-Martins, R. Lysosomal dysfunction at the centre of Parkinson’s disease and frontotemporal dementia/amyotrophic lateral sclerosis. Trends Neurosci. 2019, 42, 899–912. [Google Scholar] [CrossRef]
- Baker, M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome. Nat. Lett. 2006, 17, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; O’Reilly, M.; Gibbons, G.S.; Changolkar, L.; McBride, J.D.; Riddle, D.M.; Zhang, B.; Stieber, A.; Nirschl, J.; Kim, S.-J. In vitro amplification of pathogenic tau conserves disease-specific bioactive characteristics. Acta Neuropathol. 2021, 141, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef] [PubMed]
- Domert, J.; Rao, S.B.; Agholme, L.; Brorsson, A.-C.; Marcusson, J.; Hallbeck, M.; Nath, S. Spreading of amyloid-β peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol. Dis. 2014, 65, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Troncoso, M.; Bannoud, N.; Carvelli, L.; Asensio, J.; Seltzer, A.; Sosa, M. Hypoxia-ischemia alters distribution of lysosomal proteins in rat cortex and hippocampus. Biol. Open 2018, 7, bio036723. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, R.; Zhai, P.; Liu, Z.; Xia, R.; Zhang, Z.; Qin, X.; Li, C.; Chen, W.; Li, J. Hypoxia promotes EV secretion by impairing lysosomal homeostasis in HNSCC through negative regulation of ATP6V1A by HIF-1α. J. Extracell. Vesicles 2023, 12, e12310. [Google Scholar] [CrossRef]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012, 12, 1–10. [Google Scholar] [CrossRef]
- Muñiz-García, A.; Romero, M.; Falcόn-Perez, J.M.; Murray, P.; Zorzano, A.; Mora, S. Hypoxia-induced HIF1α activation regulates small extracellular vesicle release in human embryonic kidney cells. Sci. Rep. 2022, 12, 1443. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Cheng, S.-B.; Ikawa, M.; Yoshimori, T.; Huber, W.J.; Menon, R.; Huang, Z.; Fierce, J.; Padbury, J.F.; Sadovsky, Y. Evidence for lysosomal biogenesis proteome defect and impaired autophagy in preeclampsia. Autophagy 2020, 16, 1771–1785. [Google Scholar] [CrossRef]
- Damaghi, M.; Tafreshi, N.K.; Lloyd, M.C.; Sprung, R.; Estrella, V.; Wojtkowiak, J.W.; Morse, D.L.; Koomen, J.M.; Bui, M.M.; Gatenby, R.A. Chronic acidosis in the tumour microenvironment selects for overexpression of LAMP2 in the plasma membrane. Nat. Commun. 2015, 6, 8752. [Google Scholar] [CrossRef]
- Glunde, K.; Guggino, S.E.; Solaiyappan, M.; Pathak, A.P.; Ichikawa, Y.; Bhujwalla, Z.M. Extracellular acidification alters lysosomal trafficking in human breast cancer cells. Neoplasia 2003, 5, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Wuest, T.R.; Min, Y.; Lin, P.C. Oxygen tension regulates lysosomal activation and receptor tyrosine kinase degradation. Cancers 2019, 11, 1653. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Palmfeldt, J.; Lin, L.; Colaço, A.; Clemmensen, K.K.; Huang, J.; Xu, F.; Liu, X.; Maeda, K.; Luo, Y. STAT3 associates with vacuolar H+-ATPase and regulates cytosolic and lysosomal pH. Cell Res. 2018, 28, 996–1012. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y. Hypoxia Regulates Type II Collagen Secretion by Chondrocytes through the Autophagy-Lysosomal Pathway. Ph.D. Thesis, The Chinese University of Hong Kong, Hong Kong, 2019. [Google Scholar]
- Feng, H.; Wang, J.; Chen, W.; Shan, B.; Guo, Y.; Xu, J.; Wang, L.; Guo, P.; Zhang, Y. Hypoxia-induced autophagy as an additional mechanism in human osteosarcoma radioresistance. J. Bone Oncol. 2016, 5, 67–73. [Google Scholar] [CrossRef]
- Monti, C.; Bondi, H.; Urbani, A.; Fasano, M.; Alberio, T. Systems biology analysis of the proteomic alterations induced by MPP+, a Parkinson’s disease-related mitochondrial toxin. Front. Cell. Neurosci. 2015, 9, 14. [Google Scholar] [CrossRef]
- Chen, T.; Hou, R.; Li, C.; Wu, C.; Xu, S. MPTP/MPP+ suppresses activation of protein C in Parkinson’s disease. J. Alzheimer’s Dis. 2015, 43, 133–142. [Google Scholar] [CrossRef]
- Dong, S.-Y.; Guo, Y.-J.; Feng, Y.; Cui, X.-X.; Kuo, S.-H.; Liu, T.; Wu, Y.-C. The epigenetic regulation of HIF-1α by SIRT1 in MPP+ treated SH-SY5Y cells. Biochem. Biophys. Res. Commun. 2016, 470, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Grande, B.; Blackabey, V.; Gittens, B.; Pinteaux, E.; Denes, A. Loss of substance P and inflammation precede delayed neurodegeneration in the substantia nigra after cerebral ischemia. Brain Behav. Immun. 2013, 29, 51–61. [Google Scholar] [CrossRef]
- Arena, G.; Gelmetti, V.; Torosantucci, L.; Vignone, D.; Lamorte, G.; De Rosa, P.; Cilia, E.; Jonas, E.; Valente, E.M. PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ. 2013, 20, 920–930. [Google Scholar] [CrossRef]
- Gautier, C.; Corti, O.; Brice, A. Mitochondrial dysfunctions in Parkinson’s disease. Rev. Neurol. 2014, 170, 339–343. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-R.; Li, Y.-Q.; Hua, C.; Li, S.-J.; Zhao, G.; Song, H.-M.; Yu, M.-X.; Huang, Q. Oxidative stress inhibits growth and induces apoptotic cell death in human U251 glioma cells via the caspase-3-dependent pathway. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4068–4075. [Google Scholar] [PubMed]
- Xu, Y.; Zhi, F.; Mao, J.; Peng, Y.; Shao, N.; Balboni, G.; Yang, Y.; Xia, Y. δ-opioid receptor activation protects against Parkinson’s disease-related mitochondrial dysfunction by enhancing PINK1/Parkin-dependent mitophagy. Aging 2020, 12, 25035. [Google Scholar] [CrossRef] [PubMed]
- Hornykiewicz, O. Brain monoamines and parkinsonism. Natl. Inst. Drug Abus. Monogr. Ser. 1975, 3, 13–21. [Google Scholar]
- Reader, T.; Dewar, K. Review article Effects of denervation and hyperinnervation on dopamine and serotonin systems in the rat neostriatum: Implications for human Parkinsons disease. Neurochem. Int. 1999, 34, 1–21. [Google Scholar] [CrossRef]
- Scholtissen, B.; Verhey, F.; Steinbusch, H.; Leentjens, A. Serotonergic mechanisms in Parkinson’s disease: Opposing results from preclinical and clinical data. J. Neural Transm. 2006, 113, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, K.; Kaczyńska, K.; Zaremba, M. Serotonergic system in hypoxic ventilatory response in unilateral rat model of Parkinson’s disease. J. Biomed. Sci. 2017, 24, 1–10. [Google Scholar] [CrossRef]
- Hickner, S.; Hussain, N.; Angoa-Perez, M.; Francescutti, D.M.; Kuhn, D.M.; Mateika, J.H. Ventilatory long-term facilitation is evident after initial and repeated exposure to intermittent hypoxia in mice genetically depleted of brain serotonin. J. Appl. Physiol. 2014, 116, 240–250. [Google Scholar] [CrossRef]
- Lin, L.-F.H.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef]
- Foti, R.; Zucchelli, S.; Biagioli, M.; Roncaglia, P.; Vilotti, S.; Calligaris, R.; Krmac, H.; Girardini, J.E.; Del Sal, G.; Gustincich, S. Parkinson disease-associated DJ-1 is required for the expression of the glial cell line-derived neurotrophic factor receptor RET in human neuroblastoma cells. J. Biol. Chem. 2010, 285, 18565–18574. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.K. Hypoxia. 3. Hypoxia and neurotransmitter synthesis. Am. J. Physiol.-Cell Physiol. 2011, 300, C743–C751. [Google Scholar] [CrossRef] [PubMed]
- Kalashnyk, O.; Lykhmus, O.; Koval, L.; Uspenska, K.; Obolenskaya, M.; Chernyshov, V.; Komisarenko, S.; Skok, M. α7 Nicotinic acetylcholine receptors regulate translocation of HIF-1α to the cell nucleus and mitochondria upon hypoxia. Biochem. Biophys. Res. Commun. 2023, 657, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Rengasamy, A.; Johns, R. Characterization of endothelium-derived relaxing factor/nitric oxide synthase from bovine cerebellum and mechanism of modulation by high and low oxygen tensions. J. Pharmacol. Exp. Ther. 1991, 259, 310–316. [Google Scholar] [PubMed]
- Barodia, S.K.; Creed, R.B.; Goldberg, M.S. Parkin and PINK1 functions in oxidative stress and neurodegeneration. Brain Res. Bull. 2017, 133, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiu, L.; Liu, X.; Hou, Z.; Yu, B. PINK1 alleviates myocardial hypoxia-reoxygenation injury by ameliorating mitochondrial dysfunction. Biochem. Biophys. Res. Commun. 2017, 484, 118–124. [Google Scholar] [CrossRef]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of ubiquitin-proteasome system in neurodegenerative diseases. Front. Aging Neurosci. 2016, 8, 303. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Zhao, Y.; Yue, X.; Wu, H.; Huang, S.; Chen, J.; Tomsky, K.; Xie, H.; Khella, C.A. Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 2017, 8, 1823. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, J.; Liu, Z.; Zhou, J.; Yao, W.; Tao, J.; Shen, M.; Liu, H. FSH prevents porcine granulosa cells from hypoxia-induced apoptosis via activating mitophagy through the HIF-1α-PINK1-Parkin pathway. FASEB J. 2020, 34, 3631–3645. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Qin, Z.; Chen, X.; He, W.; Li, D.; Zhang, L.; Le, Y.; Xiong, Q.; Zhang, B.; Wang, H. HIF-1α Ameliorates Diabetic Neuropathic Pain via Parkin-Mediated Mitophagy in a Mouse Model. BioMed Res. Int. 2022, 2022, 5274375. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, Y.; Guo, Y.H.; Wang, L.; Yang, Z.; Zhai, Z.H.; Tang, L. HIF-1α alleviates high-glucose-induced renal tubular cell injury by promoting Parkin/PINK1-mediated mitophagy. Front. Med. 2022, 8, 803874. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, G.; D’Amico, A.G.; Reitano, R.; Saccone, S.; Federico, C.; Cavallaro, S.; D’Agata, V. Parkin modulates expression of HIF-1α and HIF-3α during hypoxia in gliobastoma-derived cell lines in vitro. Cell Tissue Res. 2016, 364, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Kasai, S.; Torii, S.; Kakita, A.; Sogawa, K. Inhibitory PAS domain protein is a substrate of PINK1 and Parkin and mediates cell death in a Parkinson’s disease model. Cell Death Dis. 2015, 6, e1886. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Kang, U.J. Characterization of PINK1 processing, stability, and subcellular localization. J. Neurochem. 2008, 106, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Priyadarshini, M.; Tuimala, J.; Chen, Y.; Panula, P. A zebrafish model of PINK1 deficiency reveals key pathway dysfunction including HIF signaling. Neurobiol. Dis. 2013, 54, 127–138. [Google Scholar] [CrossRef]
- Xu, Y.; Zhi, F.; Peng, Y.; Shao, N.; Khiati, D.; Balboni, G.; Yang, Y.; Xia, Y. δ-Opioid receptor activation attenuates hypoxia/MPP+-Induced downregulation of PINK1: A novel mechanism of neuroprotection against parkinsonian injury. Mol. Neurobiol. 2019, 56, 252–266. [Google Scholar] [CrossRef]
- Wadlington, N.L. The Role of PINK1 in the Hypoxic Stress Response Through the Regulation of Hif-1alpha Translation. Ph.D. Thesis, University of Chicago, Division of the Biological Sciences, and The Pritzker, Chicago, IL, USA, 2013. [Google Scholar]
- Lin, W.; Wadlington, N.L.; Chen, L.; Zhuang, X.; Brorson, J.R.; Kang, U.J. Loss of PINK1 attenuates HIF-1α induction by preventing 4E-BP1-dependent switch in protein translation under hypoxia. J. Neurosci. 2014, 34, 3079–3089. [Google Scholar] [CrossRef]
- Chiu, D.K.-C.; Tse, A.P.-W.; Law, C.-T.; Xu, I.M.-J.; Lee, D.; Chen, M.; Lai, R.K.-H.; Yuen, V.W.-H.; Cheu, J.W.-S.; Ho, D.W.-H. Hypoxia regulates the mitochondrial activity of hepatocellular carcinoma cells through HIF/HEY1/PINK1 pathway. Cell Death Dis. 2019, 10, 934. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guo, J.; Wang, Y.; Jiang, Y.; Li, S.; Liu, Y.N.; Liu, W.J. JinChan YiShen TongLuo Formula ameliorate mitochondrial dysfunction and apoptosis in diabetic nephropathy through the HIF-1α-PINK1-Parkin pathway. J. Ethnopharmacol. 2024, 328, 117863. [Google Scholar] [CrossRef] [PubMed]
- Mencke, P.; Boussaad, I.; Romano, C.D.; Kitami, T.; Linster, C.L.; Krüger, R. The role of DJ-1 in cellular metabolism and pathophysiological implications for Parkinson’s disease. Cells 2021, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Biosa, A.; Sandrelli, F.; Beltramini, M.; Greggio, E.; Bubacco, L.; Bisaglia, M. Recent findings on the physiological function of DJ-1: Beyond Parkinson’s disease. Neurobiol. Dis. 2017, 108, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Rizzu, P.; Hinkle, D.A.; Zhukareva, V.; Bonifati, V.; Severijnen, L.A.; Martinez, D.; Ravid, R.; Kamphorst, W.; Eberwine, J.H.; Lee, V.M.Y. DJ-1 colocalizes with tau inclusions: A link between parkinsonism and dementia. Ann. Neurol. 2004, 55, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Petrie, T.G.; Liu, Y.; Liu, J.; Fujioka, H.; Zhu, X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J. Neurochem. 2012, 121, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Aleyasin, H.; Rousseaux, M.W.; Phillips, M.; Kim, R.H.; Bland, R.J.; Callaghan, S.; Slack, R.S.; During, M.J.; Mak, T.W.; Park, D.S. The Parkinson’s disease gene DJ-1 is also a key regulator of stroke-induced damage. Proc. Natl. Acad. Sci. USA 2007, 104, 18748–18753. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, S.; Afzal, S.; Tardivel-Lacombe, J.; Park, D.S.; Iovanna, J.L.; Mak, T.W. DJ-1/PARK7 is an important mediator of hypoxia-induced cellular responses. Proc. Natl. Acad. Sci. USA 2009, 106, 1111–1116. [Google Scholar] [CrossRef]
- Zheng, H.; Zhou, C.; Lu, X.; Liu, Q.; Liu, M.; Chen, G.; Chen, W.; Wang, S.; Qiu, Y. DJ-1 promotes survival of human colon cancer cells under hypoxia by modulating HIF-1α expression through the PI3K-AKT pathway. Cancer Manag. Res. 2018, 10, 4615–4629. [Google Scholar] [CrossRef]
- Parsanejad, M.; Zhang, Y.; Qu, D.; Irrcher, I.; Rousseaux, M.W.; Aleyasin, H.; Kamkar, F.; Callaghan, S.; Slack, R.S.; Mak, T.W. Regulation of the VHL/HIF-1 pathway by DJ-1. J. Neurosci. 2014, 34, 8043–8050. [Google Scholar] [CrossRef]
- Orenstein, S.J.; Kuo, S.-H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Häbig, K.; Gellhaar, S.; Heim, B.; Djuric, V.; Giesert, F.; Wurst, W.; Walter, C.; Hentrich, T.; Riess, O.; Bonin, M. LRRK2 guides the actin cytoskeleton at growth cones together with ARHGEF7 and Tropomyosin 4. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2013, 1832, 2352–2367. [Google Scholar] [CrossRef] [PubMed]
- Yakhine-Diop, S.M.; Niso-Santano, M.; Rodríguez-Arribas, M.; Gómez-Sánchez, R.; Martínez-Chacón, G.; Uribe-Carretero, E.; Navarro-García, J.A.; Ruiz-Hurtado, G.; Aiastui, A.; Cooper, J.M. Impaired mitophagy and protein acetylation levels in fibroblasts from Parkinson’s disease patients. Mol. Neurobiol. 2019, 56, 2466–2481. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.-H.; Joo, H.; Bae, J.; Hyeon, S.J.; Her, S.; Ko, E.; Choi, H.G.; Ryu, H.; Hur, E.-M.; Bu, Y. Brain injury induces HIF-1α-dependent transcriptional activation of LRRK2 that exacerbates brain damage. Cell Death Dis. 2018, 9, 1125. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.-M.; Wu, C.-J.; Lin, S.-L. Physiology and pathophysiology of renal erythropoietin-producing cells. J. Formos. Med. Assoc. 2018, 117, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Xiao, K.; Zhang, Z.; Ma, L. Altered expression of EPO might underlie hepatic hemangiomas in LRRK2 knockout mice. BioMed Res. Int. 2016, 2016, 7681259. [Google Scholar] [CrossRef] [PubMed]
- Cang, C.; Aranda, K.; Seo, Y.-j.; Gasnier, B.; Ren, D. TMEM175 is an organelle K+ channel regulating lysosomal function. Cell 2015, 162, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Jian, B.; Liu, Z. Transmembrane protein 175, a lysosomal ion channel related to Parkinson’s disease. Biomolecules 2023, 13, 802. [Google Scholar] [CrossRef] [PubMed]
- Jinn, S.; Drolet, R.E.; Cramer, P.E.; Wong, A.H.-K.; Toolan, D.M.; Gretzula, C.A.; Voleti, B.; Vassileva, G.; Disa, J.; Tadin-Strapps, M. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Wie, J.; Liu, Z.; Song, H.; Tropea, T.F.; Yang, L.; Wang, H.; Liang, Y.; Cang, C.; Aranda, K.; Lohmann, J. A growth-factor-activated lysosomal K+ channel regulates Parkinson’s pathology. Nature 2021, 591, 431–437. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Wang, X.; Perry, G.; Smith, M.A.; Moreira, P.I.; Zhu, X. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, S401–S412. [Google Scholar] [CrossRef] [PubMed]
- Vives-Bauza, C.; Przedborski, S. Mitophagy: The latest problem for Parkinson’s disease. Trends Mol. Med. 2011, 17, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lu, H.; Xie, X.; Shen, H.; Li, X.; Zhang, Y.; Wu, J.; Ni, J.; Li, H.; Chen, G. TMEM175 mediates Lysosomal function and participates in neuronal injury induced by cerebral ischemia-reperfusion. Mol. Brain 2020, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Moreno, T.; García-Morales, V.; Suleiman-Martos, S.; Rivas-Domínguez, A.; Mohamed-Mohamed, H.; Ramos-Rodríguez, J.J.; Melguizo-Rodríguez, L.; González-Acedo, A. Current treatments and new, tentative therapies for Parkinson’s disease. Pharmaceutics 2023, 15, 770. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, L.; Chen, J.; Li, Q.; Huo, L.; Wang, Y.; Wang, H.; Du, J. Pharmacological modulation of Nrf2/HO-1 signaling pathway as a therapeutic target of Parkinson’s disease. Front. Pharmacol. 2021, 12, 757161. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-X.; Yang, R.; Zhang, F. Role of Nrf2 in Parkinson’s disease: Toward new perspectives. Front. Pharmacol. 2022, 13, 919233. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Ji, X.; Liu, J. Hypoxia and alpha-Synuclein: Inextricable link underlying the pathologic progression of Parkinson’s disease. Front. Aging Neurosci. 2022, 14, 919343. [Google Scholar] [CrossRef]
- You, H.; Wang, D.; Wei, L.; Chen, J.; Li, H.; Liu, Y. Deferoxamine Inhibits Acute Lymphoblastic Leukemia Progression through Repression of ROS/HIF-1α, Wnt/β-Catenin, and p38MAPK/ERK Pathways. J. Oncol. 2022, 2022, 8281267. [Google Scholar] [CrossRef]
- Guo, C.; Hao, L.-J.; Yang, Z.-H.; Chai, R.; Zhang, S.; Gu, Y.; Gao, H.-L.; Zhong, M.-L.; Wang, T.; Li, J.-Y. Deferoxamine-mediated up-regulation of HIF-1α prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice. Exp. Neurol. 2016, 280, 13–23. [Google Scholar] [CrossRef]
- Sachan, N.; Srikrishna, S.; Patel, D.K.; Singh, M.P. Deferoxamine Ameliorates Cypermethrin-Induced Iron Accumulation and Associated Alterations. Mol. Neurobiol. 2024, 61, 4178–4187. [Google Scholar] [CrossRef]
- Pieńkowska, N.; Bartosz, G.; Sadowska-Bartosz, I. Effect of 6-hydroxydopamine increase the glutathione level in SH-SY5Y human neuroblastoma cells. Acta Biochim. Pol. 2023, 70, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-H.; Song, C.-C.; Pantopoulos, K.; Wei, X.-L.; Zheng, H.; Luo, Z. Mitochondrial oxidative stress mediated Fe-induced ferroptosis via the NRF2-ARE pathway. Free. Radic. Biol. Med. 2022, 180, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Chen, Y.; Chen, D.; Reddy, M.B. The Protection of EGCG Against 6-OHDA-Induced Oxidative Damage by Regulating PPARγ and Nrf2/HO-1 Signaling. Nutr. Metab. Insights 2024, 17, 11786388241253436. [Google Scholar] [CrossRef] [PubMed]
- Yong, S.J.; Veerakumarasivam, A.; Lim, W.L.; Chew, J. Neuroprotective effects of lactoferrin in Alzheimer’s and Parkinson’s diseases: A narrative review. ACS Chem. Neurosci. 2023, 14, 1342–1355. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.P.; Pandey, A.; Vishwakarma, S.; Modi, G. A review on iron chelators as potential therapeutic agents for the treatment of Alzheimer’s and Parkinson’s diseases. Mol. Divers. 2019, 23, 509–526. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Du, J.; Wang, J. Albendazole inhibits HIF-1α-dependent glycolysis and VEGF expression in non-small cell lung cancer cells. Mol. Cell. Biochem. 2017, 428, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Kandil, E.A.; Sayed, R.H.; Ahmed, L.A.; Abd El Fattah, M.A.; El-Sayeh, B.M. Hypoxia-inducible factor 1 alpha and nuclear-related receptor 1 as targets for neuroprotection by albendazole in a rat rotenone model of Parkinson’s disease. Clin. Exp. Pharmacol. Physiol. 2019, 46, 1141–1150. [Google Scholar] [CrossRef]
- Ferlazzo, N.; Currò, M.; Giunta, M.L.; Longo, D.; Rizzo, V.; Caccamo, D.; Ientile, R. Up-regulation of HIF-1α is associated with neuroprotective effects of agmatine against rotenone-induced toxicity in differentiated SH-SY5Y cells. Amino Acids 2020, 52, 171–179. [Google Scholar] [CrossRef]
- Azar, Y.O.; Badawi, G.A.; Zaki, H.F.; Ibrahim, S.M. Agmatine-mediated inhibition of NMDA receptor expression and amelioration of dyskinesia via activation of Nrf2 and suppression of HMGB1/RAGE/TLR4/MYD88/NF-κB signaling cascade in rotenone lesioned rats. Life Sci. 2022, 311, 121049. [Google Scholar] [CrossRef]
- El-Sayed, E.; Ahmed, A.; Morsy, E.E.; Nofal, S. Neuroprotective effect of agmatine (decarboxylated l-arginine) against oxidative stress and neuroinflammation in rotenone model of Parkinson’s disease. Hum. Exp. Toxicol. 2019, 38, 173–184. [Google Scholar] [CrossRef]
- Janssen Daalen, J.M.; Meinders, M.J.; Mathur, S.; van Hees, H.W.; Ainslie, P.N.; Thijssen, D.H.; Bloem, B.R. Randomized controlled trial of intermittent hypoxia in Parkinson’s disease: Study rationale and protocol. BMC Neurol. 2024, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Krause, P.; Berking, S.; Astalosch, M.; Grünheid, R.; Kühn, A.A. Motor and non-motor improvements following short-term multidisciplinary day-clinic care in Parkinson’ s disease. J. Neural Transm. 2022, 129, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Papastergiou, D.; Kokaridas, D.; Bonotis, K.; Digelidis, N.; Patsiaouras, A. Intervention effect of supportive group therapy and physical exercise on the quality of life of cancer patients. Cent. Eur. J. Sport Sci. Med. 2019, 25, 5–13. [Google Scholar] [CrossRef]
- Aimée, N.R.; Damé-Teixeira, N.; Alves, L.S.; Borges, G.Á.; Foster Page, L.; Mestrinho, H.D.; Carvalho, J.C. Responsiveness of oral health-related quality of life questionnaires to dental caries interventions: Systematic review and meta-analysis. Caries Res. 2019, 53, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Ford, B.Q.; Lam, P.; John, O.P.; Mauss, I.B. The psychological health benefits of accepting negative emotions and thoughts: Laboratory, diary, and longitudinal evidence. J. Personal. Soc. Psychol. 2018, 115, 1075. [Google Scholar] [CrossRef] [PubMed]
- Bialkowska, M.; Zajac, D.; Mazzatenta, A.; Di Giulio, C.; Pokorski, M. Inhibition of peripheral dopamine metabolism and the ventilatory response to hypoxia in the rat. Neurotransm. Interact. Cogn. Funct. 2015, 9–17. [Google Scholar]
- Kaczyńska, K.; Orłowska, M.E.; Andrzejewski, K. Respiratory abnormalities in Parkinson’s disease: What do we know from studies in humans and animal models? Int. J. Mol. Sci. 2022, 23, 3499. [Google Scholar] [CrossRef]
- Marino, G.; Campanelli, F.; Natale, G.; De Carluccio, M.; Servillo, F.; Ferrari, E.; Gardoni, F.; Caristo, M.E.; Picconi, B.; Cardinale, A. Intensive exercise ameliorates motor and cognitive symptoms in experimental Parkinson’s disease restoring striatal synaptic plasticity. Sci. Adv. 2023, 9, eadh1403. [Google Scholar] [CrossRef] [PubMed]
- Morris, H.R.; Spillantini, M.G.; Sue, C.M.; Williams-Gray, C.H. The pathogenesis of Parkinson’s disease. Lancet 2024, 403, 293–304. [Google Scholar] [CrossRef]
- Dong-Chen, X.; Yong, C.; Yang, X.; Chen-Yu, S.; Li-Hua, P. Signaling pathways in Parkinson’s disease: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 73. [Google Scholar] [CrossRef]
- Choi, H.N.; Kim, S.-H.; Lee, B.; Yoo, H.S.; Lee, J.H.; Jo, M.G.; Seong, H.M.; Song, C.; Kim, S.J.; Park, S.W. A2-astrocyte activation by short term hypoxia rescue α-synuclein preformed fibril induced neuronal cell death. Preprint 2024. [Google Scholar] [CrossRef]
- Ogunshola, O.; Antoniou, X. Contribution of hypoxia to Alzheimer’s disease: Is HIF-1α a mediator of neurodegeneration? Cell. Mol. Life Sci. 2009, 66, 3555–3563. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhang, X.; Duan, Y.; He, Y.; Zhao, J.; Wang, Z.; Wang, J.; Li, Q.; Fan, G.; Liu, Z. Hypoxia inducible factor-1α regulates microglial innate immune memory and the pathology of Parkinson’s disease. J. Neuroinflammation 2024, 21, 80. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, A.; Ohuchi, K.; Takizawa, S.; Murakami, T.; Kurita, H.; Hozumi, I.; Wen, X.; Kitamura, Y.; Wu, Z.; Maekawa, Y. The neuroprotective effects of FG-4592, a hypoxia-inducible factor-prolyl hydroxylase inhibitor, against oxidative stress induced by alpha-synuclein in N2a cells. Sci. Rep. 2023, 13, 15629. [Google Scholar] [CrossRef] [PubMed]
- Fouché, B.; Turner, S.; Gorham, R.; Stephenson, E.J.; Gutbier, S.; Elson, J.L.; García-Beltrán, O.; Van Der Westhuizen, F.H.; Pienaar, I.S. A novel mitochondria-targeting iron chelator neuroprotects multimodally via HIF-1 modulation against a mitochondrial toxin in a dopaminergic cell model of Parkinson’s disease. Mol. Neurobiol. 2023, 60, 749–767. [Google Scholar] [CrossRef] [PubMed]
- Agani, F.H.; Pichiule, P.; Chavez, J.C.; LaManna, J.C. The role of mitochondria in the regulation of hypoxia-inducible factor 1 expression during hypoxia. J. Biol. Chem. 2000, 275, 35863–35867. [Google Scholar] [CrossRef] [PubMed]
- Janssen Daalen, J.M.; Meinders, M.J.; Giardina, F.; Roes, K.C.; Stunnenberg, B.C.; Mathur, S.; Ainslie, P.N.; Thijssen, D.H.; Bloem, B.R. Multiple N-of-1 trials to investigate hypoxia therapy in Parkinson’s disease: Study rationale and protocol. BMC Neurol. 2022, 22, 262. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Zhang, J.; Tang, T.; Liu, Z. Hypoxia Pathways in Parkinson’s Disease: From Pathogenesis to Therapeutic Targets. Int. J. Mol. Sci. 2024, 25, 10484. https://doi.org/10.3390/ijms251910484
Gao Y, Zhang J, Tang T, Liu Z. Hypoxia Pathways in Parkinson’s Disease: From Pathogenesis to Therapeutic Targets. International Journal of Molecular Sciences. 2024; 25(19):10484. https://doi.org/10.3390/ijms251910484
Chicago/Turabian StyleGao, Yuanyuan, Jiarui Zhang, Tuoxian Tang, and Zhenjiang Liu. 2024. "Hypoxia Pathways in Parkinson’s Disease: From Pathogenesis to Therapeutic Targets" International Journal of Molecular Sciences 25, no. 19: 10484. https://doi.org/10.3390/ijms251910484