1. Introduction
Sumatriptan is a drug for migraine and cluster headaches treatment [
1,
2]. It is a selective 5-hydroxytryptamine (5-HT
1B/1D) receptor agonist. The binding between sumatriptan and serotonin type-1D receptors results in vasoconstriction of extensively dilated cranial blood vessels and subsequent relief of migraine-related pain. Sumatriptan is commercially available in several dosage forms such as oral, nasal, and parenteral delivery products. The recommended dose is a single 50 mg tablet with the maximum daily dose of 100 mg. The pharmacokinetics of sumatriptan depend on the route of administration, in which the most common route is oral administration. Oral bioavailability of sumatriptan is low (approximately 14%) because of extensive hepatic first-pass metabolism and incomplete absorption. After oral administration of a sumatriptan 50 mg tablet, it shows
tmax at 1.13 h with
Cmax of 33.21 ng/mL and an elimination half-life (
t1/2) of 2.96 h [
3].
Several analytical techniques have been developed for the quantitative determination of sumatriptan, including voltammetry [
4,
5], ultraviolet (UV) spectroscopy [
6], and liquid chromatography (LC) coupled with UV [
7,
8,
9], fluorescence [
10], electrochemical [
11,
12], and mass spectrometric (MS) detectors [
3,
13,
14,
15,
16]. Electrochemical, UV–vis, and fluorescent spectroscopic techniques can be advantageous as they are fast, simple, and require inexpensive instruments. However, these methods are limited by sensitivity and/or specificity and often require a large amount of sample, which is unsuitable for analyzing trace amounts of sumatriptan in human serum samples [
4,
5,
6,
10,
11,
12]. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) has been demonstrated as a routine technique for applications in pharmaceutical analyses that affords high throughput, good sensitivity, and selectivity. A few LC-MS/MS methods were developed to quantify sumatriptan in human plasma. For example, Stewart et al. described an LC/ESI-MS/MS method for the simultaneous determination of sumatriptan, naratriptan, zolmitriptan, and rizatriptan in human serum using a solid-phase extraction (SPE) for sample preparation and bufotenine as an internal standard (IS). This method exhibited a linear range of 1–100 ng/mL with a lower limit of quantification (LLOQ) of 250 pg/mL [
16]. Patel et al. developed ultra-performance liquid chromatography–tandem mass spectrometry with SPE for the simultaneous determination of sumatriptan and naproxen in human plasma using naratriptan and indomethacin as an IS. The linear range was validated from 0.050 to 100 ng/mL [
14].
Besides the selection of an analytical technique, sample preparation is another key factor for quantitative analysis of sumatriptan in human plasma samples because the complexity of biological matrices can significantly affect accuracy and precision. This step must ensure an efficient extraction of the target analytes, adequate sample clean-up, and high sample throughput. Different extraction procedures have been used and the most common method for extracting sumatriptan is SPE [
13,
14,
15,
17]. SPE provides clean samples with good analyte recovery, but it usually requires complicated procedures and long sample preparation times, which may cause unintentional errors in the analytical results. Automated SPE systems with 96-well SPE cartridges were developed to overcome these limitations. These automated systems provide high throughput, accurate sample processing, and a reduction of labor cost. Liquid-liquid extraction (LLE) is an alternative technique for sample clean-up. This methodology is simple and robust but requires a skilled researcher. The consideration of an appropriate extraction solvent for LLE is important for achieving recovery, sufficiently clean sample, and high sample throughput. Ethyl acetate was used as a solvent for extracting sumatriptan in human plasma [
3,
10]. However, the use of ethyl acetate consumes time in an evaporation step (about 40 min per run). Ethyl acetate often extracts endogenous compounds from the sample matrix due to its polarity, which frequently results in a high background as well as ionization suppression in mass spectrometric detection. Therefore, the development of a simple, rapid, cost-effective, and sensitive method for the quantification of sumatriptan in human plasma is still in demand.
In this study, we demonstrated an LC-MS/MS with liquid-liquid extraction for the quantitative determination of sumatriptan in human plasma.
tert-Butyl methyl ether (
t-BME) was selected as an extraction solvent since it offers advantages over ethyl acetate and other organic solvents in terms of volatility and low water solubility. These properties allow it to yield high recovery and high selectivity with less evaporating time. The LC-MS/MS method was fully validated covering all validation parameters listed in the US-FDA and EMA guidelines on bioanalytical method validation [
18,
19] and then applied in pilot pharmacokinetic and bioequivalence studies of sumatriptan tablets.
3. Materials and Methods
3.1. Drugs and Chemicals
Sumatriptan succinate (lot no. G0L156, purity 98.4%) and terazosin hydrochloride dehydrate (lot no. G0F290, purity 91.9%) were purchased from U.S. Pharmacopeia (Rockville, MD, USA). The generic sumatriptan 50-mg film-coated tablets (Siam Bheasach Co., Ltd., Bangkok, Thailand) and ImigranTM 50-mg tablets (GlaxoSmithKline Co., Ltd., Poznan, Poland) were used as the test and reference products, respectively. Acetonitrile, methanol and t-BME were purchased from Scharlau (Barcelona, Spain). Formic acid and 30% ammonia solution were obtained from Carlo Erba (Milano, Italy). All reagents were of at least analytical grade and used as received. Human blank plasma containing lithium heparin as an anticoagulant was obtained from IBS Co., Ltd. (Nakhon Pathom, Thailand). The purified water (18.2 MΩ cm) used throughout this experiment was obtained from a Milli-Q water purification system (Millipore; S.A.S, France).
3.2. Instrument Conditions
The liquid chromatographic system performed on a Prominence LC system (Shimadzu, Japan) consisted of a system controller (CBM-20Alite), a DGU-20A3 degasser, LC-20AD solvent delivery units, an FCV-11AL valve unit, a SIL-20AC autosampler, and a CTO-20A column oven. An analytical separation was carried out using a reversed-phase Symmetry® C18 column (150 × 4.6 mm i.d., 5 µm) (Thermo Scientific, Tewksbury, MA, USA) placed in a CTO-20A column oven whose temperature was set at 35 °C. The elution of sample was performed in a gradient mode using a 0.2% formic acid aqueous solution (solvent A) and acetonitrile (solvent B) as a mobile phase at a flow rate of 0.5 mL/min. Gradient elution program was as follows: 0.00–0.02 min, B: 40%; 0.02–0.50 min, B: 40%; 0.50–1.00 min, B: 80%; 1.00–2.00 min, B: 80%; 2.00–2.20 min, B: 40%; 2.20–4.00 min, B: 40%. The injection volume was 10 µL. The total run time was 4 min. The entire flow was directed into the detector. Carry-over was evaluated by injecting an extracted blank sample after an upper limit of quantification (ULOQ) sample. For the acceptance criteria of the carry-over experiment, the response for the analyte and IS in a blank sample after the injection of a ULOQ standard must be lower than 20% and 5% of the analyte response in the LLOQ standard, respectively. In this study, there was no peak at the retention time of sumatriptan and terazosin presented in blank sample queued after ULOQ injection, suggesting that no carry-over effect was observed.
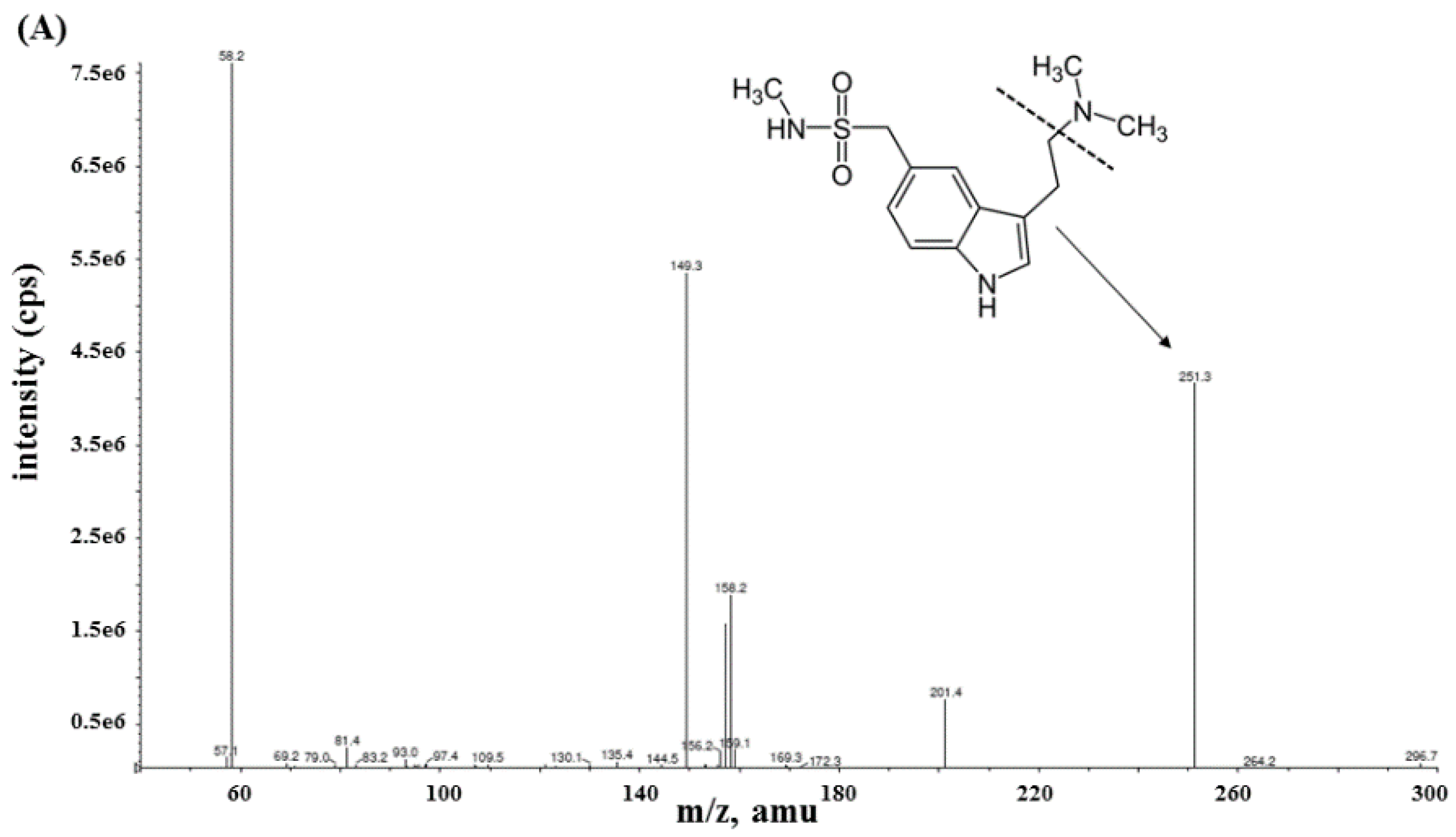
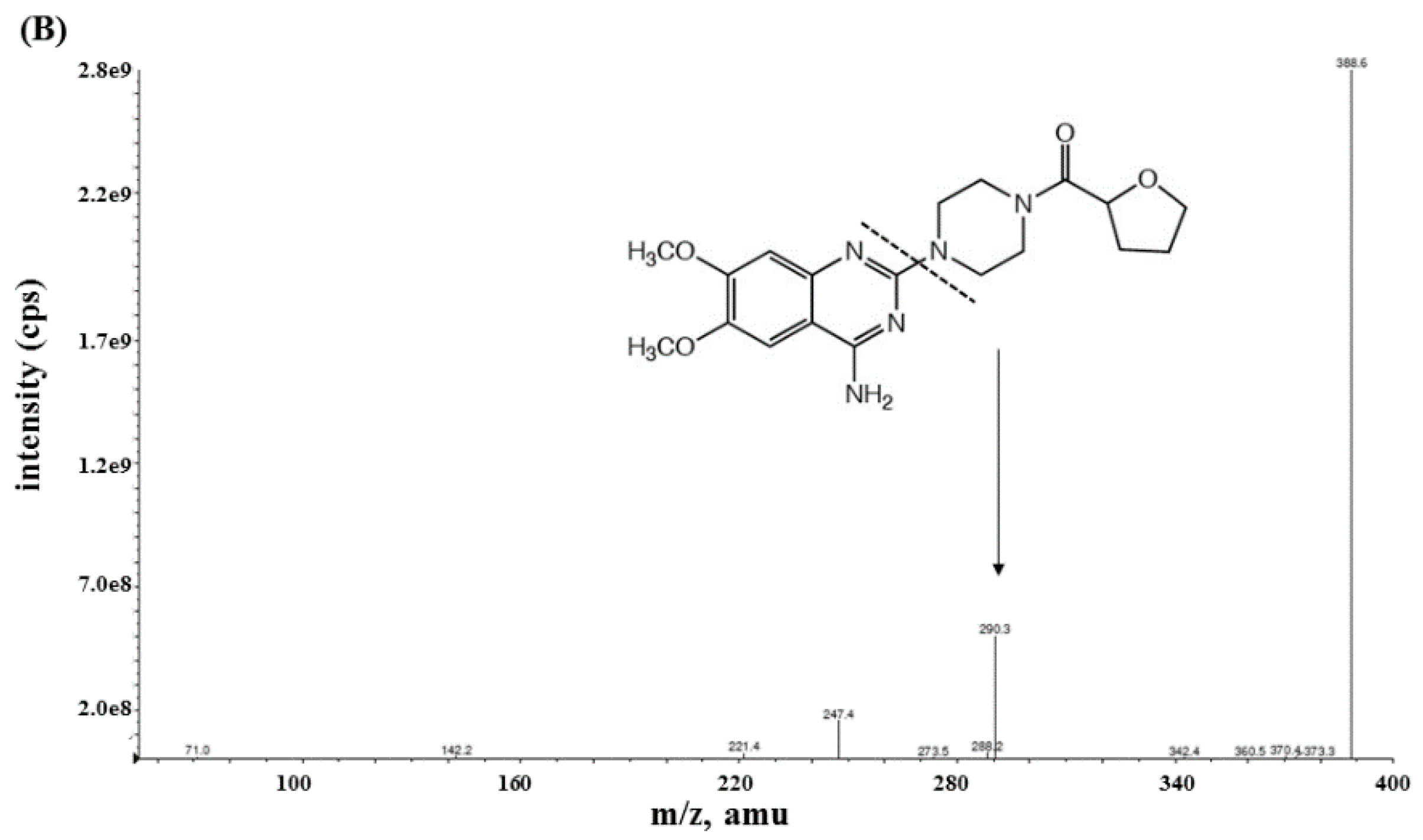
Mass spectrometric analysis of sumatriptan and terazosin (used as an IS) was achieved with MS/MS detection in a positive ion mode using an AB Sciex API-4000 mass spectrometer (Foster City, CA, USA) equipped with a Turbo ionspray™ interface which was set at 500 °C. The ion spray voltage was set at 4500 V. The source parameters including the collision gas, curtain gas, ion source gas 1 and ion source gas 2 were set at 4, 20, 50, and 50 psi, respectively. Nitrogen was designed as carrier and fragmentation gas. The compound parameters including the declustering potential (DP), entrance potential (EP), collision energy (CE), and collision cell exit potential (CXP) were 61, 10, 25, and 24 V for sumatriptan, respectively, and 116, 10, 35, and 24 V for terazosin, respectively. Detection of the ions was carried out in the multiple reaction monitoring mode (MRM) by monitoring the transition at m/z [M+H]+ 296.26→251.05 for sumatriptan and m/z [M’+H]+ 388.10→290.25 for terazosin with a 200 ms scan dwell time for all compounds. Quantitation of sumatriptan was based on the peak area ratio of sumatriptan versus terazosin. The data acquisition was processed using Analyst software™ (version 1.4.2). The calculations of validation data and plasma concentration were performed by Microsoft Excel program (Microsoft, Albuquerque, NM, USA).
3.3. Preparation of Stock Standard Solutions, Calibration Standards, and QC Samples
Stock standard solutions of sumatriptan and terazosin were prepared separately at 1 mg/mL in methanol. Working standard solutions of sumatriptan for calibration standards and QC samples were prepared from the 1 mg/mL sumatriptan stock solution. A working standard solution of terazosin at a concentration of 868.0 ng/mL was prepared from the stock solution of terazosin. All working solutions were diluted with a mixture of 0.2% formic acid solution and acetonitrile (60:40, v/v).
Seven-point calibration standard samples were prepared by spiking the working standard solutions in blank human plasma to yield final concentrations of 0.5 (LLOQ), 1.0, 2.5, 10.0, 20.0, 35.0, and 50.0 (ULOQ) ng/mL. Four concentration levels of spiked plasma QC samples were prepared at final concentrations of 0.5 (LLOQ QC), 1.5 (LQC), 25.0 (MQC), and 45.0 (HQC) ng/mL with 41.3 ng/mL of terazosin.
3.4. Sample Preparation
A 300 µL of plasma sample was transferred into a centrifuge tube. A 15 µL of terazosin working solution and 100 µL of 9.3 mol/L ammonium hydroxide solution were added to the plasma sample. The mixture was vortexed for 30 s followed by the addition of 2.0 mL of t-BME. The mixture was vortexed for 1 min and centrifuged at 1792 g, 4 °C for 10 min to make completely separated layers. A 1.7 mL of the upper layer was taken into a 2 mL microcentrifuge tube, and then evaporated to dryness at 45 °C for 10 min. After that, the residue was reconstituted with a 400 µL of a mixture of 0.2% formic acid solution and acetonitrile (60:40, v/v), followed by vortex-mixing for 30 s and centrifugation at 11,200 g, 4 °C for 10 min to remove undissolved matrix. The clear supernatant was taken out for LC-MS/MS analysis.
3.5. Method Validation
The method performance was evaluated according to US-FDA and EMA guidance for bioanalytical method validation for specificity, carry-over, linearity, accuracy, precision, LLOQ, recovery, dilution integrity, matrix effect, hemolytic and lipemic effect, and stability [
18,
19].
3.5.1. Specificity and Sensitivity
Specificity of the method was performed using eight different lots of blank plasma (six lots of normal blank plasma, one lot of hemolyzed blank plasma, and one lot of hyperlipidemic plasma). The spiked samples at LLOQ were extracted and the presence or absence of interfering peak at the same retention time of sumatriptan or terazosin was investigated. Chromatograms from each blank sample were compared with the LLOQ sample.
The sensitivity of the method was evaluated at LLOQ concentration. The LLOQ is accepted if the sumatriptan response at LLOQ level is at least five times over the baseline response with a %deviation of ±20% and a percentage coefficient of variation (%CV) of ≤20%.
3.5.2. Calibration Curve and Linearity
Calibration curves were evaluated from the analysis of spiked calibration samples at seven different concentration levels. The calibration curves were constructed by plotting the ratio of sumatriptan signal to IS signal as a function of sumatriptan concentrations. The equation model was obtained by weighted least squares linear regression analysis with a weighting factor of 1/x2. Back-calculation of the calibration standard concentration was used to confirm the suitability of equation. Each back-calculated concentration standard should be within ±15 %deviation, except for LLOQ which should be within ±20 %deviation. The coefficient of determination (r2) should be >0.99.
3.5.3. Accuracy and Precision
Accuracy and precision were evaluated by analyzing drug concentration in plasma samples at LLOQ, LQC, MQC, HQC, and ULOQ levels. Six replicates of each concentration level were analyzed on the same day for intra-day (within-run) analysis and on three different days for inter-day (between-run) analysis. Accuracy was assessed by calculating the %deviation based on the difference between the mean concentration found and concentration added. The mean accuracy of each level should be within ±15 %deviation, except for LLOQ which should be within ±20%. Precision was evaluated by calculating the %CV of the mean concentration found. The %CV of each level should be within 15%, except for LLOQ which should not be more than 20%.
3.5.4. Recovery
The recovery of extraction was evaluated from responses of sumatriptan obtained from QC samples in comparison with those obtained from post-spiked QC samples at the same concentration. The extraction recovery of sumatriptan was determined at LQC, MQC, and HQC levels. The extraction recovery of terazosin was determined at 41.3 ng/mL from MQC samples using the same procedure. The extents of recovery of analyte and IS should be consistent and reproducible.
3.5.5. Dilution Integrity
Dilution integrity was evaluated to ensure that sample dilution with the same matrix did not have any effects on the reliability of the method. The accuracy and precision on 1:1 and 1:3 ratios (70.0 and 100.0 ng/mL, respectively) were analyzed. Samples at 70.0 ng/mL and 100.0 ng/mL of sumatriptan were diluted with human blank plasma to get diluted samples before analysis. The average %deviation should be ±15% of the actual concentration and %CV should be within 15%.
3.5.6. Matrix Effect
The effect of extracted plasma matrix was evaluated in nine different sets of QC samples prepared from six different lots of human blank plasma, one lot of human hemolyzed blank plasma, one lot of human hyperlipidemic blank plasma, and one lot of human blank plasma containing 5.0 ng/mL of chlorpheniramine as a possible concomitant drug in case of drug allergic accident during study. The responses of sumatriptan obtained from post-spiked QC samples were compared with those obtained from the pure authentic standard solution at the same concentration. Each set of QC samples composed of three replicates of LQC and HQC samples. The matrix effect on terazosin was determined at 41.3 ng/mL. The effect of plasma matrix was separately determined for sumatriptan and terazosin and reported as MF. The ratio between the MF of sumatriptan and that of terazosin is calculated in term of the IS-normalized MF. The CV of IS-normalized matrix factor at each concentration level should be within 15%.
3.5.7. Hemolytic and Lipemic Effects
The hemolytic and lipemic effects were evaluated by analyzing six replicates of samples containing sumatriptan at LQC (1.5 ng/mL) and HQC (45.0 ng/mL) levels in hemolyzed plasma and hyperlipidemic plasma, respectively. The concentrations of drug in plasma samples were determined against calibration curve prepared in human normal blank plasma. The acceptable limit of both effects should be within ±15 %deviation and 15 %CV.
3.5.8. Stability
Stability experiments were performed with LQC and HQC samples under different conditions. The experiments were performed in three replicates at each QC level to determine freeze-thaw stability (4 cycles at −70 °C and room temperature), short-term stability (on bench-top at 25 °C for 4 h), post-preparation in dry state (at 2–8 °C for 4 days), autosampler (at 4 °C for 48 h), reinjection stability (at 4 °C for 48 h), and long-term stability (at −70 °C for 77 days). Additionally, stock solution stability was evaluated in different conditions (kept in refrigerator at 2–8 °C for 30 days (sumatriptan), 31 days (terazosin), and on bench-top at 25 °C for 8 h).
3.6. Pharmacokinetic and Bioequivalence Studies
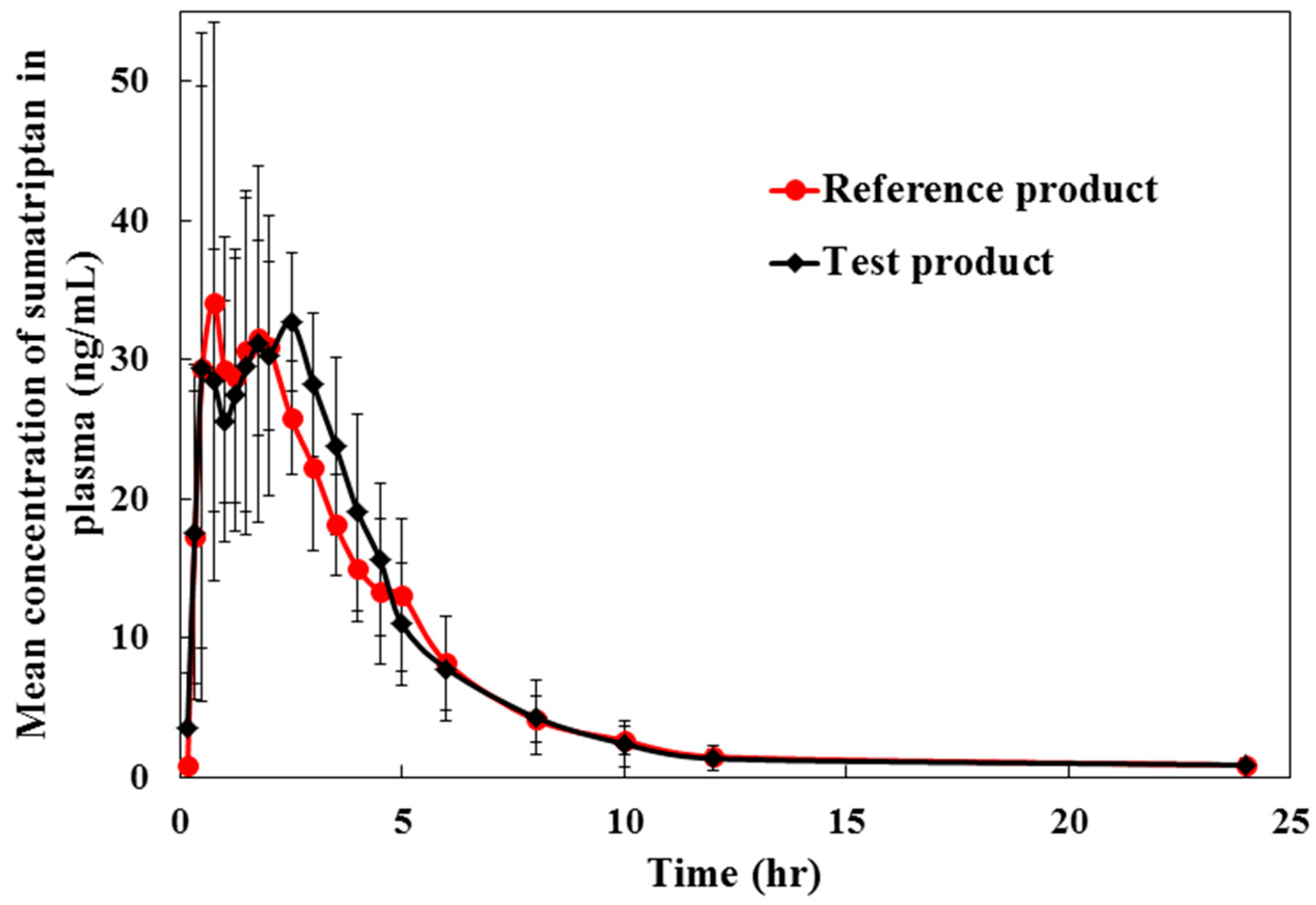
Pilot pharmacokinetic and bioequivalence studies of a single dose of 50 mg sumatriptan tablet were performed in six healthy volunteers. The study was designed as an open-label, single dose, fasting, two-treatment, two-period, two-sequence randomized crossover with 7 days washout period between period 1 and period 2 dosing. Subjects were randomly assigned to a treatment sequence (test/reference or reference/test group). Six healthy Thai volunteers with mean age group 32.08 ± 9.20 years and body mass index (BMI) 22.27 ± 1.90 kg/m2 were included in the study. Vital signs including pulse (60–90 bpm), blood pressure (SBP of 100–135 mmHg and DBP of 60–90 mmHg), and body temperature (36.5–37.5 °C) were monitored prior to and during the study. All subjects were in good health as shown by clinical laboratory screening including serology, hematology, and biochemistry tests. None of the volunteers reported a history of allergy to sumatriptan, sulfonamides or any of the excipients of this product. All subjects abstained from intake of other drugs and alcohol for two weeks prior to and throughout the study. Caffeine-containing beverages were not allowed in the 7 days prior to and during the study. All the subjects were informed of the aims and risks in the study and written consent was obtained.
In each period, blood samples (6 mL, except 12 mL at 0 h) were collected in tubes containing lithium heparin before (0 h) and at 0.17, 0.33, 0.50, 0.75, 1.00, 1.25, 1.50, 1.75, 2.00, 2.50, 3.00, 3.50, 4.00, 4.50, 5.00, 6.00, 8.00, 10.00, 12.00, and 24.00 h after sumatriptan administration. Plasma samples were separated from blood by centrifugation at 4000 ± 100 rpm, 4 °C for 10 min, aliquoted into microcentrifuge tubes under low light condition, flash frozen immediately and stored at −70 ± 10 °C until analysis. A 15 µL of IS was spiked into a 300 µL of thawed plasma and then followed the procedure described in the sample preparation section.
The pharmacokinetic parameters including the AUC,
Cmax,
Tmax,
ke, and
t1/2 were calculated using WinNonlin 6.4 software (Pharsight, NC, USA). Bioequivalence analysis for the two formulations was determined by an analysis of variance (ANOVA) at α = 0.05. Bioequivalence interpretation requires the 90% confident interval (CI) of geometric means test/reference for
Cmax, AUC
0→t, and AUC
0→∞ to be within the acceptance criteria of 80–125% [
22,
23]. The study protocol was approved by the Institute for the Development of Human Research Protections (IHRP). The study was carried out in accordance with the international guidelines for human research protection set out by the Declaration of Helsinki, The Belmont Report, CIOMS Guideline, and International Conference on Harmonization in Good Clinical Practice (ICH-GCP).
An incurred sample reanalysis demonstrated the method performance in term of the reproducibility of this assay during BE study. Incurred samples were randomly selected from every volunteer and from every period. The selection criteria included samples which were near the
Cmax and the elimination phase in the pharmacokinetic profile of the drug. The results obtained were compared with the data obtained earlier for the same sample using the same procedure. The %different should be within ±20% [
24].







{kind=link}
{kind=link}
{kind=link}
{kind=link}