
Assessment of Tumorigenic Potential in Mesenchymal-Stem/Stromal-Cell-Derived Small Extracellular Vesicles (MSC-sEV)

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. E1-MYC Does Not Form Tumors In Vivo
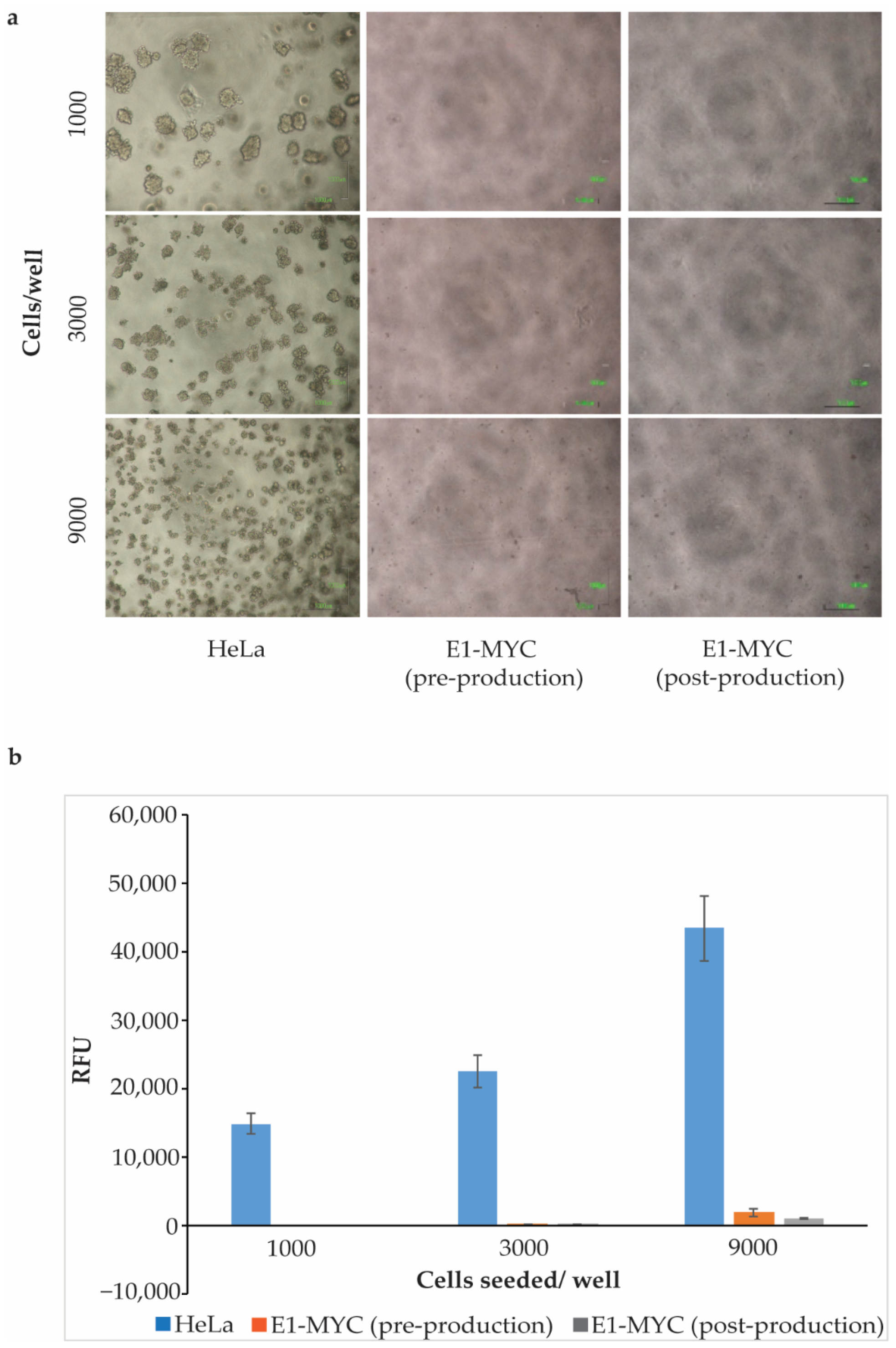
2.2. Anchorage-Independent Growth Assay
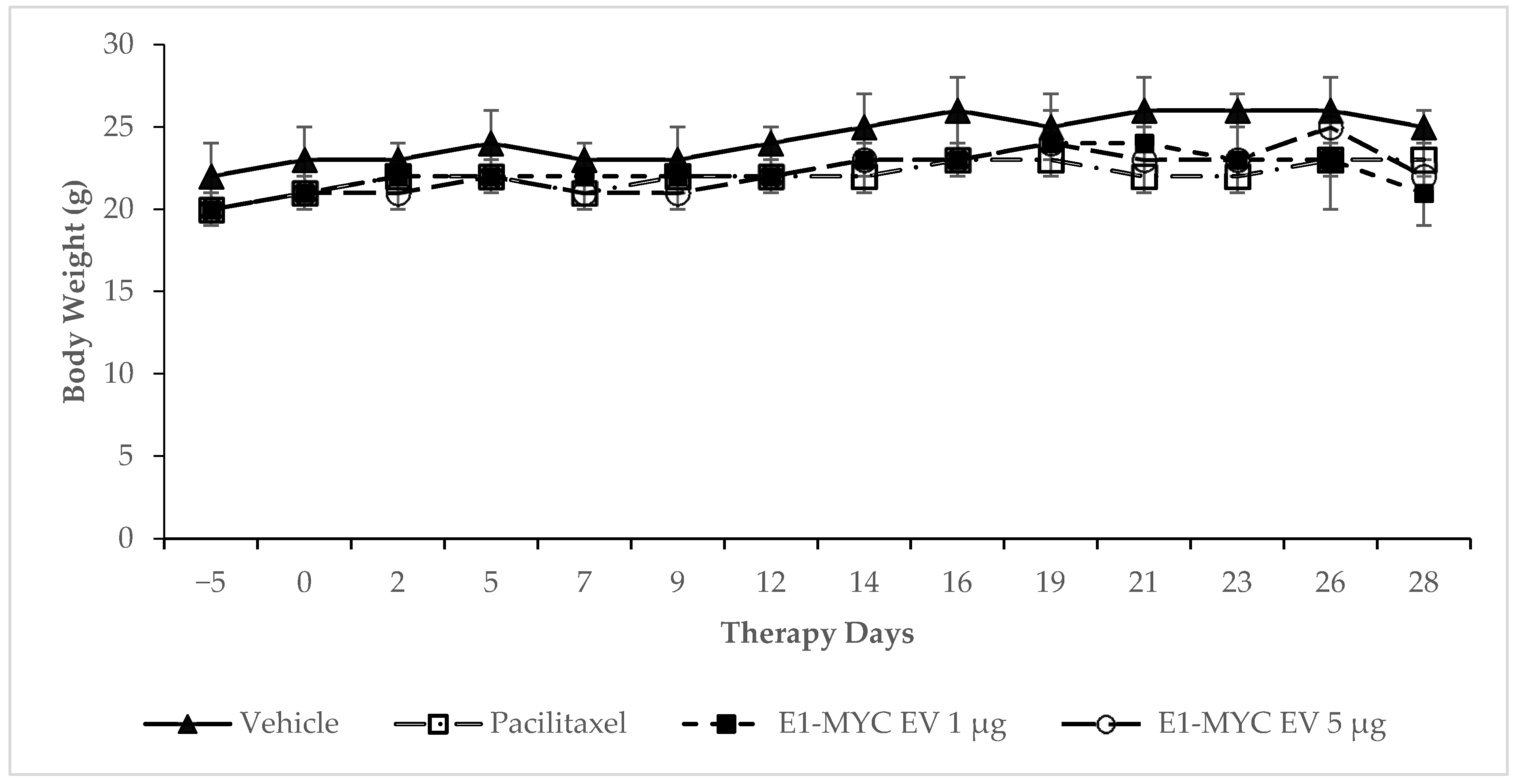
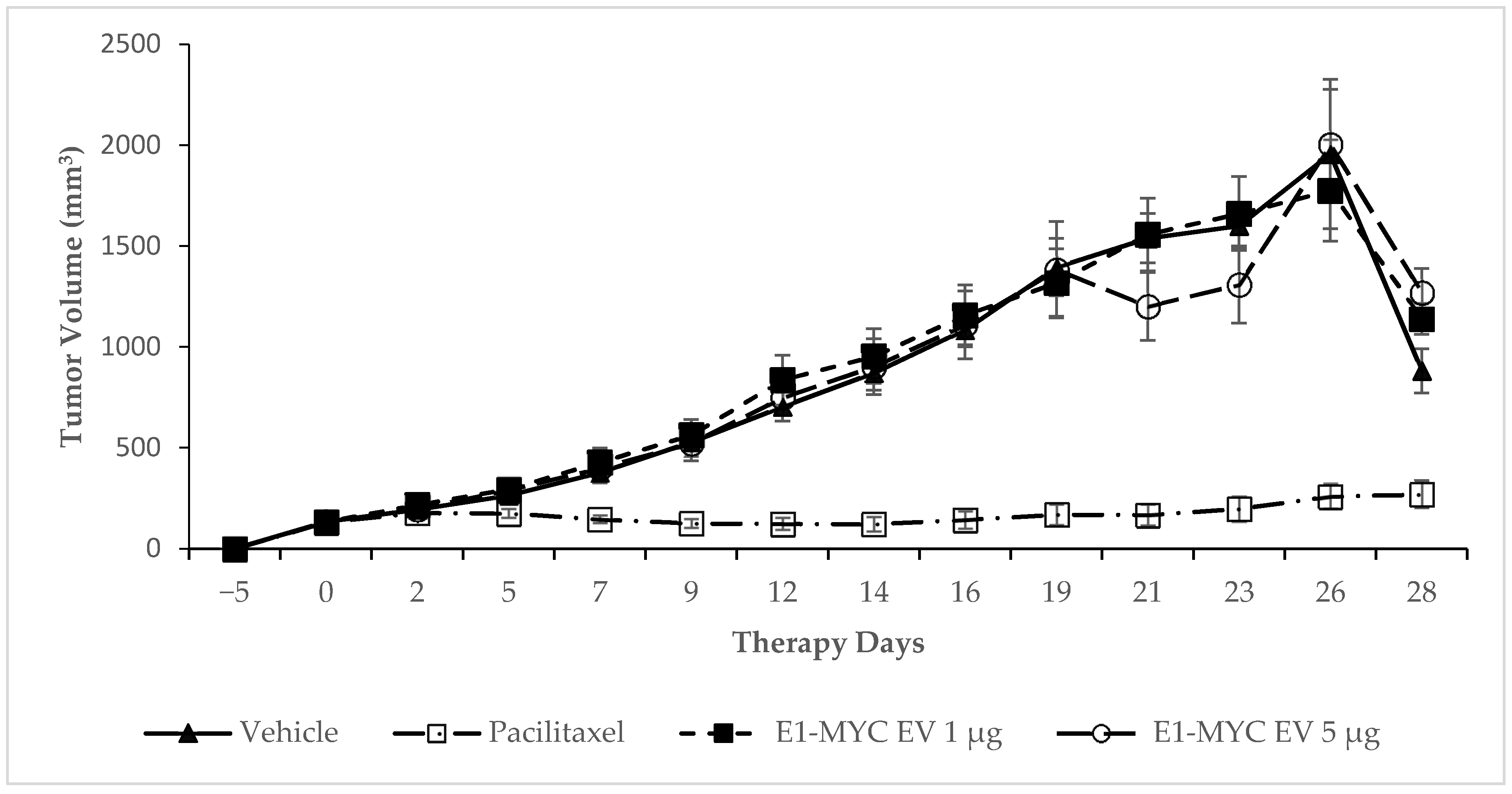
2.3. The Effects of MSC-sEV on Tumor Progression
3. Discussion
4. Materials and Methods
4.1. Culture of MSC and Preparation of MSC-Exosome
4.2. In Vitro Tumorigenicity Assay
4.3. Cell Implantation and Histological Evaluation for Tumor Formation
4.4. Pro-Tumorigenic Activity of MSC-sEV
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Caplan, A.I.; Dennis, J.E. Mesenchymal stem cells as trophic mediators. J. Cell. Biochem. 2006, 98, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Haynesworth, S.E.; Baber, M.A.; Caplan, A.I. Cytokine expression by human marrow-derived mesenchymal progenitor cells in vitro: Effects of dexamethasone and IL-1 alpha. J. Cell Physiol. 1996, 166, 585–592. [Google Scholar] [CrossRef]
- Timmers, L.; Lim, S.K.; Arslan, F.; Armstrong, J.S.; Hoefer, I.E.; Doevendans, P.A.; Piek, J.J.; El Oakley, R.M.; Choo, A.; Lee, C.N.; et al. Reduction of myocardial infarct size by human mesenchymal stem cell conditioned medium. Stem Cell Res. 2008, 1, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.K.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; El Oakley, R.M.; et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, S.; Grange, C.; Deregibus, M.C.; Calogero, R.A.; Saviozzi, S.; Collino, F.; Morando, L.; Busca, A.; Falda, M.; Bussolati, B.; et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J. Am. Soc. Nephrol. 2009, 20, 1053–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witwer, K.W.; Van Balkom, B.W.; Bruno, S.; Choo, A.; Dominici, M.; Gimona, M.; Hill, A.F.; De Kleijn, D.; Koh, M.; Lai, R.C.; et al. Defining mesenchymal stromal cell (MSC)-derived small extracellular vesicles for therapeutic applications. J. Extracell. Vesicles 2019, 8, 1609206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Lai, R.C.; Yeo, R.W.; Lim, S.K. Mesenchymal stem cell exosomes. Semin. Cell Dev. Biol. 2015, 40, 82–88. [Google Scholar] [CrossRef]
- Lai, R.C.; Tan, S.S.; Yeo, R.W.Y.; Choo, A.B.H.; Reiner, A.T.; Su, Y.; Shen, Y.; Fu, Z.; Alexander, L.; Sze, S.K.; et al. MSC secretes at least 3 EV types each with a unique permutation of membrane lipid, protein and RNA. J. Extracell. Vesicles 2016, 5, 29828. [Google Scholar] [CrossRef]
- Tan, S.S.; Yin, Y.; Lee, T.; Lai, R.C.; Yeo, R.W.Y.; Zhang, B.; Choo, A.; Lim, S.K. Therapeutic MSC exosomes are derived from lipid raft microdomains in the plasma membrane. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef]
- Reiner, A.T.; Witwer, K.W.; Van Balkom, B.W.M.; De Beer, J.; Brodie, C.; Corteling, R.L.; Gabrielsson, S.; Gimona, M.; Ibrahim, A.G.; De Kleijn, D.; et al. Concise Review: Developing Best-Practice Models for the Therapeutic Use of Extracellular Vesicles. Stem Cells Transl. Med. 2017, 6, 1730–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.S.; Arslan, F.; Yin, Y.; Tan, S.S.; Lai, R.C.; Choo, A.B.H.; Padmanabhan, J.; Lee, C.N.; De Kleijn, D.P.V.; Lim, S.K. Enabling a robust scalable manufacturing process for therapeutic exosomes through oncogenic immortalization of human ESC-derived MSCs. J. Transl. Med. 2011, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Lai, R.C.; Tan, S.S.; Teh, B.J.; Sze, S.K.; Arslan, F.; De Kleijn, D.P.; Choo, A.; Lim, S.K. Proteolytic Potential of the MSC Exosome Proteome: Implications for an Exosome-Mediated Delivery of Therapeutic Proteasome. Int. J. Proteom. 2012, 2012, 971907. [Google Scholar] [CrossRef] [Green Version]
- Lai, R.C.; Yeo, R.W.Y.; Tan, S.S.; Zhang, B.; Yin, Y.; Sze, N.S.K.; Choo, A.; Lim, S.K. Mesenchymal Stem Cell Exosomes: The Future MSC-Based Therapy? In Mesenchymal Stem Cell Therapy; Chase, L.G., Vemuri, M.C., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 39–61. [Google Scholar]
- Chen, T.S.; Lai, R.C.; Lee, M.M.; Choo, A.B.H.; Lee, C.N.; Lim, S.K. Mesenchymal stem cell secretes microparticles enriched in pre-microRNAs. Nucleic Acids Res. 2009, 38, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Accarie, A.; L’Homme, B.; Benadjaoud, M.A.; Lim, S.K.; Guha, C.; Benderitter, M.; Tamarat, R.; Sémont, A. Extracellular vesicles derived from mesenchymal stromal cells mitigate intestinal toxicity in a mouse model of acute radiation syndrome. Stem Cell Res. Ther. 2020, 11, 371. [Google Scholar] [CrossRef]
- Rangan, S.R. A new human cell line (FaDu) from a hypopharyngeal carcinoma. Cancer 1972, 29, 117–121. [Google Scholar] [CrossRef]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Long, H.J. Paclitaxel (Taxol): A novel anticancer chemotherapeutic drug. Mayo Clin. Proc. 1994, 69, 341–345. [Google Scholar] [CrossRef]
- Vilalta, M.; Dégano, I.R.; Bago, J.R.; Gould, D.; Dos Santos, M.V.F.; García-Arranz, M.; Ayats, R.; Fuster, C.; Chernajovsky, Y.; García-Olmo, D.; et al. Biodistribution, long-term survival, and safety of human adipose tissue-derived mesenchymal stem cells transplanted in nude mice by high sensitivity non-invasive bioluminescence imaging. Stem Cells Dev. 2008, 17, 993–1003. [Google Scholar] [CrossRef]
- Lopez-Iglesias, P.; Blázquez-Martínez, A.; Fernández-Delgado, J.; Regadera, J.; Nistal, M.; De Miguel, M.P. Short and long term fate of human AMSC subcutaneously injected in mice. World J. Stem Cells 2011, 3, 53–62. [Google Scholar] [CrossRef]
- Von Bahr, L.; Batsis, I.; Moll, G.; Hägg, M.; Szakos, A.; Sundberg, B.; Uzunel, M.; Ringden, O.; Le Blanc, K. Analysis of tissues following mesenchymal stromal cell therapy in humans indicates limited long-term engraftment and no ectopic tissue formation. Stem Cells 2012, 30, 1575–1578. [Google Scholar] [CrossRef]
- MacIsaac, Z.M.; Shang, H.; Agrawal, H.; Yang, N.; Parker, A.; Katz, A.J. Long-term in-vivo tumorigenic assessment of human culture-expanded adipose stromal/stem cells. Exp. Cell Res. 2012, 318, 416–423. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, Z.; Chi, Y.; Zhang, Q.; Xu, F.; Yang, Z.; Meng, L.; Yang, S.; Mao, A.; Zhang, J.; et al. Long-term cultured mesenchymal stem cells frequently develop genomic mutations but do not undergo malignant transformation. Cell Death Dis. 2013, 4, e950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheers, I.; Lombard, C.; Paganelli, M.; Campard, D.; Najimi, M.; Gala, J.-L.; Decottignies, A.; Sokal, E. Human umbilical cord matrix stem cells maintain multilineage differentiation abilities and do not transform during long-term culture. PLoS ONE 2013, 8, e71374. [Google Scholar] [CrossRef]
- Simonsen, J.L.; Rosada, C.; Serakinci, N.; Justesen, J.; Stenderup, K.; Rattan, S.I.; Jensen, T.G.; Kassem, M. Telomerase expression extends the proliferative life-span and maintains the osteogenic potential of human bone marrow stromal cells. Nat. Biotechnol. 2002, 20, 592–596. [Google Scholar] [CrossRef]
- Liu, T.M.; Ng, W.M.; Tan, H.S.; Vinitha, D.; Yang, Z.; Fan, J.B.; Zou, Y.; Hui, J.H.; Lee, E.H.; Lim, B. Molecular basis of immortalization of human mesenchymal stem cells by combination of p53 knockdown and human telomerase reverse transcriptase overexpression. Stem Cells Dev. 2013, 22, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Skarn, M.; Noordhuis, P.; Wang, M.-Y.; Veuger, M.; Kresse, S.H.; Egeland, E.V.; Micci, F.; Namløs, H.M.; Håkelien, A.-M.; Olafsrud, S.M.; et al. Generation and characterization of an immortalized human mesenchymal stromal cell line. Stem Cells Dev. 2014, 23, 2377–2389. [Google Scholar] [CrossRef] [Green Version]
- Bruno, S.; Collino, F.; Deregibus, M.C.; Grange, C.; Tetta, C.; Camussi, G. Microvesicles derived from human bone marrow mesenchymal stem cells inhibit tumor growth. Stem Cells Dev. 2013, 22, 758–771. [Google Scholar] [CrossRef]
- Lee, J.K.; Park, S.-R.; Jung, B.-K.; Jeon, Y.-K.; Lee, Y.-S.; Kim, M.-K.; Kim, Y.-G.; Jang, J.-Y.; Kim, C.-W. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS ONE 2013, 8, e84256. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Ju, G.-Q.; Du, T.; Zhu, Y.-J.; Liu, G.-H. Microvesicles derived from human umbilical cord Wharton’s jelly mesenchymal stem cells attenuate bladder tumor cell growth in vitro and in vivo. PLoS ONE 2013, 8, e61366. [Google Scholar]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.-T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542–1555. [Google Scholar] [CrossRef]
- Rodini, C.O.; Da Silva, P.B.G.; Assoni, A.F.; Carvalho, V.M.; Okamoto, O.K. Mesenchymal stem cells enhance tumorigenic properties of human glioblastoma through independent cell-cell communication mechanisms. Oncotarget 2018, 9, 24766–24777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, R.C.; Arslan, F.; Tan, S.S.; Tan, B.; Choo, A.; Lee, M.M.; Chen, T.S.; Teh, B.J.; Eng, J.K.L.; Sidik, H.; et al. Derivation and characterization of human fetal MSCs: An alternative cell source for large-scale production of cardioprotective microparticles. J. Mol. Cell. Cardiol. 2010, 48, 1215–1224. [Google Scholar] [CrossRef]
- Sze, S.K.; de Kleijn, D.P.V.; Lai, R.C.; Tan, E.K.W.; Zhao, H.; Yeo, K.S.; Low, T.Y.; Lian, Q.; Lee, C.N.; Mitchell, W.; et al. Elucidating the secretion proteome of human embryonic stem cell-derived mesenchymal stem cells. Mol. Cell. Proteom. 2007, 6, 1680–1689. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Parameter | E1-MYC | HT-1080 | MRC-5 |
|---|---|---|---|
| No. of animals | 10 | 10 | 10 |
| Lesion observed | |||
| -injection site | 0 | 10 | 0 |
| -axillary lymph | 0 | 1 | 0 |
| -lungs | 0 | 1 | 0 |
| -spleen | 0 | 0 | 0 |
| -liver | 0 | 0 | 0 |
| -kidney | 0 | 0 | 0 |
| -additional site | 0 | 4 | 0 |
| Group | No. Mice | Test Material | Dose (mg/kg) | ROA | Frequency |
|---|---|---|---|---|---|
| 1 | 8 | Vehicle | 200 µL/per mouse | IP | Daily for 4 weeks |
| 2 | 8 | Paclitaxel | 15 mg/kg | IV | Twice per week |
| 3 | 8 | E1-MYC exosomes | 1 µg/per dose | IP | Daily for 4 weeks |
| 4 | 8 | E1-MYC exosomes | 5 µg/per dose | IP | Daily for 4 weeks |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, T.T.; Lai, R.C.; Padmanabhan, J.; Sim, W.K.; Choo, A.B.H.; Lim, S.K. Assessment of Tumorigenic Potential in Mesenchymal-Stem/Stromal-Cell-Derived Small Extracellular Vesicles (MSC-sEV). Pharmaceuticals 2021, 14, 345. https://doi.org/10.3390/ph14040345
Tan TT, Lai RC, Padmanabhan J, Sim WK, Choo ABH, Lim SK. Assessment of Tumorigenic Potential in Mesenchymal-Stem/Stromal-Cell-Derived Small Extracellular Vesicles (MSC-sEV). Pharmaceuticals. 2021; 14(4):345. https://doi.org/10.3390/ph14040345
Chicago/Turabian StyleTan, Thong Teck, Ruenn Chai Lai, Jayanthi Padmanabhan, Wei Kian Sim, Andre Boon Hwa Choo, and Sai Kiang Lim. 2021. "Assessment of Tumorigenic Potential in Mesenchymal-Stem/Stromal-Cell-Derived Small Extracellular Vesicles (MSC-sEV)" Pharmaceuticals 14, no. 4: 345. https://doi.org/10.3390/ph14040345
APA StyleTan, T. T., Lai, R. C., Padmanabhan, J., Sim, W. K., Choo, A. B. H., & Lim, S. K. (2021). Assessment of Tumorigenic Potential in Mesenchymal-Stem/Stromal-Cell-Derived Small Extracellular Vesicles (MSC-sEV). Pharmaceuticals, 14(4), 345. https://doi.org/10.3390/ph14040345