
Applicability and Advantage of Mitochondrial Metagenomics and Metabarcoding in Spider Biodiversity Survey

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
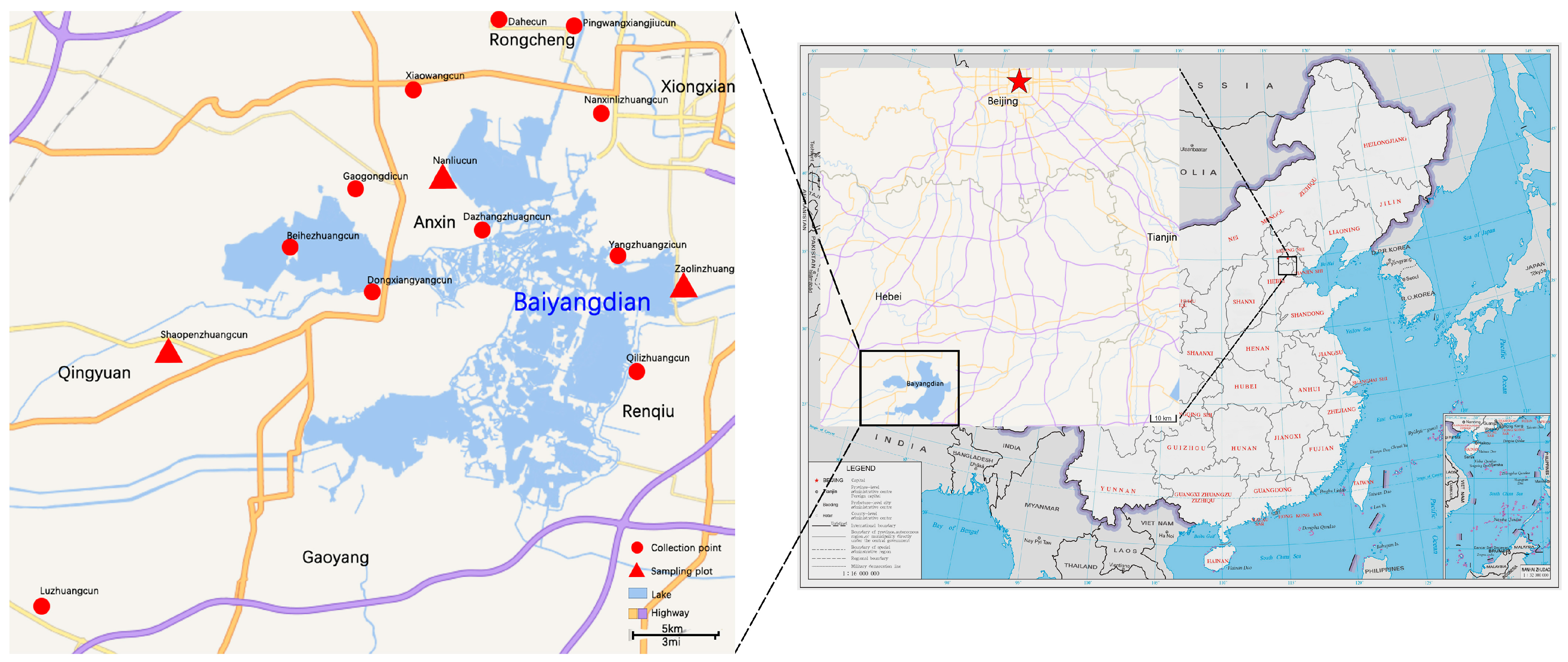
2.1. Sampling Sites and Collecting Spider Specimens
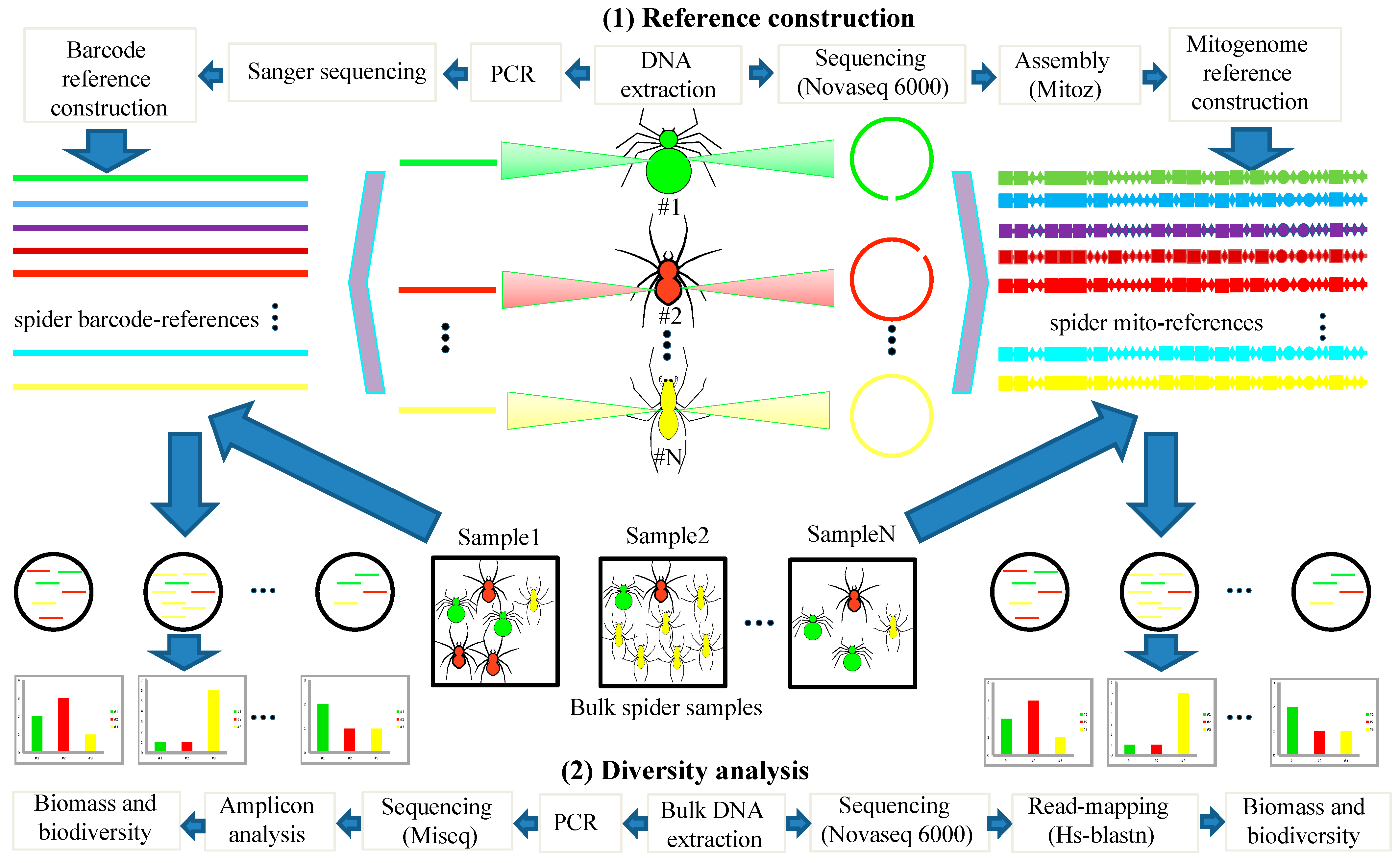
2.2. Mitogenome Assembly, Annotation, and Phylogenetic Analysis
2.3. Mitochondrial Metagenomics
2.4. PCR-Based Metabarcoding
3. Results
3.1. Establishment of Local Mitogenome and Barcode Reference Databases
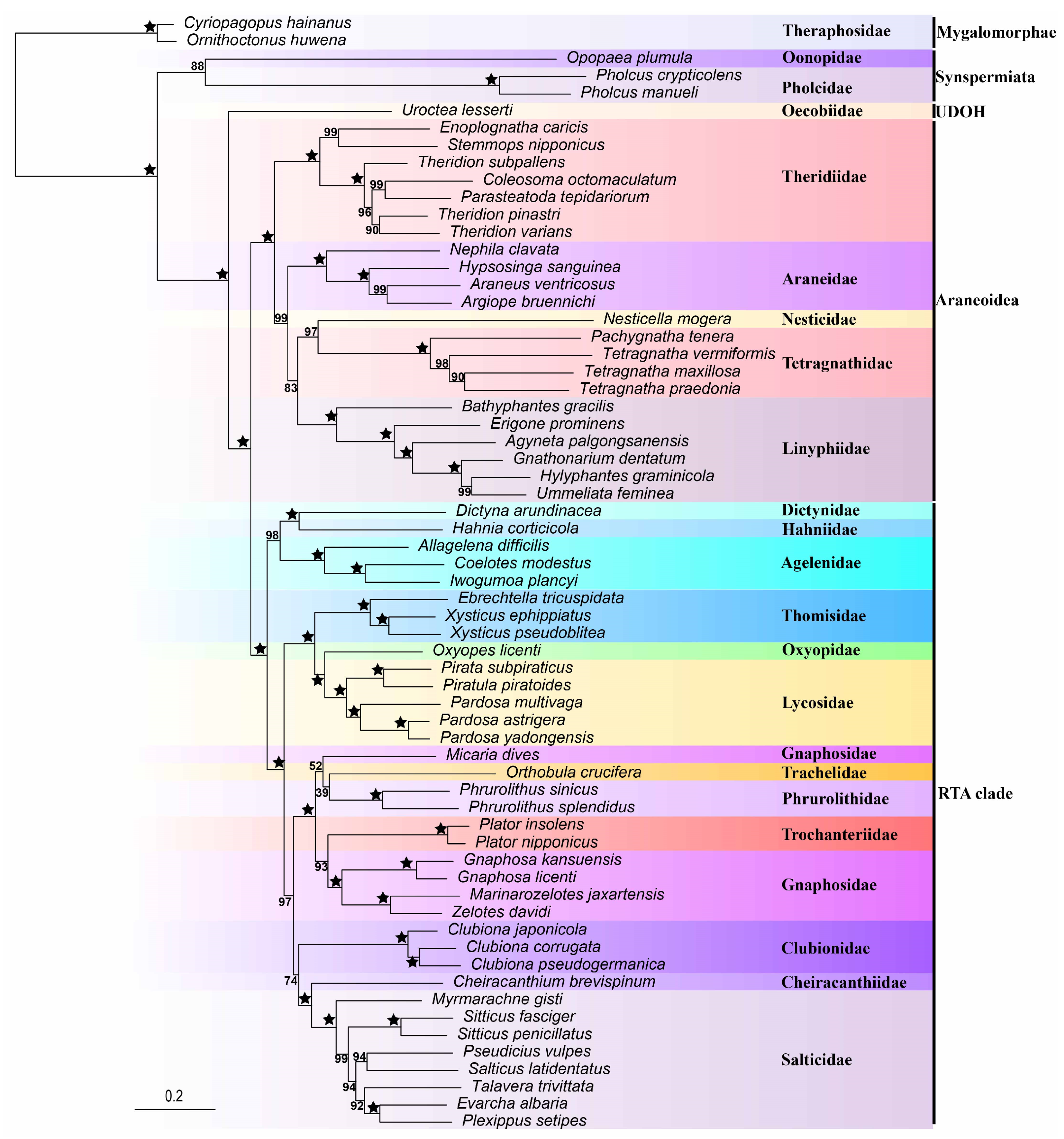
3.2. Phylogeny Based on Mitochondrial Genomes
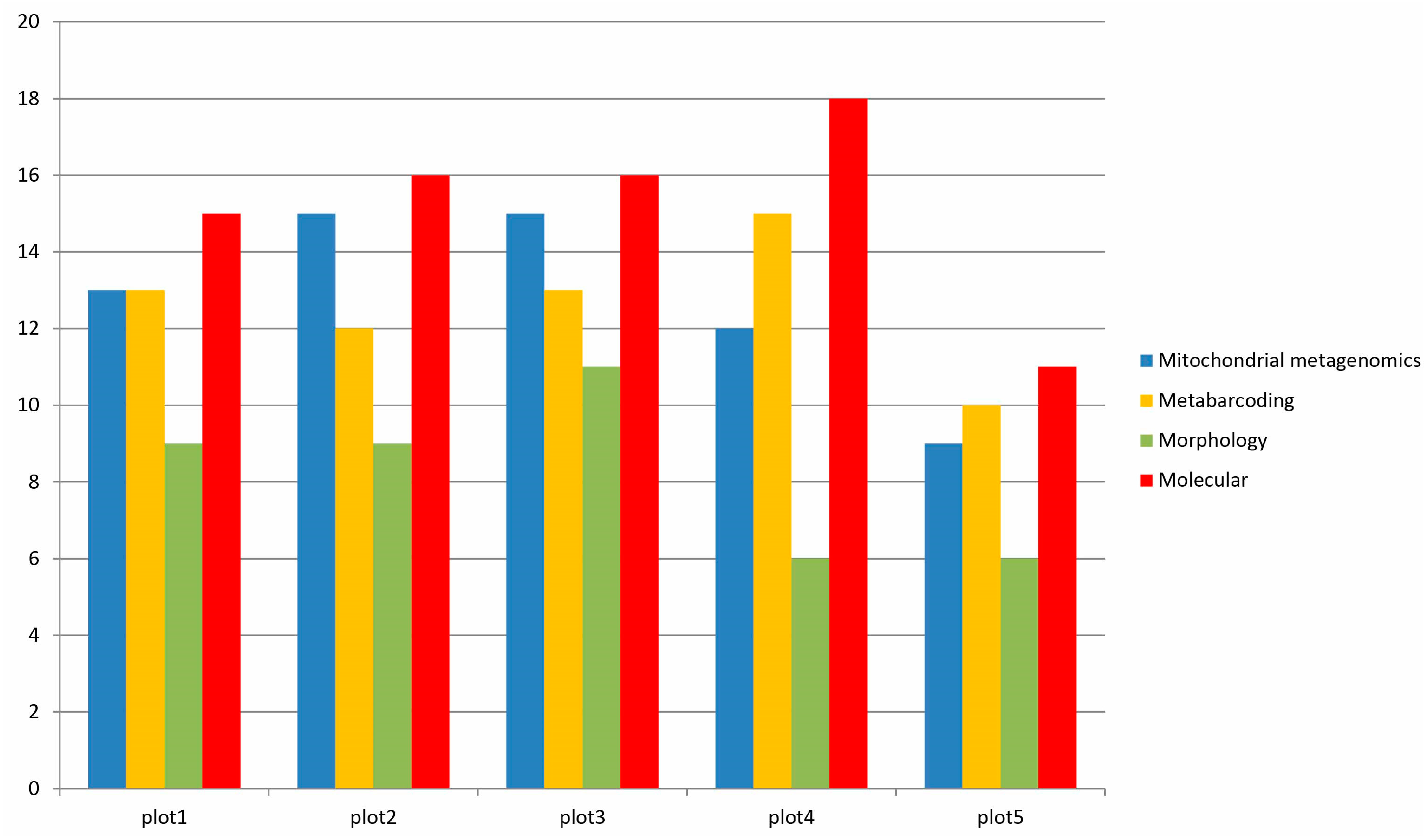
3.3. Species Detection by Morphology, Mitochondrial Metagenomics, and Metabarcoding
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cristescu, M.E. From barcoding single individuals to metabarcoding biological communities: Towards an integrative approach to the study of global biodiversity. Trends Ecol. Evol. 2014, 29, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Hardman, C.J.; Ji, Y.; Meng, G.; Liu, S.; Tan, M.; Yang, S.; Moss, E.D.; Wang, J.; Yang, C.; et al. High-throughput monitoring of wild bee diversity and abundance via mitogenomics. Methods Ecol. Evol. 2015, 6, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Blaxter, M. Molecular systematics: Counting angels with DNA. Nature 2003, 421, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Domenech, M.; Wangensteen, O.S.; Enguidanos, A.; Malumbres-Olarte, J.; Arnedo, M.A. For all audiences: Incorporating immature stages into standardised spider inventories has a major impact on the assessment of biodiversity patterns. Mol. Ecol. Resour. 2022, 22, 2319–2332. [Google Scholar] [CrossRef]
- Tautz, D.; Arctander, P.; Minelli, A.; Thomas, R.H.; Vogler, A.P. DNA points the way ahead in taxonomy. Nature 2002, 418, 479. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological identifications through DNA barcodes. Proc. Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef]
- Toju, H.; Baba, Y.G. DNA metabarcoding of spiders, insects, and springtails for exploring potential linkage between above- and below-ground food webs. Zool. Lett. 2018, 4, 4. [Google Scholar] [CrossRef]
- Piper, A.M.; Batovska, J.; Cogan, N.O.I.; Weiss, J.; Cunningham, J.P.; Rodoni, B.C.; Blacket, M.J. Prospects and challenges of implementing DNA metabarcoding for high-throughput insect surveillance. GigaScience 2019, 8, giz092. [Google Scholar] [CrossRef]
- da Silva, L.P.; Mata, V.A.; Lopes, P.B.; Lopes, R.J.; Beja, P. High-resolution multi-marker DNA metabarcoding reveals sexual dietary differentiation in a bird with minor dimorphism. Ecol. Evol. 2020, 10, 10364–10373. [Google Scholar] [CrossRef]
- Kodada, J.; Jaech, M.A.; Freitag, H.; Ciamporova-Zatovicova, Z.; Goffova, K.; Selnekovic, D.; Ciampor, F., Jr. Ancyronyx clisteri, a new spider riffle beetle species from Borneo, redescription of A. sarawacensis Jach including a description of the larva and new distribution data for A. procerus Jach using DNA barcodes (Coleoptera, Elmidae). ZooKeys 2020, 912, 25–64. [Google Scholar] [CrossRef]
- Borrell, Y.J.; Miralles, L.; Do Huu, H.; Mohammed-Geba, K.; Garcia-Vazquez, E. DNA in a bottle-Rapid metabarcoding survey for early alerts of invasive species in ports. PLoS ONE 2017, 12, e0183347. [Google Scholar] [CrossRef] [PubMed]
- Deiner, K.; Bik, H.M.; Machler, E.; Seymour, M.; Lacoursiere-Roussel, A.; Altermatt, F.; Creer, S.; Bista, I.; Lodge, D.M.; de Vere, N.; et al. Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Mol. Ecol. 2017, 26, 5872–5895. [Google Scholar] [CrossRef]
- Hatzenbuhler, C.; Kelly, J.R.; Martinson, J.; Okum, S.; Pilgrim, E. Sensitivity and accuracy of high-throughput metabarcoding methods for early detection of invasive fish species. Sci. Rep. 2017, 7, 46393. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Rodriguez, C.; Crampton-Platt, A.; Timmermans, M.J.T.N.; Baselga, A.; Vogler, A.P. Validating the power of mitochondrial metagenomics for community ecology and phylogenetics of complex assemblages. Methods Ecol. Evol. 2015, 6, 883–894. [Google Scholar] [CrossRef]
- Crampton-Platt, A.; Yu, D.W.; Zhou, X.; Vogler, A.P. Mitochondrial metagenomics: Letting the genes out of the bottle. GigaScience 2016, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Du, S.Y.; Dong, J.; Godeiro, N.N.; Wu, J.; Zhang, F. Advancing mitochondrial metagenomics: A new assembly strategy and validating the power of seed-based approach. Diversity 2022, 14, 317. [Google Scholar] [CrossRef]
- Andujar, C.; Arribas, P.; Ruzicka, F.; Crampton-Platt, A.; Timmermans, M.J.T.N.; Vogler, A.P. Phylogenetic community ecology of soil biodiversity using mitochondrial metagenomics. Mol. Ecol. 2015, 24, 3603–3617. [Google Scholar] [CrossRef]
- World Spider Catalog (2023). Available online: http://wsc.nmbe.ch (accessed on 30 April 2023).
- Blagoev, G.A.; Dondale, C.D. A new species of Alopecosa (Araneae: Lycosidae) from Canada: A morphological description supported by DNA barcoding of 19 congeners. Zootaxa 2014, 3894, 152–160. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, Y.-T.; Yao, C.; Deng, Y.-P.; Nie, Y.; Liu, G.-H. Mitochondrial phylogenomics provides insights into the taxonomy and phylogeny of fleas. Parasit. Vectors 2022, 15, 223. [Google Scholar] [CrossRef]
- Kirse, A.; Bourlat, S.J.; Langen, K.; Fonseca, V.G. Metabarcoding Malaise traps and soil eDNA reveals seasonal and local arthropod diversity shifts. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Hu, W.H.; Mei, Z.L.; Liu, Y.H.; Yu, Z.R.; Zhang, F.; Duan, M.C. Recovered grassland area rather than plantation forest could contribute more to protect epigeic spider diversity in northern China. Agric. Ecosyst. Environ. 2022, 326, 107726. [Google Scholar] [CrossRef]
- Zhang, X.Z.; Axmacher, J.C.; Wu, P.L.; Song, X.; Yu, Z.R.; Liu, Y.H. The taxon- and functional trait-dependent effects of field margin and landscape composition on predatory arthropods in wheat fields of the North China Plain. Insect Conserv. Diver. 2020, 13, 328–339. [Google Scholar] [CrossRef]
- He, X.Q.; Qiao, Y.H.; Sigsgaard, L.; Wu, X.J. The spider diversity and plant hopper control potential in the long-term organic paddy fields in sub-tropical area, China. Agric. Ecosyst. Environ. 2020, 295, 106921. [Google Scholar] [CrossRef]
- Cardoso, P.; Henriques, S.S.; Gaspar, C.; Crespo, L.C.; Carvalho, R.; Schmidt, J.B.; Sousa, P.; Szuts, T. Species richness and composition assessment of spiders in a Mediterranean scrubland. J. Insect Conserv. 2009, 13, 45–55. [Google Scholar] [CrossRef]
- Song, D.; Zhu, M.; Chen, J. The Fauna of Hebei, China: Araneae, 1st ed.; Hebei Science and Technology Publishing House: Shijiazhuang, China, 2001; p. 510. [Google Scholar]
- Zhu, M.; Zhang, B. Spider Fauna of Henan: Arachnida: Araneae, 1st ed.; Science Press: Beijing, China, 2011; p. 558. [Google Scholar]
- Meng, G.L.; Li, Y.Y.; Yang, C.T.; Liu, S.L. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef]
- Donath, A.; Juehling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.L.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Ranwez, V.; Douzery, E.J.P.; Cambon, C.; Chantret, N.; Delsuc, F. MACSE v2: Toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol. 2018, 35, 2582–2584. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Bui Quang, M.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast approximation for phylogeneticbootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Morgulis, A.; Gertz, E.M.; Schaffer, A.A.; Agarwala, R. WindowMasker: Window-based masker for sequenced genomes. Bioinformatics 2006, 22, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef]
- Shokralla, S.; Porter, T.M.; Gibson, J.F.; Dobosz, R.; Janzen, D.H.; Hallwachs, W.; Golding, G.B.; Hajibabaei, M. Massively parallel multiplex DNA sequencing for specimen identification using an Illumina MiSeq platform. Sci. Rep. 2015, 5, 9687. [Google Scholar] [CrossRef]
- Yu, D.W.; Ji, Y.; Emerson, B.C.; Wang, X.; Ye, C.; Yang, C.; Ding, Z. Biodiversity soup: Metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods Ecol. Evol. 2012, 3, 613–623. [Google Scholar] [CrossRef]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Doi, H.; Inui, R.; Matsuoka, S.; Akamatsu, Y.; Goto, M.; Kono, T. Estimation of biodiversity metrics by environmental DNA metabarcoding compared with visual and capture surveys of river fish communities. Freshwater Biol. 2021, 66, 1257–1266. [Google Scholar] [CrossRef]
- Soukainen, A.; Pajunen, T.; Korhonen, T.; Saarinen, J.; Chichorro, F.; Jalonen, S.; Kiljunen, N.; Koskivirta, N.; Kuurne, J.; Leinonen, S.; et al. Standardised spider (Arachnida, Araneae) inventory of Lammi, Finland. Biodivers. Data J. 2020, 8, e50775. [Google Scholar] [CrossRef]
- Rubio, G.D.; Moreno, C.E. Orb-weaving spider diversity in the Ibera Marshlands, Argentina. Neotrop. Entomol. 2010, 39, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Griotti, M.; Munoz-Escobar, C.; Ferretti, N.E. Linking vegetation structure and spider diversity in riparian and adjacent habitats in two rivers of Central Argentina: An analysis at two conceptual levels. Environ. Entomol. 2017, 46, 794–803. [Google Scholar] [CrossRef]
- Zheng, G.; Li, S.Q.; Wu, P.F.; Liu, S.J.; Kitching, R.L.; Yang, X.D. Diversity and assemblage structure of bark-dwelling spiders in tropical rainforest and plantations under different management intensities in Xishuangbanna, China. Insect Conserv. Divers. 2017, 10, 224–235. [Google Scholar] [CrossRef]
- Petillon, J.; Leroy, B.; Djoudi, E.A.; Vedel, V. Small and large spatial scale coexistence of ctenid spiders in a neotropical forest (French Guiana). Trop. Zool. 2018, 31, 85–98. [Google Scholar] [CrossRef]
- Garrido-Sanz, L.; Senar, M.A.; Pinol, J. Relative species abundance estimation in artificial mixtures of insects using mito-metagenomics and a correction factor for the mitochondrial DNA copy number. Mol. Ecol. Resour. 2022, 22, 153–167. [Google Scholar] [CrossRef]
- Xin, Z. Understanding biodiversity using genomics: Hooke’s microscope in the era of big data. Biodiv. Sci. 2019, 27, 475–479. [Google Scholar] [CrossRef]
- Karst, S.M.; Dueholm, M.S.; McIlroy, S.J.; Kirkegaard, R.H.; Nielsen, P.H.; Albertsen, M. Retrieval of a million high-quality, full-length microbial 16S and 18S rRNA gene sequences without primer bias. Nat. Biotechnol. 2018, 36, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Tang, M.; Hu, J.; Zhou, X. Genome-skimming provides accurate quantification for pollen mixtures. Mol. Ecol. Resour. 2019, 19, 1433–1446. [Google Scholar] [CrossRef]
- Hollingsworth, P.M.; Li, D.-Z.; van der Bank, M.; Twyford, A.D. Telling plant species apart with DNA: From barcodes to genomes. Philos. Trans. R. Soc. B 2016, 371, 20150338. [Google Scholar] [CrossRef] [PubMed]
- Cicconardi, F.; Borges, P.A.V.; Strasberg, D.; Oromi, P.; Lopez, H.; Perez-Delgado, A.J.; Casquet, J.; Caujape-Castells, J.; Maria Fernandez-Palacios, J.; Thebaud, C.; et al. MtDNA metagenomics reveals large-scale invasion of belowground arthropod communities by introduced species. Mol. Ecol. 2017, 26, 3104–3115. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.L.; Burgess, K.S.; Botsch, J.C.; Dobbs, E.K.; Read, T.D.; Brosi, B.J. Quantitative and qualitative assessment of pollen DNA metabarcoding using constructed species mixtures. Mol. Ecol. 2019, 28, 431–455. [Google Scholar] [CrossRef]
- Arribas, P.; Andujar, C.; Hopkins, K.; Shepherd, M.; Vogler, A.P. Metabarcoding and mitochondrial metagenomics of endogean arthropods to unveil the mesofauna of the soil. Methods Ecol. Evol. 2016, 7, 1071–1081. [Google Scholar] [CrossRef]
- Bista, I.; Carvalho, G.R.; Tang, M.; Walsh, K.; Zhou, X.; Hajibabaei, M.; Shokralla, S.; Seymour, M.; Bradley, D.; Liu, S.; et al. Performance of amplicon and shotgun sequencing for accurate biomass estimation in invertebrate community samples. Mol. Ecol. Resour. 2018, 18, 1020–1034. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D.N. BOLD: The Barcode of Life Data System (www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Y.; Zhang, F.; Zhang, J. Applicability and Advantage of Mitochondrial Metagenomics and Metabarcoding in Spider Biodiversity Survey. Diversity 2023, 15, 711. https://doi.org/10.3390/d15060711
Ding Y, Zhang F, Zhang J. Applicability and Advantage of Mitochondrial Metagenomics and Metabarcoding in Spider Biodiversity Survey. Diversity. 2023; 15(6):711. https://doi.org/10.3390/d15060711
Chicago/Turabian StyleDing, Yuhui, Feng Zhang, and Junxia Zhang. 2023. "Applicability and Advantage of Mitochondrial Metagenomics and Metabarcoding in Spider Biodiversity Survey" Diversity 15, no. 6: 711. https://doi.org/10.3390/d15060711