
Determination of Potential Lead Compound from Magnolia officinalis for Alzheimer’s Disease through Pharmacokinetic Prediction, Molecular Docking, Dynamic Simulation, and Experimental Validation



Abstract
:1. Introduction
2. Results
2.1. Drug-Like Properties of the Compounds in M. officinalis
2.2. Pharmacokinetic Properties of Compounds in M. officinalis
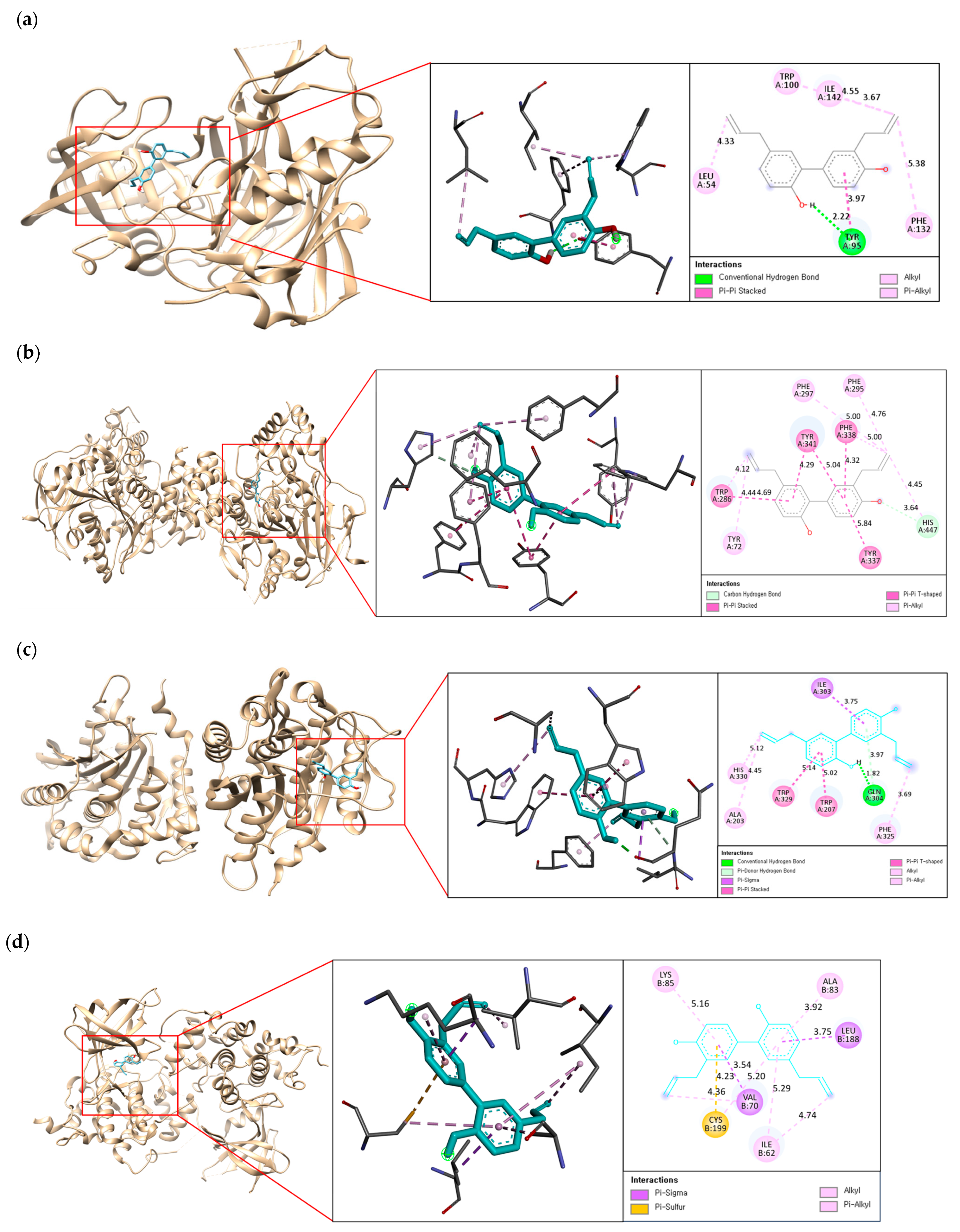
2.3. Molecular Docking on Enzymatic Targets: hBACE1, hAChE, hQC, and hGSK-3ß
2.4. Binding Interaction of the Best Compound with Multi-Enzyme Targets
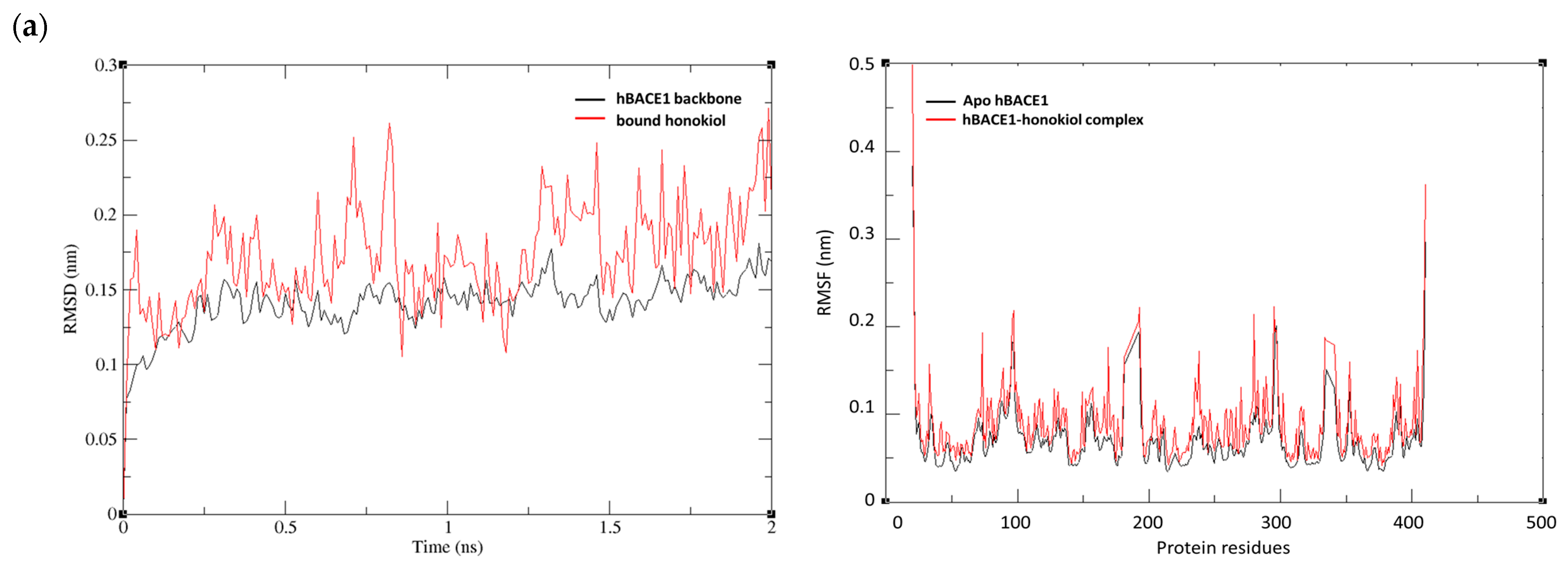
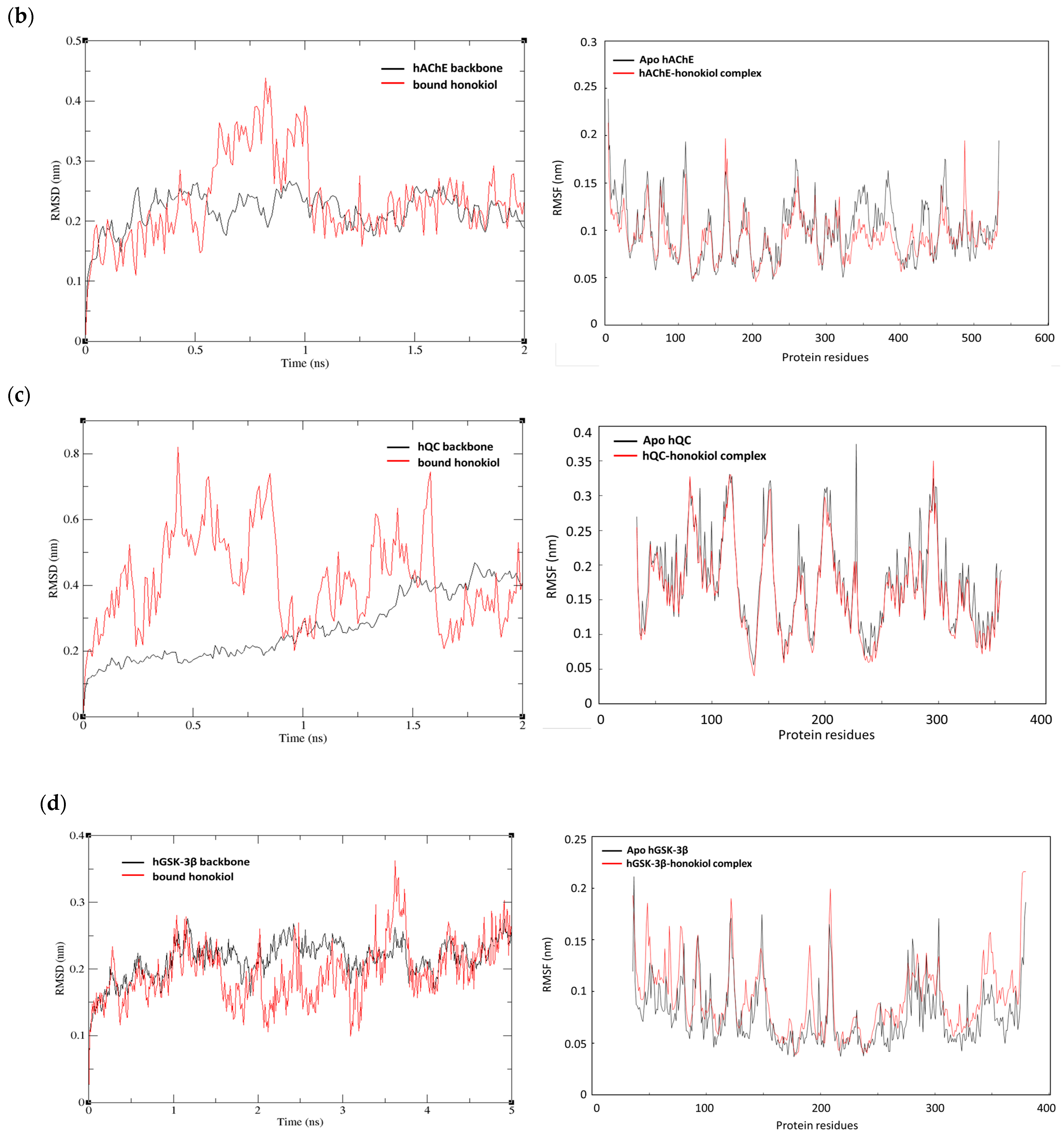
2.5. MD Simulations of Honokiol with hBACE1, hAChE, hQC, and hGSK-3ß
2.6. Inhibitory Effects of Honokiol on hBACE1, AChE, hQC, and hGSK-3β
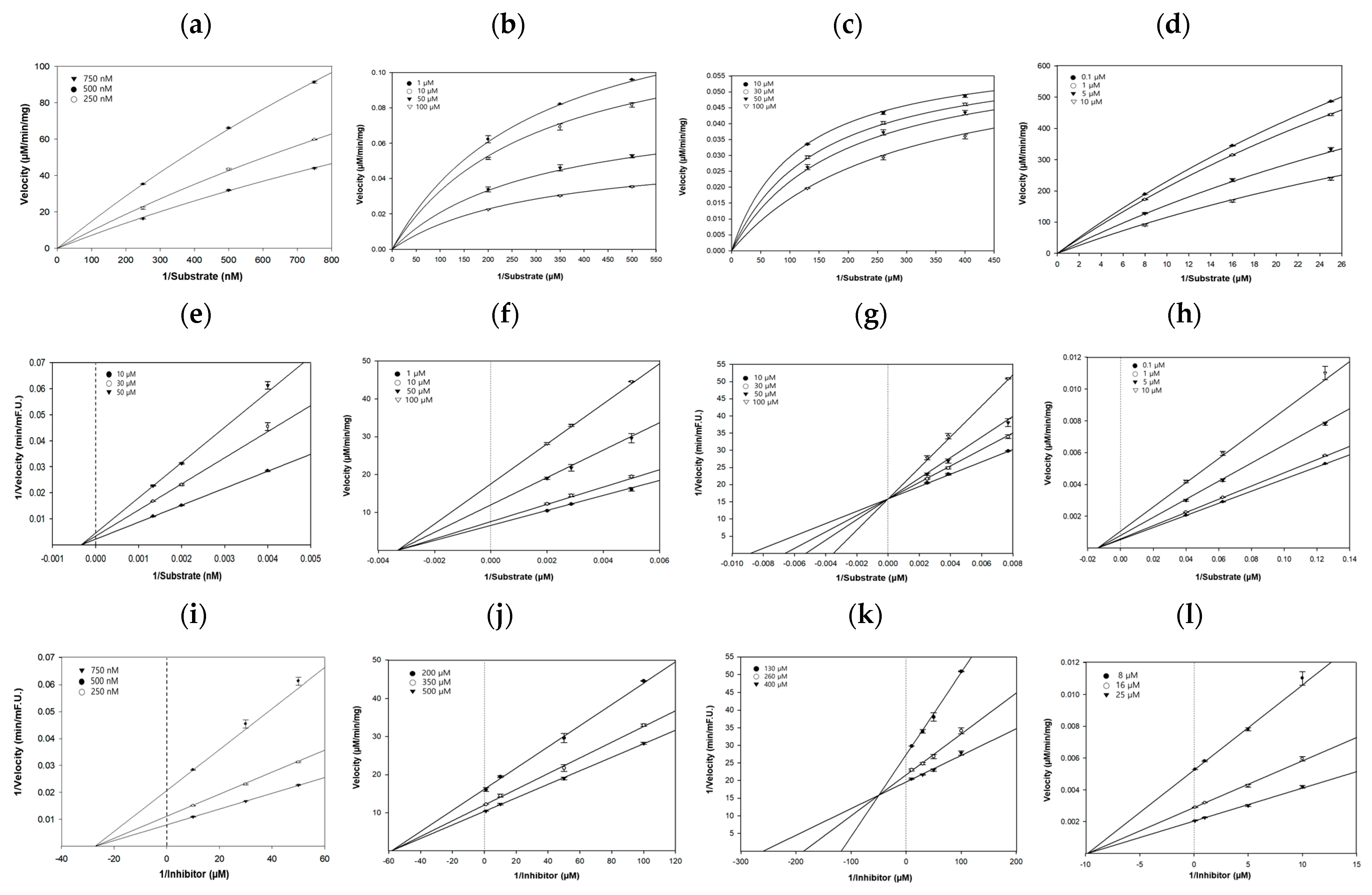
2.7. Kinetic Studies of hBACE1, AChE, hQC, and hGSK-3β Inhibition by Honokiol
3. Discussion
4. Materials and Methods
4.1. Collection of M. officinalis Compounds
4.2. Drug-Likeness and ADMET Analysis
4.3. Molecular Docking Simulation
4.4. MD Simulation
4.5. Reagents
4.6. hBACE1 Inhibitory Activity
4.7. AChE Inhibitory Activity
4.8. hQC Inhibitory Activity
4.9. hGSK-3β Activity
4.10. Enzymatic Kinetics on hBACE1, hAChE, hQC, and hGSK-3β
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 27 February 2024).
- Puzzo, D.; Fiorito, J.; Purgatorio, R.; Gulisano, W.; Palmeri, A.; Arancio, O.; Nicholls, R. Molecular mechanisms of learning and memory. In Genes, Environment and Alzheimer’s Disease; Lazarov, O., Tesco, G., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 1–27. ISBN 9780128028513. [Google Scholar]
- Mohamed, T.; Shakeri, A.; Rao, P.P. Amyloid cascade in Alzheimer’s disease: Recent advances in medicinal chemistry. Eur. J. Med. Chem. 2016, 113, 258–272. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P.; Watling, M. Investigational BACE inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2019, 28, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wehle, S.; Kuzmanovic, N.; Merget, B.; Holzgrabe, U.; König, B.; Sotriffer, C.A.; Decker, M. Acetylcholinesterase inhibitors with photoswitchable inhibition of beta-amyloid aggregation. ACS Chem. Neurosci. 2014, 5, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, K.; Girek, M.; Kręcisz, P.; Skibiński, R.; Łątka, K.; Jończyk, J.; Bajda, M.; Szymczyk, P.; Galita, G.; Kabziński, J.; et al. New cyclopentaquinoline and 3,5-dichlorobenzoic acid hybrids with neuroprotection against oxidative stress for the treatment of Alzheimer’s disease. J. Enzym. Inhib. Med. Chem. 2023, 38, 2158822. [Google Scholar] [CrossRef]
- Subramanian, N.; Watson, B.; Li, C.Z.; Moss, M.; Liu, C. Patterning amyloid-β aggregation under the effect of acetylcholinesterase using a biological nanopore—An in vitro study. Sens. Actuators Rep. 2023, 6, 100170. [Google Scholar] [CrossRef]
- Dinamarca, M.C.; Sagal, J.P.; Quintanilla, R.A.; Godoy, J.A.; Arrázola, M.S.; Inestrosa, N.C. Amyloid-beta-acetylcholinesterase complexes potentiate neurodegenerative changes induced by the abeta peptide. Mol. Neurodegener. 2010, 5, 4. [Google Scholar] [CrossRef]
- Barykin, E.P.; Mitkevich, V.A.; Kozin, S.A.; Makarov, A.A. Amyloid β modification: A key to the sporadic Alzheimer’s Disease? Front. Genet. 2017, 8, 58. [Google Scholar] [CrossRef]
- Gunn, A.P.; Wong, B.X.; Johanssen, T.; Griffith, J.C.; Masters, C.L.; Bush, A.I.; Barnham, K.J.; Duce, J.A.; Cherny, R.A. Amyloid-β peptide aβ3pE-42 induces lipid peroxidation, membrane permeabilization, and calcium influx in neurons. J. Biol. Chem. 2016, 291, 6134–6145. [Google Scholar] [CrossRef]
- Jawhar, S.; Wirths, O.; Schilling, S.; Graubner, S.; Demuth, H.U.; Bayer, T.A. Overexpression of glutaminyl cyclase, the enzyme responsible for pyroglutamate A{beta} formation, induces behavioral deficits, and glutaminyl cyclase knock-out rescues the behavioral phenotype in 5XFAD mice. J. Biol. Chem. 2011, 286, 4454–4460. [Google Scholar] [CrossRef]
- Ochalek, A.; Mihalik, B.; Avci, H.X.; Chandrasekaran, A.; Téglási, A.; Bock, I.; Giudice, M.L.; Táncos, Z.; Molnár, K.; László, L.; et al. Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimer’s Res. Ther. 2017, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and neurons at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Song, W. Increased NF-κB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef]
- Ly, P.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investig. 2013, 123, 224–235. [Google Scholar] [CrossRef]
- Poivre, M.; Duez, P. Biological activity and toxicity of the Chinese herb Magnolia officinalis Rehder & E. Wilson (Houpo) and its constituents. J. Zhejiang Univ. Sci. B 2017, 18, 194–214. [Google Scholar] [PubMed]
- Lee, S.; Khoo, C.; Halstead, C.W.; Huynh, T.; Bensoussan, A. Liquid chromatographic determination of honokiol and magnolol in hou po (Magnolia officinalis) as the raw herb and dried aqueous extract. J. AOAC Int. 2007, 90, 1210–1218. [Google Scholar] [CrossRef]
- Sarrica, A.; Kirika, N.; Romeo, M.; Salmona, M.; Diomede, L. Safety and toxicology of magnolol and honokiol. Planta Med. 2018, 84, 1151–1164. [Google Scholar] [CrossRef]
- Lee, Y.J.; Choi, D.Y.; Yun, Y.P.; Han, S.B.; Kim, H.M.; Lee, K.; Choi, S.H.; Yang, M.P.; Jeon, H.S.; Jeong, J.H.; et al. Ethanol extract of Magnolia officinalis prevents llipopolysaccharide-induced memory deficiency via its antineuroinflammatory and antiamyloidogenic effects. Phytother. Res. 2013, 27, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M. Search of neurotrophin-mimic natural products for prevention and treatment of neurodegenerative disease. Yakugaku Zasshi 2015, 135, 1147–1152. [Google Scholar] [CrossRef]
- Li, N.; Liang, Y.; Zhang, L.; Xu, C.; Wang, L. Neolignans in Magnolia officinalis as natural anti-Alzheimer’s disease agents: A systematic review. Ageing Res. Rev. 2024, 99, 102398. [Google Scholar] [CrossRef]
- Zhu, S.; Liu, F.; Zhang, R.; Xiong, Z.; Zhang, Q.; Hao, L.; Chen, S. Neuroprotective potency of neolignans in Magnolia officinalis cortex against brain disorders. Front. Pharmacol. 2022, 13, 857449. [Google Scholar] [CrossRef]
- Talarek, S.; Listos, J.; Barreca, D.; Tellone, E.; Sureda, A.; Nabavi, S.F.; Braidy, N.; Nabavi, S.M. Neuroprotective effects of honokiol: From chemistry to medicine. Biofactors 2017, 43, 760–769. [Google Scholar] [CrossRef]
- Kim, S.K.; Lee, M.K.; Jang, H.; Lee, J.J.; Lee, S.; Jang, Y.; Jang, H.; Kim, A. TM-MC 2.0: An enhanced chemical database of medicinal materials in Northeast Asian traditional medicine. BMC Complement. Med. Ther. 2024, 24, 40. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids. Barriers. CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Shi, S.; Zhao, S.; Tian, X.; Liu, F.; Lu, X.; Zang, H.; Li, F.; Xiang, L.; Li, L.; Jiang, S. Molecular and metabolic mechanisms of bufalin against lung adenocarcinoma: New and comprehensive evidences from network pharmacology, metabolomics and molecular biology experiment. Comput. Biol. Med. 2023, 157, 106777. [Google Scholar] [CrossRef]
- Barman, A.; Prabhakar, R. Elucidating the catalytic mechanism of β-secretase (BACE1): A quantum mechanics/molecular mechanics (QM/MM) approach. J. Mol. Graph. Model. 2013, 40, 1–9. [Google Scholar] [CrossRef]
- Pandolfi, F.; Vita, D.D.; Bortolami, M.; Coluccia, A.; Santo, R.D.; Costi, R.; Andrisano, V.; Alabiso, F.; Bergamini, C.; Fato, R.; et al. New pyridine derivatives as inhibitors of acetylcholinesterase and amyloid aggregation. Eur. J. Med. Chem. 2017, 141, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.F.; Liu, Y.L.; Cheng, W.J.; Ko, T.P.; Wang, A.H. Crystal structures of human glutaminyl cyclase, an enzyme responsible for protein N-terminal pyroglutamate formation. Proc. Natl. Acad. Sci. USA 2005, 102, 13117–13122. [Google Scholar] [CrossRef]
- Kramer, T.; Schmidt, B.; Monte, F.L. Small-molecule inhibitors of GSK-3: Structural insights and their application to Alzheimer’s disease models. Int. J. Alzheimer’s Dis. 2012, 2012, 381029. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLoS ONE 2015, 10, e0119264. [Google Scholar] [CrossRef] [PubMed]
- Baier, A.; Szyszka, R. Compounds from Natural Sources as Protein Kinase Inhibitors. Biomolecules 2020, 10, 1546. [Google Scholar] [CrossRef] [PubMed]
- Paudel, P.; Seong, S.H.; Zhou, Y.; Park, C.H.; Yokozawa, T.; Jung, H.A.; Choi, J.S. Rosmarinic acid derivatives’ inhibition of glycogen synthase kinase-3β is the pharmacological basis of Kangen-karyu in Alzheimer’s Disease. Molecules 2018, 23, 2919. [Google Scholar] [CrossRef]
- Ahmad, F.; Gupta, A.; Marzook, H.; Woodgett, J.R.; Saleh, M.A.; Qaisar, R. Natural compound screening predicts novel GSK-3 isoform-specific inhibitors. Biochimie 2024, 225, 68–80. [Google Scholar] [CrossRef]
- Shimmyo, Y.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. Flavonols and flavones as BACE-1 inhibitors: Structure-activity relationship in cell-free, cell-based and in silico studies reveal novel pharmacophore features. Biochim. Biophys. Acta 2008, 1780, 819–825. [Google Scholar] [CrossRef]
- Jung, H.A.; Lee, E.J.; Kim, J.S.; Kang, S.S.; Lee, J.H.; Min, B.S.; Choi, J.S. Cholinesterase and BACE1 inhibitory diterpenoids from Aralia cordata. Arch. Pharm. Res. 2009, 32, 1399–1408. [Google Scholar] [CrossRef]
- Naushad, M.; Durairajan, S.S.K.; Bera, A.K.; Senapati, S.; Li, M. Natural compounds with anti-BACE1 activity as promising therapeutic drugs for treating Alzheimer’s Disease. Planta Med. 2019, 85, 1316–1325. [Google Scholar] [PubMed]
- Gajendra, K.; Pratap, G.K.; Poornima, D.V.; Shantaram, M.; Ranjita, G. Natural acetylcholinesterase inhibitors: A multi-targeted therapeutic potential in Alzheimer’s disease. Eur. J. Med. Chem. Rep. 2024, 11, 100154. [Google Scholar] [CrossRef]
- Chen, D.; Chen, Q.; Qin, X.; Tong, P.; Peng, L.; Zhang, T.; Xia, C. Development and evolution of human glutaminyl cyclase inhibitors (QCIs): An alternative promising approach for disease-modifying treatment of Alzheimer’s disease. Front. Aging Neurosci. 2023, 15, 1209863. [Google Scholar] [CrossRef]
- Hielscher-Michael, S.; Griehl, C.; Buchholz, M.; Demuth, H.U.; Arnold, N.; Wessjohann, L.A. Natural products from microalgae with potential against Alzheimer’s disease: Sulfolipids are potent glutaminyl cyclase inhibitors. Mar. Drugs 2019, 14, 203. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Dong, Y.; Yu, X.; Zou, Y.; Zheng, Y.; Bu, X.; Quan, J.; He, Z.; Wu, H. Inhibitory effect of flavonoids on human glutaminyl cyclase. Bioorg. Med. Chem. 2016, 24, 2280–2286. [Google Scholar] [CrossRef]
- Kumar, A.; Roy, S.; Tripathi, S.; Sharma, A. Molecular docking based virtual screening of natural compounds as potential BACE1 inhibitors: 3D QSAR pharmacophore mapping and molecular dynamics analysis. J. Biomol. Struct. Dyn. 2016, 34, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Makarian, M.; Gonzalez, M.; Salvador, S.M.; Lorzadeh, S.; Hudson, P.K.; Pecic, S. Synthesis, kinetic evaluation and molecular docking studies of donepezil-based acetylcholinesterase inhibitors. J. Mol. Struct. 2022, 1247, 131425. [Google Scholar] [CrossRef] [PubMed]
- Mirza, F.J.; Zahid, S.; Amber, S.; Sumera; Jabeen, H.; Asim, N.; Ali Shah, S.A. Multitargeted molecular docking and dynamic simulation studies of bioactive compounds from Rosmarinus officinalis against Alzheimer’s Disease. Molecules 2022, 27, 7241. [Google Scholar] [CrossRef]
- Fukuyama, Y.; Kubo, M.; Harada, K. The search for, and chemistry and mechanism of, neurotrophic natural products. J. Nat. Med. 2020, 74, 648–671. [Google Scholar] [CrossRef]
- Rauf, A.; Olatunde, A.; Imran, M.; Alhumaydhi, F.A.; Aljohani, A.S.M.; Khan, S.A.; Uddin, M.S.; Mitra, S.; Emran, T.B.; Khayrullin, M.; et al. Honokiol: A review of its pharmacological potential and therapeutic insights. Phytomedicine 2021, 90, 53647. [Google Scholar] [CrossRef]
- Tan, Y.; Yu, H.; Sun, S.; Gan, S.; Gong, R.; Mou, K.J.; Xue, J.; Xu, S.; Wu, J.; Ma, L. Honokiol exerts protective effects on neural myelin sheaths after compressed spinal cord injury by inhibiting oligodendrocyte apoptosis through regulation of ER-mitochondrial interactions. J. Spinal Cord Med. 2022, 45, 595–604. [Google Scholar] [CrossRef]
- Zhou, Y.; Tang, J.; Lan, J.; Zhang, Y.; Wang, H.; Chen, Q.; Kang, Y.; Sun, Y.; Feng, X.; Wu, L.; et al. Honokiol alleviated neurodegeneration by reducing oxidative stress and improving mitochondrial function in mutant SOD1 cellular and mouse models of amyotrophic lateral sclerosis. Acta Pharm. Sin. B 2023, 13, 577–597. [Google Scholar] [CrossRef]
- Matsui, N.; Takahashi, K.; Takeichi, M.; Kuroshita, T.; Noguchi, K.; Yamazaki, K.; Tagashira, H.; Tsutsui, K.; Okada, H.; Kido, Y.; et al. Magnolol and honokiol prevent learning and memory impairment and cholinergic deficit in SAMP8 mice. Brain Res. 2009, 1305, 108–117. [Google Scholar] [CrossRef]
- Xian, Y.F.; Ip, S.P.; Mao, Q.Q.; Su, Z.R.; Chen, J.N.; Lai, X.P.; Lin, Z.X. Honokiol improves learning and memory impairments induced by scopolamine in mice. Eur. J. Pharmacol. 2015, 760, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Jangra, A.; Dwivedi, S.; Sriram, C.S.; Gurjar, S.S.; Kwatra, M.; Sulakhiya, K.; Baruah, C.C.; Lahkar, M. Honokiol abrogates chronic restraint stress-induced cognitive impairment and depressive-like behavior by blocking endoplasmic reticulum stress in the hippocampus of mice. Eur. J. Pharmacol. 2016, 770, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Bao, W.; Gao, Y.; Chen, J.; Song, G. Honokiol improves cognitive impairment in APP/PS1 mice through activating mitophagy and mitochondrial unfolded protein response. Chem. Biol. Interact. 2022, 351, 109741. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Youn, K.; Ji, Y.; Lee, S.; Lim, G.; Lee, J.; Ho, C.T.; Leem, S.H.; Jun, M. Biological and computational studies for dual cholinesterases inhibitory effect of zerumbone. Nutrients 2020, 12, 1215. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Fan, Y.G.; Zhao, L.X.; Zhang, Q.; Wang, Z.Y. The metal ion hypothesis of Alzheimer’s disease and the anti-neuroinflammatory effect of metal chelators. Bioorg. Chem. 2023, 131, 106301. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jiang, L.; Hu, Y.; Tang, N.; Liang, N.; Li, X.F.; Chen, Y.W.; Qin, H.; Wu, L. Ferritin reduction is essential for cerebral ischemia-induced hippocampal neuronal death through p53/SLC7A11-mediated ferroptosis. Brain Res. 2021, 1752, 147216. [Google Scholar] [CrossRef]
- Wang, M.; Li, Y.; Ni, C.; Song, G. Honokiol attenuates oligomeric amyloid β1-42-induced Alzheimer’s disease in mice through attenuating mitochondrial apoptosis and inhibiting the nuclear factor kappa-B signaling pathway. Cell Physiol. Biochem. 2017, 43, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Kantham, S.; Chan, S.; McColl, G.; Miles, J.A.; Veliyath, S.K.; Deora, G.S.; Dighe, S.N.; Khabbazi, S.; Parat, M.O.; Ross, B.P. Effect of the Biphenyl Neolignan Honokiol on Aβ42-Induced Toxicity in Caenorhabditis elegans, Aβ42 Fibrillation, Cholinesterase Activity, DPPH Radicals, and Iron(II) Chelation. ACS Chem. Neurosci. 2017, 8, 1901–1912. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Drug-Likeness Property | Pharmacokinetic Profiles | ||
|---|---|---|---|---|
| Lipinski’s Rule | Veber’s Rule | BBB Penetration | P-gp Substrate | |
| (+)-Syringaresinol (1) | Yes | Yes | No | Yes |
| Khusilol (2) | Yes | Yes | Yes | No |
| Asimilobine (3) | Yes | Yes | Yes | Yes |
| Magnoflorine (4) | Yes | Yes | Yes | Yes |
| Tembetarine (5) | Yes | Yes | Yes | Yes |
| Ferulic acid methyl ester (6) | Yes | Yes | Yes | No |
| 3-Pinanone (9) | Yes | Yes | Yes | No |
| 6,6-Dimethyl-2-methylene-bicyclo[2.2.1]-heptan-3-one (11) | Yes | Yes | Yes | Yes |
| Xanthoplanine (13) | Yes | Yes | Yes | Yes |
| N-Methylbulbocapnine (14) | Yes | Yes | Yes | Yes |
| N-Methylcoclaurine (15) | Yes | Yes | Yes | Yes |
| N-Nornuciferine (16) | Yes | Yes | Yes | Yes |
| Anonaine (18) | Yes | Yes | Yes | Yes |
| Borneol (19) | Yes | Yes | Yes | No |
| Borneol acetate (20) | Yes | Yes | Yes | No |
| Caffeic acid methyl ester (21) | Yes | Yes | Yes | No |
| Camphor (23) | Yes | Yes | Yes | No |
| Caryophyllene oxide (25) | Yes | Yes | Yes | No |
| cis-Linalool oxide (26) | Yes | Yes | Yes | No |
| Corytuberine (28) | Yes | Yes | Yes | Yes |
| Dienestrol (29) | Yes | Yes | Yes | No |
| Esculetin (32) | Yes | Yes | No | No |
| Gallic acid (34) | Yes | Yes | No | No |
| Glaucine (36) | Yes | Yes | Yes | Yes |
| Hexanal (38) | Yes | Yes | Yes | No |
| Honokiol (39) | Yes | Yes | Yes | No |
| Isomagnolol (42) | Yes | Yes | Yes | No |
| Limonene (43) | Yes | Yes | Yes | No |
| Lirinidine (44) | Yes | Yes | Yes | Yes |
| Liriodenine (45) | Yes | Yes | Yes | Yes |
| Lotusine (46) | Yes | Yes | Yes | Yes |
| Magnaldehyde D (47) | Yes | Yes | Yes | No |
| Magnocurarine (48) | Yes | Yes | Yes | Yes |
| Magnolignan A (49) | Yes | Yes | No | No |
| Magnolignan C (50) | Yes | Yes | No | No |
| Magnolol (51) | Yes | Yes | Yes | No |
| Michelalbine (56) | Yes | Yes | Yes | Yes |
| Nandigerine (58) | Yes | Yes | Yes | Yes |
| Nornuciferine (61) | Yes | Yes | Yes | Yes |
| Obovatol (62) | Yes | Yes | Yes | No |
| Palmidin B (65) | Yes | Yes | No | No |
| Phenol (67) | Yes | Yes | Yes | No |
| Puterine (69) | Yes | Yes | Yes | Yes |
| Randaiol (70) | Yes | Yes | Yes | No |
| Reticuline (71) | Yes | Yes | Yes | Yes |
| Roemerine (72) | Yes | Yes | Yes | Yes |
| Syringin (73) | Yes | Yes | No | No |
| Xanthoplanine (74) | Yes | Yes | Yes | Yes |
| α-Eudesmol (76) | Yes | Yes | Yes | No |
| α-Terpineol (78) | Yes | Yes | Yes | No |
| β-Eudesmol (79) | Yes | Yes | Yes | No |
| γ-Eudesmol (80) | Yes | Yes | Yes | No |
| γ-Gurjunene epoxide (81) | Yes | Yes | Yes | No |
| Ligand | AMES Toxicity | Maximum Tolerated Dose in Human | Oral Rat Acute Toxicity (LD50, mol/kg) | Oral Rat Chronic Toxicity (log mg/kg) |
|---|---|---|---|---|
| Khusilol (2) | No | −0.279 | 1.673 | 1.335 |
| Ferulic acid methyl ester (6) | No | 0.309 | 1.653 | 1.908 |
| 3-Pinanone (9) | No | 0.374 | 1.692 | 1.932 |
| Borneol (19) | No | 0.577 | 1.707 | 1.877 |
| Borneol acetate (20) | No | 0.526 | 1.904 | 1.875 |
| Caffeic acid methyl ester (21) | No | −0.154 | 2.023 | 1.594 |
| Camphor (23) | No | 0.473 | 1.653 | 1.981 |
| Caryophyllene oxide (25) | No | 0.148 | 1.548 | 1.224 |
| cis-Linalool oxide (26) | No | 0.891 | 1.917 | 2.221 |
| Dienestrol (29) | No | 0.191 | 2.324 | 1.840 |
| Hexanal (38) | No | 0.833 | 1.762 | 1.922 |
| Honokiol (39) | No | 0.305 | 2.184 | 1.791 |
| Isomagnolol (42) | Yes | 0.724 | 2.025 | 1.929 |
| Limonene (43) | No | 0.777 | 1.880 | 2.336 |
| Magnaldehyde D (47) | No | 0.462 | 1.827 | 1.772 |
| Magnolol (51) | No | 0.468 | 1.976 | 1.851 |
| Obovatol (62) | No | 0.497 | 1.776 | 1.586 |
| Phenol (67) | No | 0.540 | 2.153 | 2.011 |
| Randaiol (70) | No | 0.391 | 2.383 | 1.457 |
| α-Eudesmol (76) | No | 0.131 | 1.680 | 1.231 |
| α-Terpineol (78) | No | 0.886 | 1.923 | 1.945 |
| β-Eudesmol (79) | No | −0.220 | 1.727 | 1.304 |
| γ-Eudesmol (80) | No | 0.055 | 1.681 | 1.249 |
| γ-Gurjunene epoxide (81) | No | 0.347 | 1.647 | 1.428 |
| Ligand | CID No. | Binding Energy (kcal/mol) | |||
|---|---|---|---|---|---|
| hBACE1 (6JSE) | hAChE (4EY7) | hQC (3PBB) | hGS3-β (1Q5K) | ||
| Khusilol (2) | 556427 | −6.939 | −8.324 | −8.288 | −7.686 |
| Ferulic acid methyl ester (6) | 5357283 | −6.146 | −7.533 | −6.973 | −6.403 |
| 3-Pinanone (9) | 11038 | −5.956 | −7.226 | −5.301 | −5.509 |
| Borneol (19) | 1201518 | −5.303 | −6.524 | −4.472 | −5.129 |
| Borneol acetate (20) | 93009 | −5.862 | −7.834 | −5.122 | −5.788 |
| Caffeic acid methyl ester (21) | 689075 | −6.129 | −7.489 | −7.261 | −6.393 |
| Camphor (23) | 2537 | −5.220 | −6.983 | −4.877 | −5.014 |
| Caryophyllene oxide (25) | 1742210 | −7.275 | −8.564 | −5.876 | −6.721 |
| cis-Linalool oxide (26) | 6428573 | −5.685 | −6.551 | −5.801 | −5.434 |
| Dienestrol (29) | 667476 | −7.371 | −9.494 | −7.730 | −8.395 |
| Hexanal (38) | 6184 | −3.791 | −4.576 | −4.641 | −3.992 |
| Honokiol (39) | 72303 | −7.784 | −9.442 | −8.403 | −8.303 |
| Limonene (43) | 22311 | −5.887 | −7.000 | −6.650 | −6.386 |
| Magnaldehyde D (47) | 5319189 | −7.531 | −9.390 | −7.660 | −7.079 |
| Magnolol (51) | 72300 | −7.836 | −9.825 | −7.875 | −7.284 |
| Obovatol (62) | 100771 | −7.418 | −9.827 | −7.685 | −7.675 |
| Phenol (67) | 996 | −4.415 | −5.250 | −5.684 | −4.501 |
| Randaiol (70) | 13337243 | −7.733 | −8.558 | −7.251 | −7.382 |
| α-Eudesmol (76) | 92762 | −7.719 | −9.856 | −7.222 | −7.884 |
| α-Terpineol (78) | 17100 | −5.818 | −7.086 | −6.530 | −6.173 |
| β-Eudesmol (79) | 91457 | −7.829 | −8.482 | −7.496 | −7.739 |
| γ-Eudesmol (80) | 6432005 | −7.692 | −9.668 | −8.062 | −7.473 |
| γ-Gurjunene epoxide (81) | 91750423 | −6.940 | −7.956 | −6.669 | −6.362 |
| C6R 1 | 138857908 | −10.400 | - | - | - |
| E20 1 | 1150567 | - | −12.200 | - | - |
| PDB 1 | 6539196 | - | - | −6.300 | -- |
| TMU 1 | 68670561 | - | - | −8.000 | |
| Compound | Target Marker | IC50 1 | Ki 2 | Inhibition Mode 3 |
|---|---|---|---|---|
| Honokiol | hBACE1 4 | 20.64 ± 0.80 µM | 27.3 µM | Non-competitive |
| AChE 4 | 78.85 ± 1.23 µM | 58.2 µM | Non-competitive | |
| hQC 4 | 90.74 ± 1.07 µM | 49.4 µM | Competitive | |
| hGSK-3β 4 | 6.70 ± 0.03 µM | 9.8 µM | Non-competitive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Youn, K.; Jun, M. Determination of Potential Lead Compound from Magnolia officinalis for Alzheimer’s Disease through Pharmacokinetic Prediction, Molecular Docking, Dynamic Simulation, and Experimental Validation. Int. J. Mol. Sci. 2024, 25, 10507. https://doi.org/10.3390/ijms251910507
Youn K, Jun M. Determination of Potential Lead Compound from Magnolia officinalis for Alzheimer’s Disease through Pharmacokinetic Prediction, Molecular Docking, Dynamic Simulation, and Experimental Validation. International Journal of Molecular Sciences. 2024; 25(19):10507. https://doi.org/10.3390/ijms251910507
Chicago/Turabian StyleYoun, Kumju, and Mira Jun. 2024. "Determination of Potential Lead Compound from Magnolia officinalis for Alzheimer’s Disease through Pharmacokinetic Prediction, Molecular Docking, Dynamic Simulation, and Experimental Validation" International Journal of Molecular Sciences 25, no. 19: 10507. https://doi.org/10.3390/ijms251910507