
Monogenic Defects of Beta Cell Function: From Clinical Suspicion to Genetic Diagnosis and Management of Rare Types of Diabetes

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Methods
3. Discussion
3.1. Monogenic Neonatal Diabetes Mellitus
3.1.1. Case Scenario 1
3.1.2. Neonatal Diabetes Mellitus
3.2. Maturity-Onset Diabetes of the Young (MODY)
3.2.1. Case Scenario 2
3.2.2. MODY Types
3.2.3. HNF1A MODY
3.2.4. GCK MODY
3.2.5. HNF4A MODY
3.2.6. HNF1B MODY
3.2.7. Rarer Types of MODY
3.2.8. Distinguishing MODY from Other Types of Diabetes
3.3. Syndromic Monogenic Diabetes Mellitus
3.3.1. Case Scenario 3
3.3.2. Diabetes Mellitus as Part of a Syndrome
3.4. Limitations
4. Conclusive Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Fajans, S.S.; Bell, G.I. MODY: History, genetics, pathophysiology, and clinical decision making. Diabetes Care 2011, 34, 1878–1884. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Classification of Diabetes Mellitus. 2019. Available online: https://iris.who.int/handle/10665/325182 (accessed on 15 August 2024).
- Bonnefond, A.; Unnikrishnan, R.; Doria, A.; Vaxillaire, M.; Kulkarni, R.N.; Mohan, V.; Trischitta, V.; Froguel, P. Monogenic diabetes. Nat. Rev. Dis. Primers. 2023, 9, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.H.; Srinivasan, S.; Chen, L.; Todd, J.; Mercader, J.M.; Jensen, E.T.; Divers, J.; Mottl, A.K.; Pihoker, C.; Gandica, R.G.; et al. Genetic architecture and biology of youth-onset type 2 diabetes. Nat. Metab. 2024, 6, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Riddle, M.C.; Philipson, L.H.; Rich, S.S.; Carlsson, A.; Franks, P.W.; Greeley, S.A.W.; Nolan, J.J.; Pearson, E.R.; Zeitler, P.S.; Hattersley, A.T. Monogenic Diabetes: From Genetic Insights to Population-Based Precision in Care. Reflections From a Diabetes Care Editors’ Expert Forum. Diabetes Care 2020, 43, 3117–3128. [Google Scholar] [CrossRef]
- Melvin, A.; O’Rahilly, S.; Savage, D.B. Genetic syndromes of severe insulin resistance. Curr. Opin. Genet. Dev. 2018, 50, 60–67. [Google Scholar] [CrossRef]
- Bagias, C.; Xiarchou, A.; Bargiota, A.; Tigas, S. Familial Partial Lipodystrophy (FPLD): Recent Insights. Diabetes Metab. Syndr. Obes. 2020, 13, 1531–1544. [Google Scholar] [CrossRef]
- Grulich-Henn, J.; Wagner, V.; Thon, A.; Schober, E.; Marg, W.; Kapellen, T.M.; Haberland, H.; Raile, K.; Ellard, S.; Flanagan, S.E.; et al. Entities and frequency of neonatal diabetes: Data from the diabetes documentation and quality management system (DPV). Diabet. Med. 2010, 27, 709–712. [Google Scholar] [CrossRef]
- De Franco, E.; Flanagan, S.E.; Houghton, J.A.L.; Allen, H.L.; MacKay, D.J.G.; Temple, I.K.; Ellard, S.; Hattersley, A.T. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: An international cohort study. Lancet 2015, 386, 957–963. [Google Scholar] [CrossRef]
- Greeley, S.A.W.; Polak, M.; Njølstad, P.R.; Barbetti, F.; Williams, R.; Castano, L.; Raile, K.; Chi, D.V.; Habeb, A.; Hattersley, A.T.; et al. ISPAD Clinical Practice Consensus Guidelines 2022: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr. Diabetes 2022, 23, 1188–1211. [Google Scholar] [CrossRef]
- Docherty, L.E.; Kabwama, S.; Lehmann, A.; Hawke, E.; Harrison, L.; Flanagan, S.E.; Ellard, S.; Hattersley, A.T.; Shield, J.P.H.; Ennis, S.; et al. Clinical presentation of 6q24 transient neonatal diabetes mellitus (6q24 TNDM) and genotype-phenotype correlation in an international cohort of patients. Diabetologia 2013, 56, 758–762. [Google Scholar] [CrossRef]
- Garin, I.; Edghill, E.L.; Akerman, I.; Rubio-Cabezas, O.; Rica, I.; Locke, J.M.; Maestro, M.A.; Alshaikh, A.; Bundak, R.; del Castillo, G.; et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3105–3110. [Google Scholar] [CrossRef] [PubMed]
- Lemelman, M.B.; Letourneau, L.; Greeley, S.A.W. Neonatal Diabetes Mellitus: An Update on Diagnosis and Management. Clin. Perinatol. 2018, 45, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.E.; MacKay, D.J.G.; Greeley, S.A.W.; McDonald, T.J.; Mericq, V.; Hassing, J.; Richmond, E.J.; Martin, W.R.; Acerini, C.; Kaulfers, A.M.; et al. Hypoglycaemia following diabetes remission in patients with 6q24 methylation defects: Expanding the clinical phenotype. Diabetologia 2013, 56, 218–221. [Google Scholar] [CrossRef]
- Novak, A.; Bowman, P.; Kraljevic, I.; Tripolski, M.; Houghton, J.A.L.; De Franco, E.; Shepherd, M.H.; Skrabic, V.; Patel, K.A. Transient Neonatal Diabetes: An Etiologic Clue for the Adult Diabetologist. Can. J. Diabetes 2020, 44, 128–130. [Google Scholar] [CrossRef]
- Edghill, E.L.; Flanagan, S.E.; Ellard, S. Permanent neonatal diabetes due to activating mutations in ABCC8 and KCNJ11. Rev. Endocr. Metab. Disord. 2010, 11, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Quan, H.; Chen, K.; Chen, D.; Lin, D.; Fang, T. ABCC8-Related Maturity-Onset Diabetes of the Young (MODY12): A Report of a Chinese Family. Front. Endocrinol. 2020, 11, 645–652. [Google Scholar] [CrossRef]
- De Franco, E.; Saint-Martin, C.; Brusgaard, K.; Knight Johnson, A.E.; Aguilar-Bryan, L.; Bowman, P.; Arnoux, J.B.; Larsen, A.R.; Sanyoura, M.; Greeley, S.A.W.; et al. Update of variants identified in the pancreatic β-cell KATP channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum. Mutat. 2020, 41, 884–905. [Google Scholar] [CrossRef]
- Edghill, E.L.; Flanagan, S.E.; Patch, A.M.; Boustred, C.; Parrish, A.; Shields, B.; Shepherd, M.H.; Hussain, K.; Kapoor, R.R.; Malecki, M.; et al. Insulin mutation screening in 1,044 patients with diabetes: Mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes 2008, 57, 1034–1042. [Google Scholar] [CrossRef]
- Støy, J.; Edghill, E.L.; Flanagan, S.E.; Ye, H.; Paz, V.P.; Pluzhnikov, A.; Below, J.E.; Hayes, M.J.; Cox, N.J.; Lipkind, G.M.; et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc. Natl. Acad. Sci. USA 2007, 104, 15040–15044. [Google Scholar] [CrossRef]
- Colombo, C.; Porzio, O.; Liu, M.; Massa, O.; Vasta, M.; Salardi, S.; Beccaria, L.; Monciotti, C.; Toni, S.; Pedersen, O.; et al. Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus. J. Clin. Invest. 2008, 118, 2148–2156. [Google Scholar] [CrossRef]
- Bennett, K.; James, C.; Mutair, A.; Al-Shaikh, H.; Sinani, A.; Hussain, K. Four novel cases of permanent neonatal diabetes mellitus caused by homozygous mutations in the glucokinase gene. Pediatr. Diabetes. 2011, 12, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Beltrand, J.; Busiah, K.; Vaivre-Douret, L.; Fauret, A.L.; Berdugo, M.; Cavé, H.; Polak, M. Neonatal Diabetes Mellitus. Front. Pediatr. 2020, 8, 540718. [Google Scholar] [CrossRef] [PubMed]
- Letourneau, L.R.; Carmody, D.; Wroblewski, K.; Denson, A.M.; Sanyoura, M.; Naylor, R.N.; Philipson, L.H.; Greeley, S.A.W. Diabetes Presentation in Infancy: High Risk of Diabetic Ketoacidosis. Diabetes Care 2017, 40, e147–e148. [Google Scholar] [CrossRef] [PubMed]
- Pinney, S.E.; Oliver-Krasinski, J.; Ernst, L.; Hughes, N.; Patel, P.; Stoffers, D.A.; Russo, P.; De León, D.D. Neonatal diabetes and congenital malabsorptive diarrhea attributable to a novel mutation in the human neurogenin-3 gene coding sequence. J. Clin. Endocrinol. Metab. 2011, 96, 1960–1965. [Google Scholar] [CrossRef]
- McDonald, T.J.; Ellard, S. Maturity onset diabetes of the young: Identification and diagnosis. Ann. Clin. Biochem. 2013, 50, 403–415. [Google Scholar] [CrossRef]
- Rubio-Cabezas, O.; Ellard, S. Diabetes mellitus in neonates and infants: Genetic heterogeneity, clinical approach to diagnosis, and therapeutic options. Horm. Res. Paediatr. 2013, 80, 137–146. [Google Scholar] [CrossRef]
- Niculae, A.; Ștefan Bolba, C.; Grama, A.; Mariş, A.; Bodea, L.; Căinap, S.; Mititelu, A.; Fufezan, O.; Pop, T.L. Wolcott-Rallison Syndrome, a Rare Cause of Permanent Diabetes Mellitus in Infants—Case Report. Pediatr. Rep. 2023, 15, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Aldrian, D.; Bochdansky, C.; Kavallar, A.M.; Mayerhofer, C.; Deeb, A.; Habeb, A.; Rabasa, A.R.; Khadilkar, A.; Uçar, A.; Knoppke, B.; et al. Natural history of Wolcott-Rallison syndrome: A systematic review and follow-up study. Liver Int. 2024, 44, 811–822. [Google Scholar] [CrossRef]
- Bacchetta, R.; Roncarolo, M.G. IPEX syndrome from diagnosis to cure, learning along the way. J. Allergy Clin. Immunol. 2024, 153, 595–605. [Google Scholar] [CrossRef]
- Shimomura, K.; Hörster, F.; De Wet, H.; Flanagan, S.E.; Ellard, S.; Hattersley, A.T.; Wolf, N.I.; Ashcroft, F.; Ebinger, F. A novel mutation causing DEND syndrome: A treatable channelopathy of pancreas and brain. Neurology 2007, 69, 1342–1349. [Google Scholar] [CrossRef]
- Shah, R.P.; Spruyt, K.; Kragie, B.C.; Greeley, S.A.W.; Msall, M.E. Visuomotor performance in KCNJ11-related neonatal diabetes is impaired in children with DEND-associated mutations and may be improved by early treatment with sulfonylureas. Diabetes Care 2012, 35, 2086–2088. [Google Scholar] [CrossRef] [PubMed]
- Beshlawi, I.; Zadjali, S.A.L.; Bashir, W.; Elshinawy, M.; Alrawas, A.; Wali, Y. Thiamine responsive megaloblastic anemia: The puzzling phenotype. Pediatr. Blood Cancer 2014, 61, 528–531. [Google Scholar] [CrossRef]
- Mitchell, J.; Punthakee, Z.; Lo, B.; Bernard, C.; Chong, K.; Newman, C.; Cartier, L.; Desilets, V.; Cutz, E.; Hansen, I.L.; et al. Neonatal diabetes, with hypoplastic pancreas, intestinal atresia and gall bladder hypoplasia: Search for the aetiology of a new autosomal recessive syndrome. Diabetologia 2004, 47, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Imaki, S.; Iizuka, K.; Horikawa, Y.; Yasuda, M.; Kubota, S.; Kato, T.; Liu, Y.; Takao, K.; Mizuno, M.; Hirota, T.; et al. A novel RFX6 heterozygous mutation (p.R652X) in maturity-onset diabetes mellitus: A case report. J. Diabetes Investig. 2021, 12, 1914–1918. [Google Scholar] [CrossRef] [PubMed]
- Babiker, T.; Vedovato, N.; Patel, K.; Thomas, N.; Finn, R.; Männikkö, R.; Chakera, A.J.; Flanagan, S.E.; Shepherd, M.H.; Ellard, S.; et al. Successful transfer to sulfonylureas in KCNJ11 neonatal diabetes is determined by the mutation and duration of diabetes. Diabetologia 2016, 59, 1162–1166. [Google Scholar] [CrossRef]
- Pearson, E.R.; Flechtner, I.; Njølstad, P.R.; Malecki, M.T.; Flanagan, S.E.; Larkin, B.; Ashcroft, F.M.; Klimes, I.; Codner, E.; Iotova, V.; et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N. Engl. J. Med. 2006, 355, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Shields, B.M.; Hicks, S.; Shepherd, M.H.; Colclough, K.; Hattersley, A.T.; Ellard, S. Maturity-onset diabetes of the young (MODY): How many cases are we missing? Diabetologia 2010, 53, 2504–2508. [Google Scholar] [CrossRef]
- Pihoker, C.; Gilliam, L.K.; Ellard, S.; Dabelea, D.; Davis, C.; Dolan, L.M.; Greenbaum, C.J.; Imperatore, G.; Lawrence, J.M.; Marcovia, S.M.; et al. Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: Results from the SEARCH for Diabetes in Youth. J. Clin. Endocrinol. Metab. 2013, 98, 4055–4062. [Google Scholar] [CrossRef] [PubMed]
- Irgens, H.U.; Molnes, J.; Johansson, B.B.; Ringdal, M.; Skrivarhaug, T.; Undlien, D.E.; Søvik, O.; Joner, G.; Molven, A.; Njølstad, P.R. Prevalence of monogenic diabetes in the population-based Norwegian Childhood Diabetes Registry. Diabetologia 2013, 56, 1512–1519. [Google Scholar] [CrossRef]
- Fendler, W.; Borowiec, M.; Baranowska-Jazwiecka, A.; Szadkowska, A.; Skala-Zamorowska, E.; Deja, G.; Jarosz-Chobot, P.; Techmanska, I.; Bautembach-Minkowska, J.; Mysliwiec, M.; et al. Prevalence of monogenic diabetes amongst Polish children after a nationwide genetic screening campaign. Diabetologia 2012, 55, 2631–2635. [Google Scholar] [CrossRef]
- Aloi, C.; Salina, A.; Minuto, N.; Tallone, R.; Lugani, F.; Mascagni, A.; Mazza, O.; Cassanello, M.; Maghnie, M.; d’Annunzio, G. Glucokinase mutations in pediatric patients with impaired fasting glucose. Acta Diabetol. 2017, 54, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.; Rego, T.; Armas, J.B. Insights into the Genetics and Signaling Pathways in Maturity-Onset Diabetes of the Young. Int. J. Mol. Sci. 2022, 23, 12910. [Google Scholar] [CrossRef] [PubMed]
- Miyachi, Y.; Miyazawa, T.; Ogawa, Y. HNF1A Mutations and Beta Cell Dysfunction in Diabetes. Int. J. Mol. Sci. 2022, 23, 3222–3238. [Google Scholar] [CrossRef]
- Ludwig-Słomczyńska, A.H.; Seweryn, M.T.; Radkowski, P.; Kapusta, P.; Machlowska, J.; Pruhova, S.; Gasperikova, D.; Bellanne-Chantelot, C.; Hattersley, A.; Kandasamy, B.; et al. Variants influencing age at diagnosis of HNF1A-MODY. Mol. Med. 2022, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Stride, A.; Shepherd, M.; Frayling, T.M.; Bulman, M.P.; Ellard, S.; Hattersley, A.T. Intrauterine hyperglycemia is associated with an earlier diagnosis of diabetes in HNF-1alpha gene mutation carriers. Diabetes Care 2002, 25, 2287–2291. [Google Scholar] [CrossRef] [PubMed]
- Nowak, N.; Hohendorff, J.; Solecka, I.; Szopa, M.; Skupien, J.; Kiec-Wilk, B.; Mlynarski, W.; Malecki, M.T. Circulating ghrelin level is higher in HNF1A–MODY and GCK–MODY than in polygenic forms of diabetes mellitus. Endocrine 2015, 50, 643–649. [Google Scholar] [CrossRef]
- Mcdonald, T.J.; Shields, B.M.; Lawry, J.; Owen, K.R.; Gloyn, A.L.; Ellard, S.; Hattersley, A.T. High-sensitivity CRP discriminates HNF1A-MODY from other subtypes of diabetes. Diabetes Care 2011, 34, 1860–1862. [Google Scholar] [CrossRef]
- Bacon, S.; Kyithar, M.P.; Rizvi, S.R.; Donnelly, E.; McCarthy, A.; Burke, M.; Colclough, K.; Ellard, S.; Byrne, M.M. Successful maintenance on sulphonylurea therapy and low diabetes complication rates in a HNF1A-MODY cohort. Diabet. Med. 2016, 33, 976–984. [Google Scholar] [CrossRef]
- Delvecchio, M.; Pastore, C.; Giordano, P. Treatment Options for MODY Patients: A Systematic Review of Literature. Diabetes Ther. 2020, 11, 1667–1685. [Google Scholar] [CrossRef]
- Becker, M.; Galler, A.; Raile, K. Meglitinide analogues in adolescent patients with HNF1A-MODY (MODY 3). Pediatrics 2014, 133, e775–e779. [Google Scholar] [CrossRef]
- Fantasia, K.L.; Steenkamp, D.W. Optimal Glycemic Control in a Patient With HNF1A MODY With GLP-1 RA Monotherapy: Implications for Future Therapy. J. Endocr. Soc. 2019, 3, 2286–2289. [Google Scholar] [CrossRef] [PubMed]
- Hohendorff, J.; Szopa, M.; Skupien, J.; Kapusta, M.; Zapala, B.; Platek, T.; Mrozinska, S.; Parpan, T.; Glodzik, W.; Ludwig-Galezowska, A.; et al. A single dose of dapagliflozin, an SGLT-2 inhibitor, induces higher glycosuria in GCK- and HNF1A-MODY than in type 2 diabetes mellitus. Endocrine 2017, 57, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Osbak, K.K.; Colclough, K.; Saint-Martin, C.; Beer, N.L.; Bellanné-Chantelot, C.; Ellard, S.; Gloyn, A.L. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum. Mutat. 2009, 30, 1512–1526. [Google Scholar] [CrossRef]
- Njølstad, P.R.; Søvik, O.; Cuesta-Muñoz, A.; Bjørkhaug, L.; Massa, O.; Barbetti, F.; Undlien, D.E.; Shiota, C.; Magnuson, M.A.; Molven, A.; et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. N. Engl. J. Med. 2001, 344, 1588–1592. [Google Scholar] [CrossRef]
- Hughes, A.E.; De Franco, E.; Globa, E.; Zelinska, N.; Hilgard, D.; Sifianou, P.; Hattersley, A.T.; Flanagan, S.E. Identification of GCK-MODY in cases of neonatal hyperglycemia: A case series and review of clinical features. Pediatr. Diabetes 2021, 22, 876–881. [Google Scholar] [CrossRef]
- Cuesta-Mũnoz, A.L.; Tuomi, T.; Cobo-Vuilleumier, N.; Koskela, H.; Odili, S.; Stride, A.; Buettger, C.; Otonkoski, T.; Froguel, P.; Grimsby, J.; et al. Clinical heterogeneity in monogenic diabetes caused by mutations in the glucokinase gene (GCK-MODY). Diabetes Care 2010, 33, 290–292. [Google Scholar] [CrossRef]
- Stride, A.; Vaxillaire, M.; Tuomi, T.; Barbetti, F.; Njølstad, P.R.; Hansen, T.; Costa, A.; Conget, I.; Pedersen, O.; Søvik, O.; et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia 2002, 45, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.M.; Wensley, K.J.; Ellard, S.; Murphy, R.; Shepherd, M.; Colclough, K.; Hattersley, A.T.; Shields, B.M. Use of HbA1c in the identification of patients with hyperglycaemia caused by a glucokinase mutation: Observational case control studies. PLoS ONE 2013, 8, e65326. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.M.; Shields, B.M.; Wensley, K.J.; Colclough, K.; Ellard, S.; Hattersley, A.T. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA 2014, 311, 279–286. [Google Scholar] [CrossRef]
- Carmody, D.; Naylor, R.N.; Bell, C.D.; Berry, S.; Montgomery, J.T.; Tadie, E.C.; Hwang, J.L.; Greeley, S.A.W.; Philipson, L.H. GCK-MODY in the US National Monogenic Diabetes Registry: Frequently misdiagnosed and unnecessarily treated. Acta Diabetol. 2016, 53, 703–708. [Google Scholar] [CrossRef]
- Dickens, L.T.; Naylor, R.N. Clinical Management of Women with Monogenic Diabetes During Pregnancy. Curr. Diab Rep. 2018, 18, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.E.; Houghton, J.A.L.; Bunce, B.; Chakera, A.J.; Spyer, G.; Shepherd, M.H.; Flanagan, S.E.; Hattersley, A.T. Bringing precision medicine to the management of pregnancy in women with glucokinase-MODY: A study of diagnostic accuracy and feasibility of non-invasive prenatal testing. Diabetologia 2023, 66, 1997–2006. [Google Scholar] [CrossRef]
- Fajans, S.S.; Bell, G.I.; Polonsky, K.S. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N. Engl. J. Med. 2001, 345, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Ng, N.H.J.; Ghosh, S.; Bok, C.M.; Ching, C.; Low, B.S.J.; Chen, J.T.; Lim, E.; Miserendino, M.C.; Tan, Y.S.; Hoon, S.; et al. HNF4A and HNF1A exhibit tissue specific target gene regulation in pancreatic beta cells and hepatocytes. Nat. Commun. 2024, 15, 4288–4309. [Google Scholar] [CrossRef]
- Harries, L.W.; Locke, J.M.; Shields, B.; Hanley, N.A.; Hanley, K.P.; Steele, A.; Njølstad, P.R.; Ellard, S.; Hattersley, A.T. The diabetic phenotype in HNF4A mutation carriers is moderated by the expression of HNF4A isoforms from the P1 promoter during fetal development. Diabetes 2008, 57, 1745–1752. [Google Scholar] [CrossRef]
- Pearson, E.R.; Boj, S.F.; Steele, A.M.; Barrett, T.; Stals, K.; Shield, J.P.; Ellard, S.; Ferrer, J.; Hattersley, A.T. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med. 2007, 4, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Dusatkova, P.; Pruhova, S.; Sumnik, Z.; Kolouskova, S.; Obermannova, B.; Cinek, O.; Lebl, J. HNF1A mutation presenting with fetal macrosomia and hypoglycemia in childhood prior to onset of overt diabetes. J. Pediatr. Endocrinol. Metab. 2011, 24, 187–189. [Google Scholar] [CrossRef]
- Broome, D.T.; Pantalone, K.M.; Kashyap, S.R.; Philipson, L.H. Approach to the Patient with MODY-Monogenic Diabetes. J. Clin. Endocrinol. Metab. 2021, 106, 237–250. [Google Scholar] [CrossRef]
- Clissold, R.L.; Hamilton, A.J.; Hattersley, A.T.; Ellard, S.; Bingham, C. HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat. Rev. Nephrol. 2015, 11, 102–112. [Google Scholar] [CrossRef]
- Amaral, S.; Palha, A.; Bogalho, P.; Silva-Nunes, J. Maturity-onset diabetes of the young secondary to HNF1B variants (HNF1B-MODY): A series of 10 patients from a single diabetes center. Diabetol. Metab. Syndr. 2023, 15, 21–29. [Google Scholar] [CrossRef]
- Ge, S.; Yang, M.; Cui, Y.; Wu, J.; Xu, L.; Dong, J.; Liao, L. The Clinical Characteristics and Gene Mutations of Maturity-Onset Diabetes of the Young Type 5 in Sixty-One Patients. Front. Endocrinol. 2022, 13, 911526. [Google Scholar] [CrossRef] [PubMed]
- Siomou, E.; Mitsioni, A.G.; Giapros, V.; Bouba, I.; Noutsopoulos, D.; Georgiou, I. Copy-number variation analysis in familial nonsyndromic congenital anomalies of the kidney and urinary tract: Evidence for the causative role of a transposable element-associated genomic rearrangement. Mol. Med. Rep. 2017, 15, 3631–3636. [Google Scholar] [CrossRef] [PubMed]
- Laffargue, F.; Bourthoumieu, S.; Llanas, B.; Baudouin, V.; Lahoche, A.; Morin, D.; Bessenay, L.; De Parscau, L.; Cloarec, S.; Delrue, M.-A.; et al. Towards a new point of view on the phenotype of patients with a 17q12 microdeletion syndrome. Arch. Dis. Child. 2015, 100, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Almutair, A.; Almulhem, B. Semaglutide as a potential therapeutic alternative for HNF1B-MODY: A case study. Front Endocrinol. 2024, 15, 1294264. [Google Scholar] [CrossRef] [PubMed]
- Faguer, S.; Decramer, S.; Chassaing, N.; Bellanné-Chantelot, C.; Calvas, P.; Beaufils, S.; Bessenay, L.; Lengelé, J.-P.; Dahan, K.; Ronco, P.; et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int. 2011, 80, 768–776. [Google Scholar] [CrossRef]
- Jonsson, J.; Carlsson, L.; Edlund, T.; Edlund, H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 1994, 371, 606–609. [Google Scholar] [CrossRef]
- Wang, X.; Sterr, M.; Ansarullah; Burtscher, I.; Böttcher, A.; Beckenbauer, J.; Siehler, J.; Meitinger, T.; Häring, H.-U.; Staiger, H.; et al. Point mutations in the PDX1 transactivation domain impair human β-cell development and function. Mol. Metab. 2019, 24, 80–97. [Google Scholar] [CrossRef]
- Bonnefond, A.; Philippe, J.; Durand, E.; Dechaume, A.; Huyvaert, M.; Montagne, L.; Marre, M.; Balkau, B.; Fajardy, I.; Vambergue, A.; et al. Whole-exome sequencing and high throughput genotyping identified KCNJ11 as the thirteenth MODY gene. PLoS ONE 2012, 7, e37423. [Google Scholar] [CrossRef]
- Ovsyannikova, A.K.; Rymar, O.D.; Shakhtshneider, E.V.; Klimontov, V.V.; Koroleva, E.A.; Myakina, N.E.; Voevoda, M.I. ABCC8-Related Maturity-Onset Diabetes of the Young (MODY12): Clinical Features and Treatment Perspective. Diabetes Ther. 2016, 7, 591–600. [Google Scholar] [CrossRef]
- Tatsi, E.B.; Kanaka-Gantenbein, C.; Scorilas, A.; Chrousos, G.P.; Sertedaki, A. Next generation sequencing targeted gene panel in Greek MODY patients increases diagnostic accuracy. Pediatr. Diabetes 2020, 21, 28–39. [Google Scholar] [CrossRef]
- Shields, B.M.; McDonald, T.J.; Ellard, S.; Campbell, M.J.; Hyde, C.; Hattersley, A.T. The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia 2012, 55, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Orban, T.; Sosenko, J.M.; Cuthbertson, D.; Krischer, J.P.; Skyler, J.S.; Jackson, R.; Yu, L.; Palmer, J.P.; Schatz, D.; Eisenbarth, G. Pancreatic Islet Autoantibodies as Predictors of Type 1 Diabetes in the Diabetes Prevention Trial–Type 1. Diabetes Care 2009, 32, 2269–2274. [Google Scholar] [CrossRef]
- Carlsson, A.; Shepherd, M.; Ellard, S.; Weedon, M.; Lernmark, A.; Forsander, G.; Colclough, K.; Brahimi, O.; Valtonen-Andre, C.; Ivarsson, S.A.; et al. Absence of Islet Autoantibodies and Modestly Raised Glucose Values at Diabetes Diagnosis Should Lead to Testing for MODY: Lessons From a 5-Year Pediatric Swedish National Cohort Study. Diabetes Care 2020, 43, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Hameed, S.; Ellard, S.; Woodhead, H.J.; Neville, K.A.; Walker, J.L.; Craig, M.E.; Armstrong, T.; Yu, L.; Eisenbarth, G.H.; Hattersley, A.T.; et al. Persistently autoantibody negative (PAN) type 1 diabetes mellitus in children. Pediatr. Diabetes 2011, 12, 142–149. [Google Scholar] [CrossRef]
- Sørgjerd, E.P.; Thorsby, P.M.; Torjesen, P.A.; Skorpen, F.; Kvaløy, K.; Grill, V. Presence of anti-GAD in a non-diabetic population of adults; time dynamics and clinical influence: Results from the HUNT study. BMJ Open Diabetes Res. Care 2015, 3, e000076. [Google Scholar] [CrossRef]
- Leighton, E.; Sainsbury, C.A.; Jones, G.C. A Review of C-Peptide Testing in Diabetes. Diabetes Ther. 2017, 8, 475–487. [Google Scholar] [CrossRef]
- Serbis, A.; Giapros, V.; Kotanidou, E.P.; Galli-Tsinopoulou, A.; Siomou, E. Diagnosis, treatment and prevention of type 2 diabetes mellitus in children and adolescents. World J. Diabetes 2021, 12, 344–365. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.C.; Lee, H.F.; Yue, C.T.; Chi, C.S. Clinical Characteristics of Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes. Life 2011, 11, 1111. [Google Scholar] [CrossRef]
- Romo, L.; Gold, N.B.; Walker, M.A. Endocrine features of primary mitochondrial diseases. Curr. Opin. Endocrinol. Diabetes Obes. 2024, 31, 34–42. [Google Scholar] [CrossRef]
- Serbis, A.; Rallis, D.; Giapros, V.; Galli-Tsinopoulou, A.; Siomou, E. Wolfram Syndrome 1: A Pediatrician’s and Pediatric Endocrinologist’s Perspective. Int. J. Mol. Sci. 2023, 24, 3690–3704. [Google Scholar] [CrossRef] [PubMed]
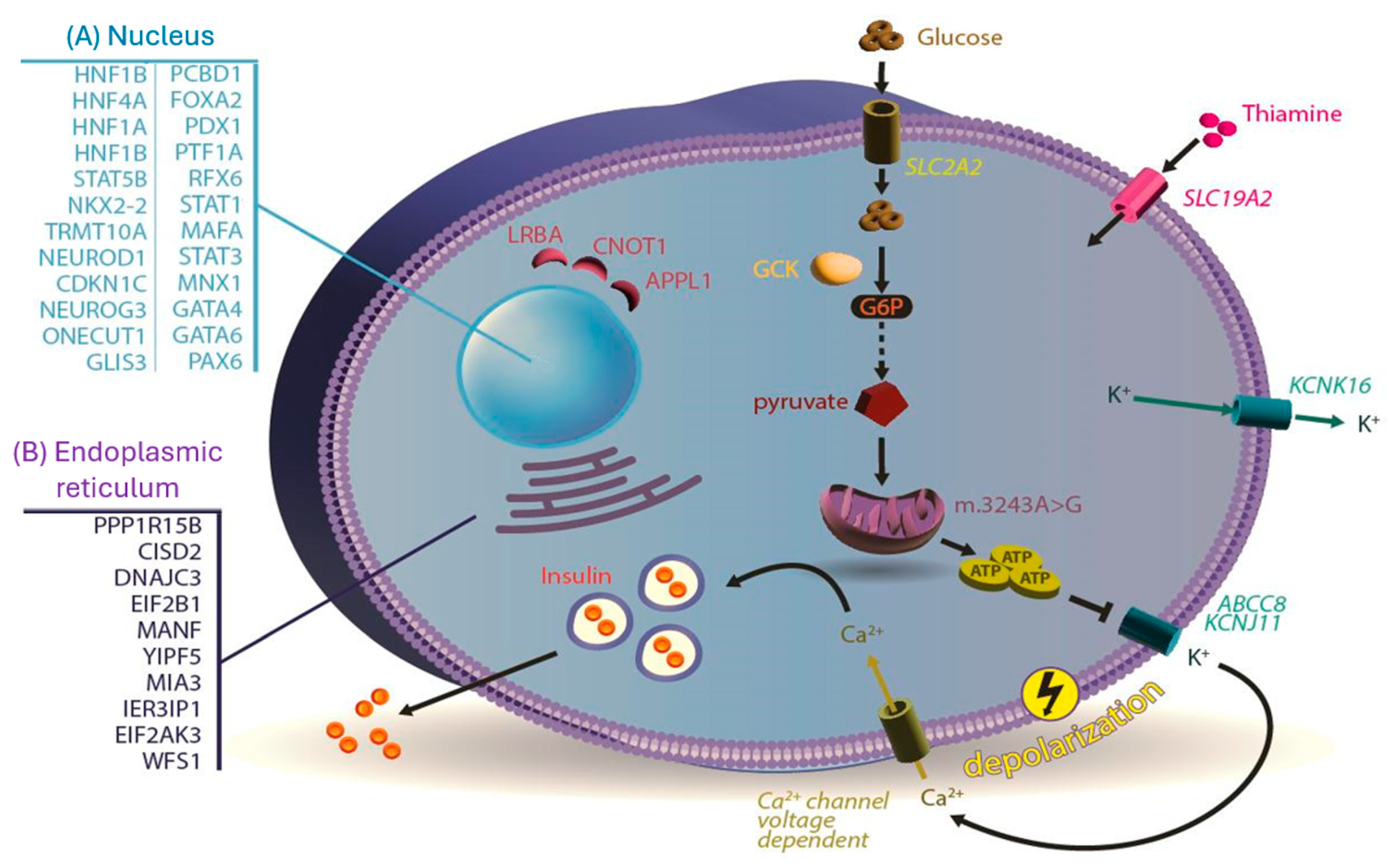
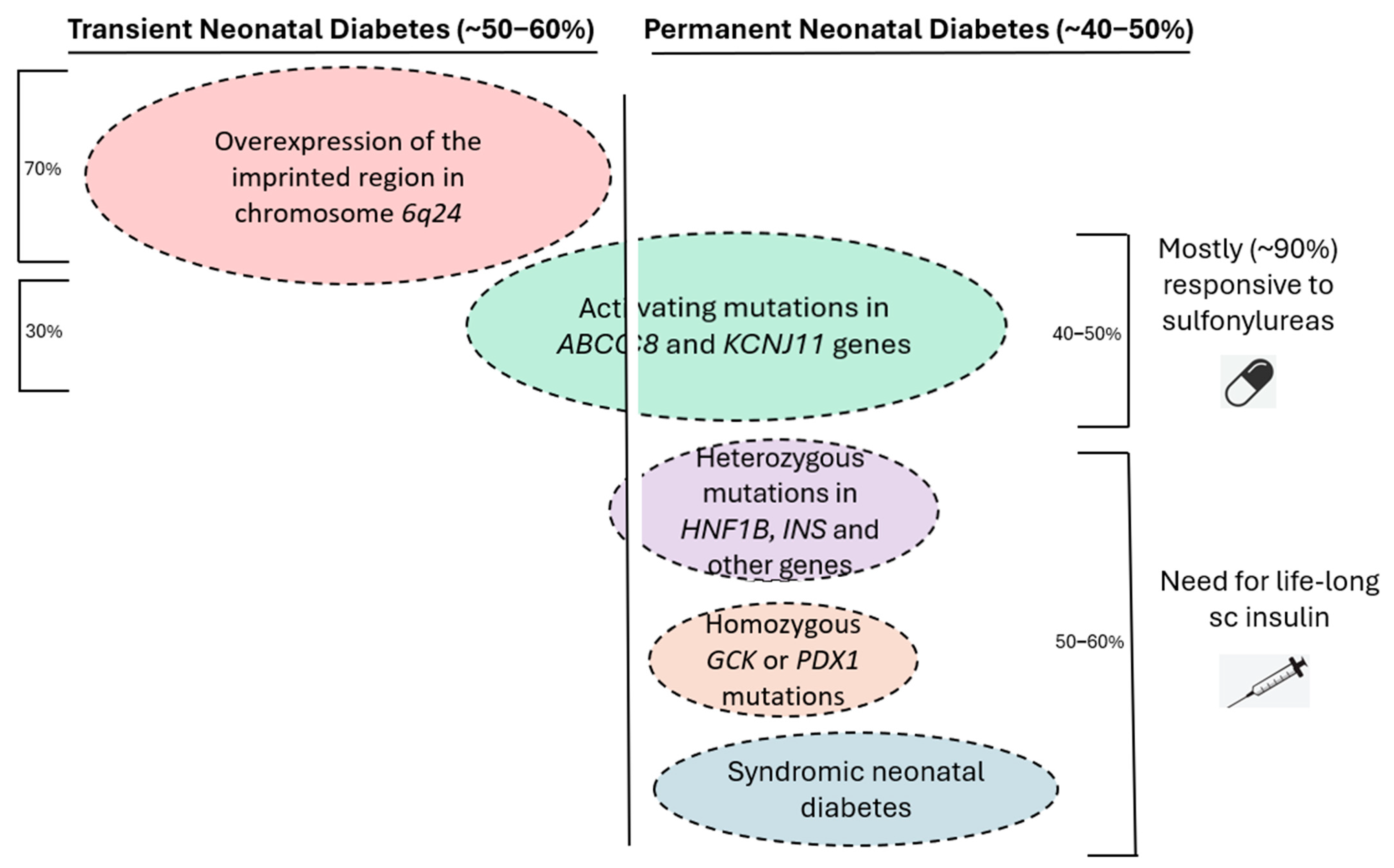
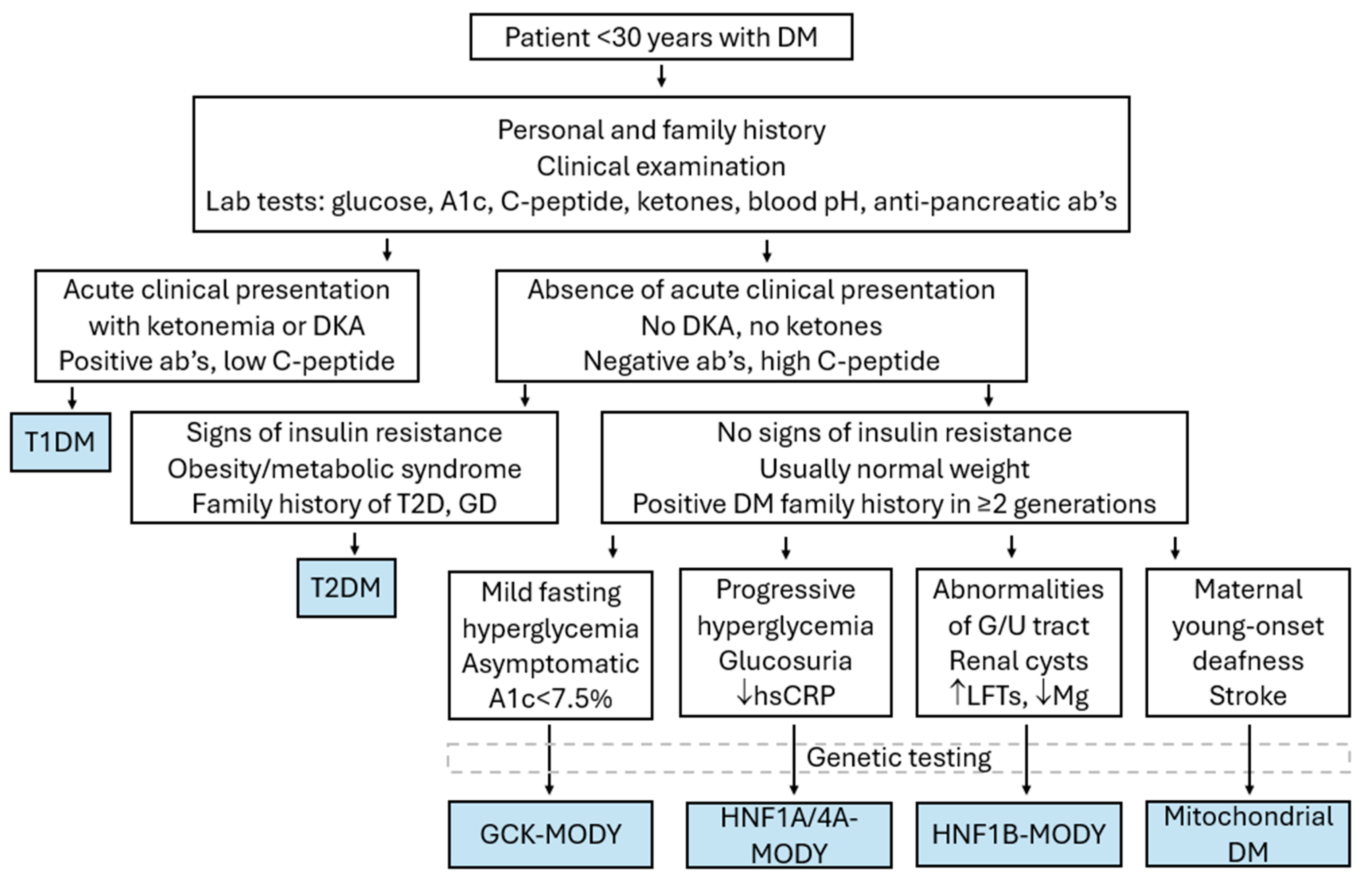

{kind=link}
{kind=link}
{kind=link}
| Gene | Previous Name | Chromosome | Associated Beta Cell Dysfunction |
|---|---|---|---|
| HNF1A | MODY 3 | 12q24.2 | Impaired glucose-mediated insulin secretion |
| GCK | MODY 2 | 7p15-p13 | Normal beta cell secretion capacity but with higher glucose threshold for insulin secretion |
| HNF4A | MODY 1 | 20q13.2 | Impaired insulin secretion with progressive deterioration in beta cell function |
| HNF1B | MODY 5 | 17q12 | Beta cell dysfunction accompanied by pancreatic agenesis or atrophy |
| PDX1 | MODY 4 | 13q12.2 | Defective differentiation of beta cells; impaired insulin secretion after glucose load |
| NEUROD1 | MODY 6 | 2q32 | Dysfunction of beta cells accompanied by insulinopenia or insulin resistance |
| KLF11 | MODY 7 | 2q25 | Insufficient insulin synthesis; impaired glucose-mediated insulin secretion |
| CEL | MODY 8 | 9q34 | Beta cell dysfunction due to protein misfolding leading to its aggregation; pancreatic atrophy |
| PAX4 | MODY 9 | 7q32 | Defective maturation of beta cells; impaired glucose-mediated insulin secretion |
| INS | MODY 10 | 11p15.5 | Apoptosis of beta cells; gradual reduction in beta cell mass |
| BLK | MODY 11 | 8p23 | Reduced beta cell mass; defective glucose-mediated insulin secretion |
| ABCC8 | MODY 12 | 11p15.1 | Insufficient insulin secretion |
| KCNJ11 | MODY 13 | 11p15 | Insufficient insulin secretion |
| APPL1 | MODY 14 | 3p14.3 | Decreased beta cell survival; defective glucose-mediated insulin secretion |
| Parameter | MODY Diabetes | T1D | T2D |
|---|---|---|---|
| Prevalence | Rare, stable | Common, increasing | Rare, increasing |
| Ethnicity | All | Mainly Caucasian | Mainly minority groups |
| Inheritance | Autosomal dominant | Multigenic | Multigenic |
| Family history | Almost 100% positive for MODY | 5%-10% positive for T1D | 75%-90% positive for T2D |
| Sex | Male = Female | Male = Female | Male < Female |
| Age at presentation | Before 25 yr of age | Childhood-adolescence | Adolescence |
| Body habitus | Various | Usually normal weight | Mostly obese |
| Acanthosis nigricans | Absent | Rare | Very common |
| Onset | Insidious | Usually acute, severe | Usually insidious, rarely acute |
| Onset with ketosis | Rare | Common | 5%–10% |
| Insulin, C-peptide | Detectable | Decreased or absent | Variable |
| Insulin sensitivity | Normal | Normal | Decreased |
| HLA-DR3/4 association | None | Strong | None |
| Pancreatic autoantibodies | Rare | 85%–100% | <10% |
| Insulin dependence | Rare | Permanent | Variable |
| Associated disorders | Depending on type, exocrine pancreas insufficiency, urogenital malformations, etc. | Autoimmune disorders (e.g., Hashimoto’s thyroiditis, vitiligo, celiac disease) | MetS components (e.g., lipid disorders, hypertension, PCOS, sleep apnea, etc.) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serbis, A.; Kantza, E.; Siomou, E.; Galli-Tsinopoulou, A.; Kanaka-Gantenbein, C.; Tigas, S. Monogenic Defects of Beta Cell Function: From Clinical Suspicion to Genetic Diagnosis and Management of Rare Types of Diabetes. Int. J. Mol. Sci. 2024, 25, 10501. https://doi.org/10.3390/ijms251910501
Serbis A, Kantza E, Siomou E, Galli-Tsinopoulou A, Kanaka-Gantenbein C, Tigas S. Monogenic Defects of Beta Cell Function: From Clinical Suspicion to Genetic Diagnosis and Management of Rare Types of Diabetes. International Journal of Molecular Sciences. 2024; 25(19):10501. https://doi.org/10.3390/ijms251910501
Chicago/Turabian StyleSerbis, Anastasios, Evanthia Kantza, Ekaterini Siomou, Assimina Galli-Tsinopoulou, Christina Kanaka-Gantenbein, and Stelios Tigas. 2024. "Monogenic Defects of Beta Cell Function: From Clinical Suspicion to Genetic Diagnosis and Management of Rare Types of Diabetes" International Journal of Molecular Sciences 25, no. 19: 10501. https://doi.org/10.3390/ijms251910501