
Computational Molecular Docking and Simulation-Based Assessment of Anti-Inflammatory Properties of Nyctanthes arbor-tristis Linn Phytochemicals

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
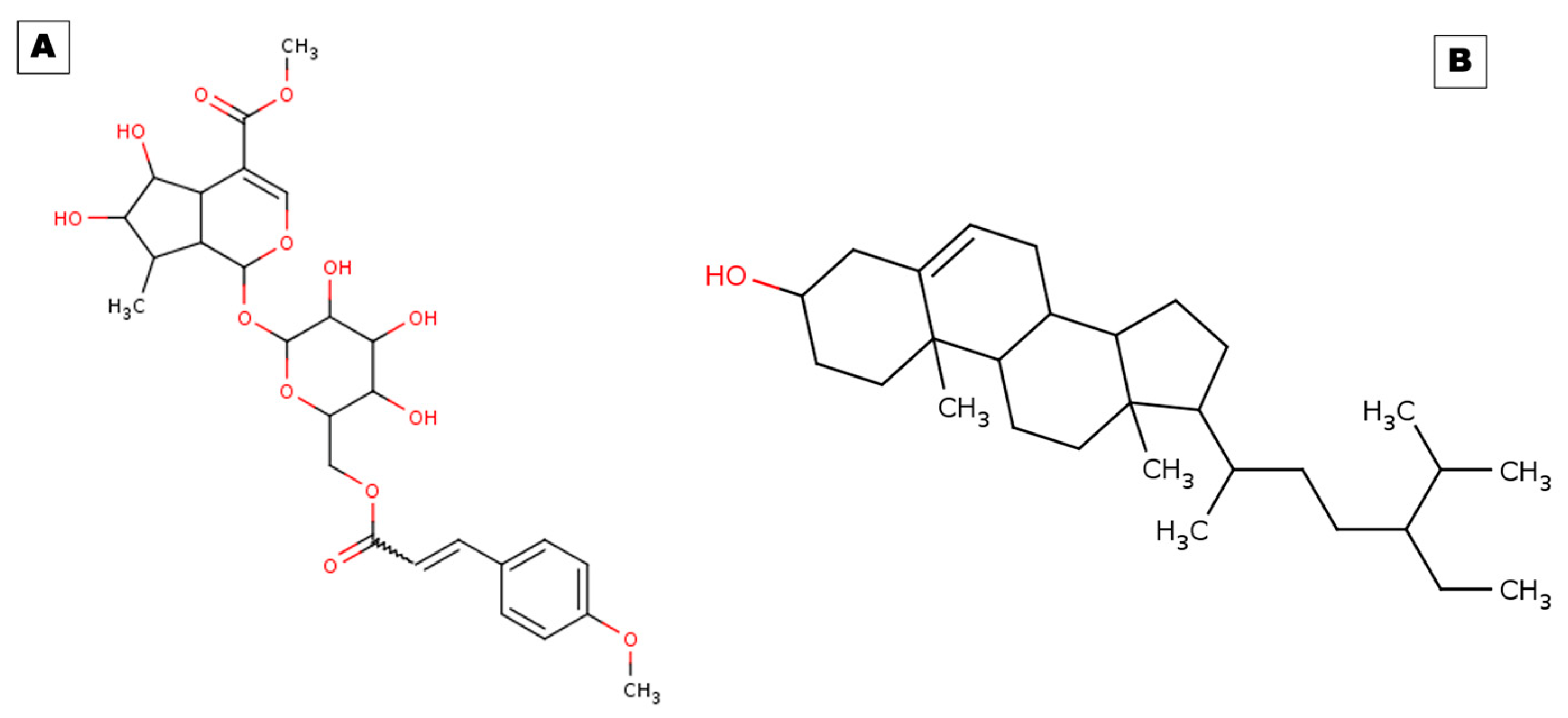
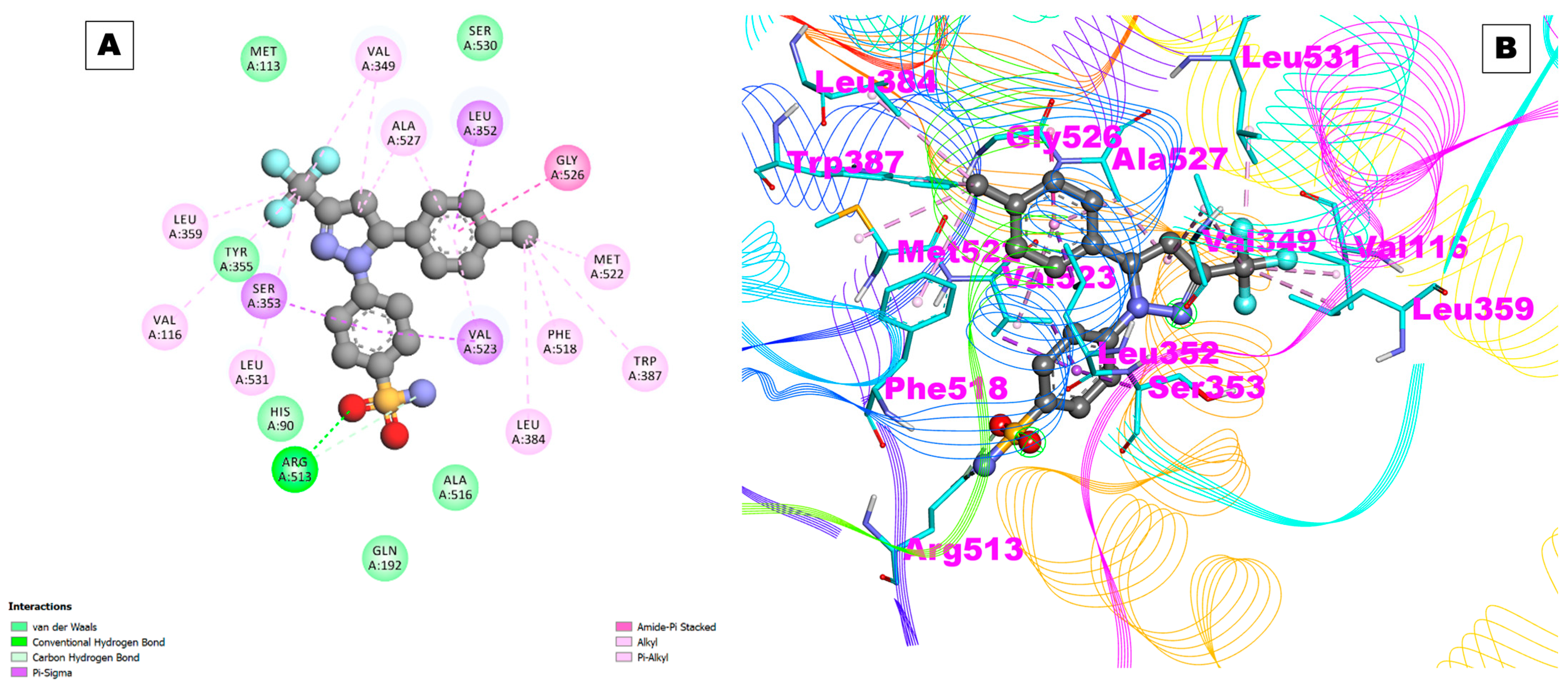
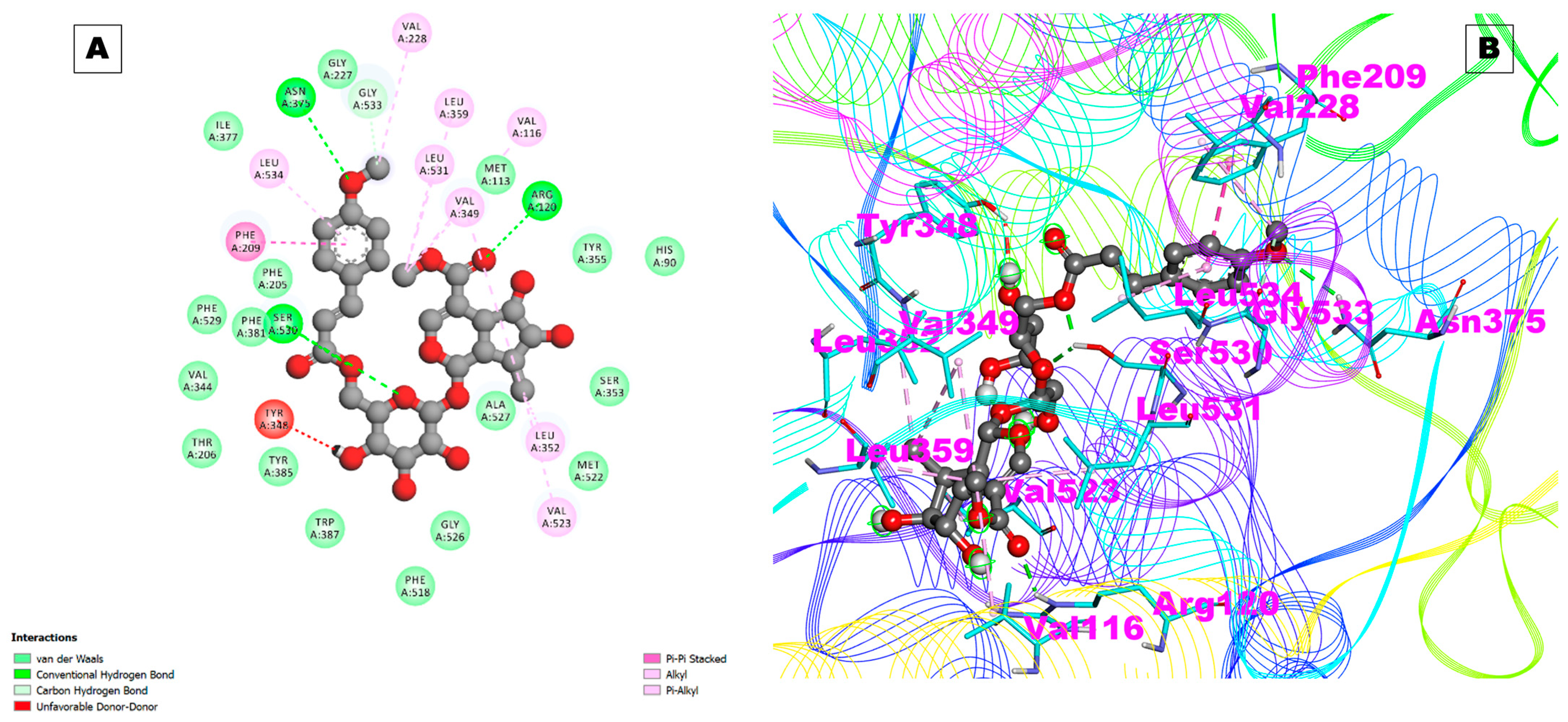
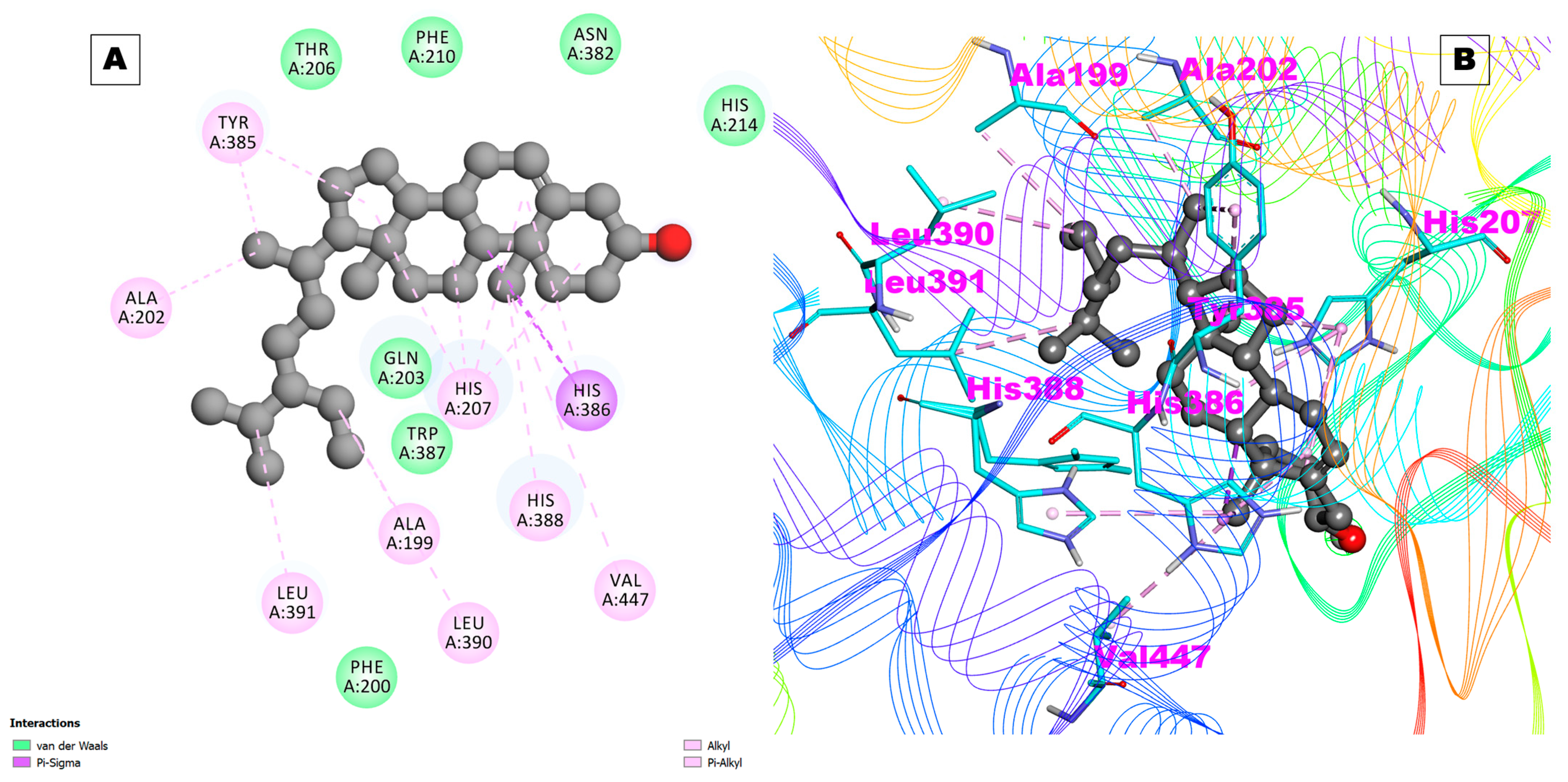
2.1. Docking Results
2.2. ADMET Results
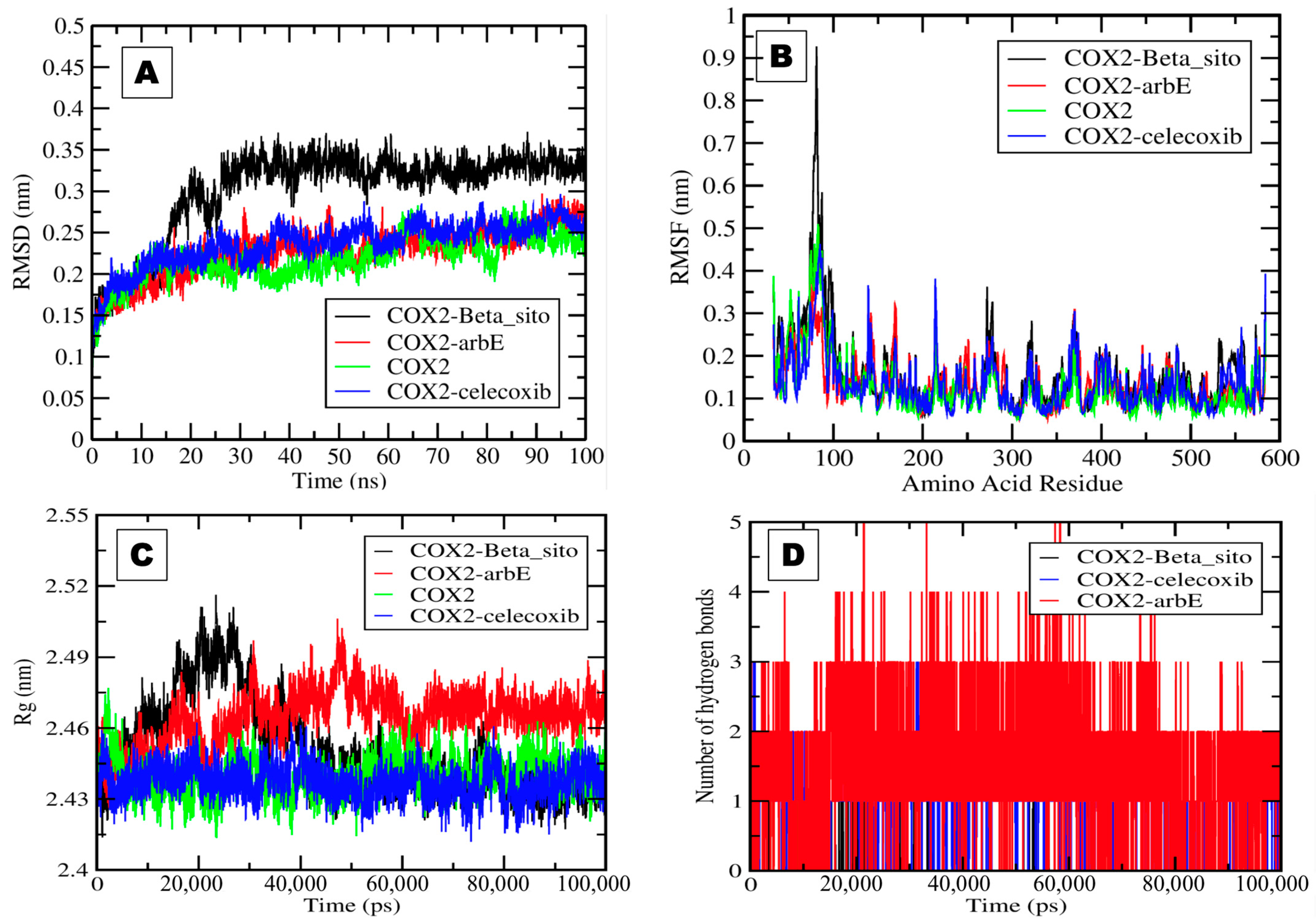
2.3. Molecular Dynamics and Simulation Analysis
2.4. MM-PBSA Results
3. Materials and Methods
3.1. Ligand Preparation
3.2. Receptor Preparation
3.3. AutoDock 4.2 Tool Receptor–Ligand Docking
3.4. Drug-Likeness and ADMET
3.5. MDS

3.6. Molecular Mechanics-Poisson–Boltzmann Surface Area (MM-PBSA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sana, T.; Khan, M.; Siddiqui, B.S.; Baig, T.A.; Jabeen, A.; Begum, S.; Hadda, T.B.; Shah, L. Anti-inflammatory and urease inhibitory iridoid glycosides from Nyctanthes arbor-tristis Linn. J. Ethnopharmacol. 2024, 319, 117368. [Google Scholar] [CrossRef]
- Chakraborty, R.; De, S.D. A Brief Overview on the Health Benefits of Nyctanthes arbor-tristis Linn.-A Wonder of Mother Nature. Indo Glob. J. Pharm. Sci. 2022, 12, 197–204. [Google Scholar] [CrossRef]
- Barua, A.; Junaid, M.; Shamsuddin, T.; Alam, M.S.; Mouri, N.J.; Akter, R.; Sharmin, T.; Hosen, S. Nyctanthes arbor-tristis Linn.: A Review on its Traditional Uses, Phytochemistry, Pharmacological Activities, and Toxicity. Curr. Tradit. Med. 2023, 9, 10–22. [Google Scholar]
- Alisherovna, K.M.; Rustamovich, T.D.; Baxtiyorovich, U.J.; Sobirovna, S.M. Diabetes Mellitus and Hyperglycemia in Patients with Rheumatoid Arthritis. Tex. J. Med. Sci. 2022, 13, 99–103. [Google Scholar]
- Gahtori, R.; Tripathi, A.H.; Chand, G.; Pande, A.; Joshi, P.; Rai, R.C.; Upadhyay, S.K. Phytochemical Screening of Nyctanthes arbor-tristis Plant Extracts and Their Antioxidant and Antibacterial Activity Analysis. Appl. Biochem. Biotechnol. 2023, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Pamuk, F.; Kantarci, A. Inflammation as a link between periodontal disease and obesity. Periodontology 2000 2022, 90, 186–196. [Google Scholar] [CrossRef]
- Pundir, S.; Gautam, G.K.; Zaidi, S. A Review on Pharmacological Activity of Nyctanthes arbor-tristis. Res. J. Pharmacogn. Phytochem. 2022, 14, 69–72. [Google Scholar] [CrossRef]
- Mendie, L.E.; Hemalatha, S. Bioactive Compounds from Nyctanthes arbor tristis Linn as potential inhibitors of janus kinases (JAKs) involved in rheumatoid arthritis. Appl. Biochem. Biotechnol. 2023, 195, 314–330. [Google Scholar] [CrossRef]
- Saxena, R.; Gupta, B.; Saxena, K.; Singh, R.; Prasad, D. Study of anti-inflammatory activity in the leaves of Nyctanthes arbor tristis Linn.—An Indian medicinal plant. J. Ethnopharmacol. 1984, 11, 319–330. [Google Scholar] [CrossRef]
- Mittal, I.; Sarvanan, K.; Singh, A. Formulation and Evaluation of anti-osteoarthritic and anti-inflammatory activity of Nyctanthes arbor tristis Linn as Emulgel. Int. J. Pharm. Res. Appl. 2023, 8, 1788–1798. [Google Scholar]
- Kim, M.-S.; Kim, S.-H. Inhibitory effect of astragalin on expression of lipopolysaccharide-induced inflammatory mediators through NF-κB in macrophages. Arch. Pharmacal Res. 2011, 34, 2101–2107. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Abuzinadah, M.F.; Alkreathy, H.M.; Kutbi, H.I.; Shaik, N.A.; Ahmad, V.; Saleem, S.; Husain, A. A novel polyherbal formulation containing thymoquinone attenuates carbon tetrachloride-induced hepatorenal injury in a rat model. Asian Pac. J. Trop. Biomed. 2020, 10, 147–155. [Google Scholar] [CrossRef]
- Altemimi, A.; Lakhssassi, N.; Baharlouei, A.; Watson, D.G.; Lightfoot, D.A. Phytochemicals: Extraction, isolation, and identification of bioactive compounds from plant extracts. Plants 2017, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Dinore, J.M.; Patil, H.S.; Dobhal, B.S.; Farooqui, M. Phytochemical analysis by GC-MS, LC-MS complementary approaches and antimicrobial activity investigation of Vigna unguiculata (L.) Walp. leaves. Nat. Prod. Res. 2022, 36, 5631–5637. [Google Scholar] [CrossRef] [PubMed]
- Yatoo, M.; Gopalakrishnan, A.; Saxena, A.; Parray, O.R.; Tufani, N.A.; Chakraborty, S.; Tiwari, R.; Dhama, K.; Iqbal, H. Anti-inflammatory drugs and herbs with special emphasis on herbal medicines for countering inflammatory diseases and disorders-a review. Recent Pat. Inflamm. Allergy Drug Discov. 2018, 12, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Osei Akoto, C.; Acheampong, A.; Boakye, Y.D.; Naazo, A.A.; Adomah, D.H. Anti-inflammatory, antioxidant, and anthelmintic activities of Ocimum basilicum (Sweet Basil) fruits. J. Chem. 2020, 2020, 2153534. [Google Scholar] [CrossRef]
- Gandhi, Y.; Kumar, R.; Grewal, J.; Rawat, H.; Mishra, S.K.; Kumar, V.; Shakya, S.K.; Jain, V.; Babu, G.; Sharma, P. Advances in anti-inflammatory medicinal plants and phytochemicals in the management of arthritis: A comprehensive review. Food Chem. Adv. 2022, 1, 100085. [Google Scholar] [CrossRef]
- Vaou, N.; Stavropoulou, E.; Voidarou, C.; Tsigalou, C.; Bezirtzoglou, E. Towards advances in medicinal plant antimicrobial activity: A review study on challenges and future perspectives. Microorganisms 2021, 9, 2041. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, Y.; Liu, M.; Qin, X.; Yu, X.; Zhao, H.; Li, X.; Li, W. COX-2 is required to mediate crosstalk of ROS-dependent activation of MAPK/NF-κB signaling with pro-inflammatory response and defense-related NO enhancement during challenge of macrophage-like cell line with Giardia duodenalis. PLoS Neglected Trop. Dis. 2022, 16, e0010402. [Google Scholar] [CrossRef]
- Zakiyah, W.; Wibowo, S.P.S.; Elyyana, N.; Darmawan, S.A.N.; Lestari, S.A.; Sa’diyyah, N.; Malau, J.; Mulki, M.A. Literature Review: Study of molecular mechanism level of NSAID class of drugs as COX-2 inhibitors. J. EduHealth 2022, 13, 572–580. [Google Scholar]
- Muthal, A.P.; Kulkarni, R.; Kumar, D.; Bagul, C.; Mukherjee-Kandhare, A.A.; Kandhare, A.D.; Ambavade, S.D.; Wagh, V.; Bodhankar, S.L. Cyclic adenosine monophosphate: Recent and future perspectives on various diseases. J. Appl. Pharm. Sci. 2022, 12, 001–015. [Google Scholar]
- Orhan, I.E.; Rauf, A.; Saleem, M.; Khalil, A.A. Natural Molecules as Talented Inhibitors of Nucleotide Pyrophosphatases/Phosphodiesterases (PDEs). Curr. Top. Med. Chem. 2022, 22, 209–228. [Google Scholar] [CrossRef] [PubMed]
- Paroli, M.; Spadea, L.; Caccavale, R.; Spadea, L.; Paroli, M.P.; Nante, N. The role of Interleukin-17 in juvenile idiopathic arthritis: From pathogenesis to treatment. Medicina 2022, 58, 1552. [Google Scholar] [CrossRef] [PubMed]
- Bi, Z.; Zhang, W.; Yan, X. Anti-inflammatory and immunoregulatory effects of icariin and icaritin. Biomed. Pharmacother. 2022, 151, 113180. [Google Scholar] [CrossRef] [PubMed]
- Arockia Babu, M.; Shukla, R.; Nath, C.; Kaskhedikar, S. Synthesis and biological evaluation of ester derivatives of indomethacin as selective COX-2 inhibitors. Med. Chem. Res. 2012, 21, 2223–2228. [Google Scholar] [CrossRef]
- Lin, D.; Xu, X.; Chen, L.; Chen, L.; Deng, M.; Chen, J.; Ren, Z.; Lei, L.; Wang, J.; Deng, J. Supramolecular nanofiber of indomethacin derivative confers highly cyclooxygenase-2 (COX-2) selectivity and boosts anti-inflammatory efficacy. J. Control. Release 2023, 364, 272–282. [Google Scholar] [CrossRef]
- Kulesza, A.; Paczek, L.; Burdzinska, A. The role of COX-2 and PGE2 in the regulation of immunomodulation and other functions of mesenchymal stromal cells. Biomedicines 2023, 11, 445. [Google Scholar] [CrossRef]
- Jogpal, V.; Sanduja, M.; Dutt, R.; Garg, V.; Tinku. Advancement of nanomedicines in chronic inflammatory disorders. Inflammopharmacology 2022, 30, 355–368. [Google Scholar] [CrossRef]
- Rashed, K. Beta-sitosterol medicinal properties: A review article. J. Sci. Innov. Technol 2020, 9, 208–212. [Google Scholar]
- Saeidnia, S.; Manayi, A.; Gohari, A.R.; Abdollahi, M. The story of beta-sitosterol-a review. Eur. J. Med. Plants 2014, 4, 590. [Google Scholar] [CrossRef]
- Jin, W.; Dong, C. IL-17 cytokines in immunity and inflammation. Emerg. Microbes Infect. 2013, 2, e60. [Google Scholar] [CrossRef] [PubMed]
- Tracey, D.; Klareskog, L.; Sasso, E.H.; Salfeld, J.G.; Tak, P.P. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 2008, 117, 244–279. [Google Scholar] [CrossRef] [PubMed]
- Banner, D.W.; D’Arcy, A.; Janes, W.; Gentz, R.; Schoenfeld, H.-J.; Broger, C.; Loetscher, H.; Lesslauer, W. Crystal structure of the soluble human 55 kd TNF receptor-human TNFβ complex: Implications for TNF receptor activation. Cell 1993, 73, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, Q.; Ding, L.; Song, S.; Li, Y.; Shi, L.; Wang, T.; Zhao, D.; Wang, Z.; Li, X. Therapeutic effects and molecular mechanisms of bioactive compounds against respiratory diseases: Traditional chinese medicine theory and high-frequency use. Front. Pharmacol. 2021, 12, 734450. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.E.; Haider, M.K. Hydrogen Bonds in Proteins: Role and Strength. In Encyclopedia of Life Sciences (ELS); John Wiley & Sons, Ltd.: Chichester, UK, 2010. [Google Scholar]
- Kim, S. Exploring chemical information in PubChem. Curr. Protoc. 2021, 1, e217. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Duarte, J.M.; Dutta, S.; Fayazi, M.; Feng, Z. RCSB Protein Data Bank: Celebrating 50 years of the PDB with new tools for understanding and visualizing biological macromolecules in 3D. Protein Sci. 2022, 31, 187–208. [Google Scholar] [CrossRef]
- Abuzinadah, M.F.; Ahmad, V.; Al-Thawdi, S.; Zakai, S.A.; Jamal, Q.M.S. Exploring the Binding Interaction of Active Compound of Pineapple against Foodborne Bacteria and Novel Coronavirus (SARS-CoV-2) Based on Molecular Docking and Simulation Studies. Nutrients 2022, 14, 3045. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks III, C.L.; Mackerell Jr, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Shakya, S.; Khan, I.M.; Shakya, B.; Siddique, Y.H.; Varshney, H.; Jyoti, S. Protective effect of the newly synthesized and characterized charge transfer (CT) complex against arecoline induced toxicity in third-instar larvae of transgenic Drosophila melanogaster (hsp70-lacZ) Bg 9: Experimental and theoretical mechanistic insights. J. Mater. Chem. B 2023, 11, 1262–1278. [Google Scholar] [CrossRef]
- Lucido, M.J.; Orlando, B.J.; Vecchio, A.J.; Malkowski, M.G. Crystal structure of aspirin-acetylated human cyclooxygenase-2: Insight into the formation of products with reversed stereochemistry. Biochemistry 2016, 55, 1226–1238. [Google Scholar] [CrossRef]
- Biovia, D. Discovery Studio Visualizer Version 21.1.0.20298, San Diego. 2021. Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 19 December 2023).
- Bitencourt-Ferreira, G.; Pintro, V.O.; de Azevedo, W.F. Docking with autodock4. In Docking Screens for Drug Discovery; Humana: New York, NY, USA, 2019; pp. 125–148. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using autodock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Jamal, Q.M.S.; Khan, M.I.; Alharbi, A.H.; Ahmad, V.; Yadav, B.S. Identification of Natural Compounds of the Apple as Inhibitors against Cholinesterase for the Treatment of Alzheimer’s Disease: An In Silico Molecular Docking Simulation and ADMET Study. Nutrients 2023, 15, 1579. [Google Scholar] [CrossRef]
- Malik, M.S.; Faazil, S.; Alsharif, M.A.; Sajid Jamal, Q.M.; Al-Fahemi, J.H.; Banerjee, A.; Chattopadhyay, A.; Pal, S.K.; Kamal, A.; Ahmed, S.A. Antibacterial Properties and Computational Insights of Potent Novel Linezolid-Based Oxazolidinones. Pharmaceuticals 2023, 16, 516. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. Platinum: A database of experimentally measured effects of mutations on structurally defined protein–ligand complexes. Nucleic Acids Res. 2015, 43, D387–D391. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Alhomrani, M.; Alsanie, W.F.; Alamri, A.S.; Alyami, H.; Habeeballah, H.; Alkhatabi, H.A.; Felimban, R.I.; Haynes, J.M.; Shakya, S.; Raafat, B.M. Enhancing the antipsychotic effect of risperidone by increasing its binding affinity to serotonin receptor via picric acid: A molecular dynamics simulation. Pharmaceuticals 2022, 15, 285. [Google Scholar] [CrossRef]
- Gupta, S.; Tiwari, N.; Verma, J.; Waseem, M.; Subbarao, N.; Munde, M. Estimation of a stronger heparin binding locus in fibronectin domain III 14 using thermodynamics and molecular dynamics. RSC Adv. 2020, 10, 20288–20301. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Homol. Model. Methods Protoc. 2012, 857, 231–257. [Google Scholar]
- Kuzmanic, A.; Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Turner, P. XMGRACE, Version 5.1; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptors | PDB ID | Binding Energy (kcal/mol) | Inhibition Constant (Ki) | Hydrogen Bond Details | Hydrogen Bond Length (Angstrom) | Residues Involved in Hydrophobic Interaction |
|---|---|---|---|---|---|---|
| Control (Celecoxib) | Cox-2 | −9.29 | 155.52 nM | A:ARG513:HH11—:UNK0:O | 2.04571 | Val116,Leu359,Tyr335,Ser353,Leu531,His90,Gln192,ALa516,Leu384,Val523,Phe518,Trp387,Met522,Gly526,Leu352,Ala527,Val349,Ser530,Met113 |
| A:ARG513:CD—:UNK0:O | 3.00729 | |||||
| A:ARG513:CD—:UNK0:N | 3.62419 | |||||
| COX-1 | 6Y3C | −3.05 | 5.81 mM | A:TRP387:HN—:UNK1:O36 | 1.64895 | Ala199,Ala202,Gln203,Thr206,His207,Phe210,Phe381,Asn382,Tyr385,His386,Trp387,His388,Leu390,Met391,Ile444 |
| UNK1:H49—A:ASN382:O | 2.56551 | |||||
| UNK1:H50—A:ASN382:OD1 | 2.53999 | |||||
| UNK1:H71—A:TYR385:O | 1.92435 | |||||
| UNK1:H70—A:TYR385:O | 2.23812 | |||||
| A:HIS388:CA—:UNK1:O23 | 3.75406 | |||||
| UNK1:C40—A:PHE210:O | 3.26066 | |||||
| COX-2 | 5F1A | −10.26 | 30.07 nM | A:ARG120:HE—:UNK1:O38 | 2.11946 | Val116,Arg120,Phe205,Phe209,Gly227,Val228,Tyr348,Val349,Leu352,Ser353,Tyr355, Leu359,Asn375,Ile377,Phe381,Tyr385,Trp387,Phe518,Met522,Val523,Gly526,Ala527,Phe529,Ser530,Leu531,Gly533,Leu534 |
| A:ASN375:HD22—:UNK1:O32 | 2.79305 | |||||
| A:SER530:HG—:UNK1:O19 | 2.24954 | |||||
| A:SER530:HG—:UNK1:O21 | 1.89473 | |||||
| UNK1:C33—A:GLY533:O | 2.89113 | |||||
| PDE4 | 2QYK | −9.00 | 251.54 nM | A:HIS416:HE2—:UNK1:O38 | 2.03017 | Asp413,His416,Ser420,Asn421,Gln422,Leu441,Glu442,His445,Asp484,Met485,Ser486,Asn533,Thr545,Ile548,Phe552,Gln555,Ser567,Met569,Gln581,Phe584 |
| A:GLN555:HE22—:UNK1:O36 | 2.64094 | |||||
| UNK1:H49—A:ASP484:OD1 | 1.91855 | |||||
| UNK1:H50—A:GLU442:OE2 | 1.92009 | |||||
| A:SER420:CB—:UNK1:O13 | 3.0371 | |||||
| UNK1:C40—A:ASP413:OD1 | 3.165 | |||||
| PDE7 | 1ZKL | −6.74 | 11.43 uM | A:HIS256:HE2—:UNK1:O34 | Tyr211,His212, His216,Asp253,His256,Gly258,Asn260,Gln261,Leu281,Glu282,His285,Ile323,Asp362,Asn365,Trp376,Ser377,Val380,Glu383,Phe384,Gln387,Pro400,Leu401,Cys402,Gln413,Phe416 | |
| A:HIS256:HE2—:UNK1:O35 | 3.03088 | |||||
| A:GLN413:HE22—:UNK1:O32 | 2.10147 | |||||
| UNK1:H69—A:ASP253:OD1 | 2.53525 | |||||
| UNK1:H71—A:GLU282:OE2 | 2.16071 | |||||
| UNK1:H49—A:GLU383:OE2 | 2.69909 | |||||
| UNK1:C40—A:PRO400:O | 2.37041 | |||||
| IL-17A | 5HI4 | −5.81 | 54.74 uM | UNK1:H50—A:VAL65:O | 1.98348 | Leu53,Tyr62,Pro63,Val65,Ile66,Trp67,Gln94,Ile96,Leu97,Val98,Leu99,Val117,Ser118,Val119 |
| UNK1:H69—A:TYR62:O | 2.4814 | |||||
| A:PRO63:CD—:UNK1:O35 | 2.97263 | |||||
| A:VAL119:CA—:UNK1:O32 | 3.01728 | |||||
| UNK1:C33—A:TRP67:O | 3.37552 | |||||
| IL-17D | Modeled from MODBASE server | −6.70 | 12.36 uM | UNK1:H69—A:VAL141:O | 2.64013 | Ala78,Arg80,Tyr96,Tyr105,Pro106,Tyr108,Leu109,Pro110,Ala112,Thr140,Val141,Val142,Ile163,Pro164,Val165 |
| UNK1:C40—A:ALA78:O | 2.95779 | |||||
| UNK1:C33—A:PRO110:O | 3.29094 | |||||
| UNK1:C33—A:PRO164:O | 3.0518 | |||||
| TNF-α | 1A8M | −4.70 | 360.94 uM | UNK1:C20—A:LEU142:O | 3.23102 | Pro20,Ala22,Gly24,Lys65,Gly66,Gln67,Asp140,Leu142,Phe144,Ala145 |
| UNK1:C33—A:GLN67:OE1 | 3.04695 | |||||
| IL-1β | 6Y8M | −4.59 | 431.16 uM | A:LYS103:HZ2—:UNK1:O7 | 2.66882 | Lys103,Asn108,Lys109,Leu110,Phe146,Thr147,Met148,Gln149,Phe150 |
| UNK1:C33—A:PHE150:O | 3.21082 | |||||
| Prostaglandin E2 | 4YHL | −7.23 | 4.98 uM | UNK1:H49—A:THR168:OG1 | 2.63075 | Ile23,Pro24,Met27,Val72,Val75,Thr76,Thr79,Tyr80,Leu99,Thr168,Trp169,Cys170,Arg316,Ser319,Val320 |
| UNK1:H50—A:THR168:O | 1.8278 | |||||
| UNK1:C40—A:CYS170:O | 3.05882 | |||||
| Prostaglandin F synthase | 1RY0 | −10.19 | 33.66 nM | A:TYR24:HN—:UNK1:O7 | 2.28139 | Thr251,Gln279,Asn280,ALA253,Arg276,Leu219,Leu236,Ala269,Leu268,Ala218,Ser221,Gly22,Tyr216,Thr23,Ser51,Lys84,His117 |
| A:SER217:HN—:UNK1:O36 | 2.31396 | |||||
| A:LYS270:HN—:UNK1:O35 | 2.95418 | |||||
| A:LYS270:HZ2—:UNK1:O23 | 2.7668 | |||||
| A:LYS270:HZ3—:UNK1:O23 | 2.84067 | |||||
| UNK1:H49—A:ASP50:OD2 | 2.45111 | |||||
| UNK1:H49—A:ASP50:O | 2.87918 | |||||
| UNK1:H50—A:TYR55:OH | 2.08453 | |||||
| UNK1:C20—A:LYS270:O | 2.79899 | |||||
| UNK1:C33—A:THR251:OG1 | 3.62729 |
| Receptors | PDB ID | Binding Energy (kcal/mol) | Inhibition Constant (Ki) | Hydrogen Bond Details | Hydrogen Bond Length (Angstrom) | Residues Involved in Hydrophobic Interaction |
|---|---|---|---|---|---|---|
| Control (Celecoxib) | Cox-2 | −9.29 | 155.52 nM | A:ARG513:HH11—:UNK0:O | 2.04571 | Val116,Leu359,Tyr335,Ser353,Leu531,His90,Gln192,ALa516,Leu384,Val523,Phe518,Trp387,Met522,Gly526,Leu352,Ala527,Val349,Ser530,Met113 |
| A:ARG513:CD—:UNK0:O | 3.00729 | |||||
| A:ARG513:CD—:UNK0:N | 3.62419 | |||||
| COX-1 | 6Y3C | −4.91 | 253.57 uM | NA | NA | Ala199,Phe200,Ala202,Gln203,Thr206,His207,Leu295,Tyr385,His386,Trp387,His388, Leu390,Met391,Tyr404,Leu408,Ile444 |
| COX-2 | 5F1A | −8.86 | 320.37 nM | NA | NA | Ala199,Ala202,Gln203,Thr206,His207,Phe210,His214, Asn382,Tyr385,His386,Trp387,His388,Leu390,Leu391 |
| PDE4 | 2QYK | −8.66 | 448.17 nM | NA | NA | Tyr371,His372,Asp413,His416,Asn421,Leu441,Glu442, His445,Thr483,Met485,Asp530,Leu531,Ile548,Phe552,Met569,Phe584,Ile588 |
| PDE7 | 1ZKL | −7.48 | 3.68 uM | A:HIS256:HE2—:UNK1:O25 | 2.68409 | Tyr211,His212,His216,His252,Asp253,His256,Leu281,Glu282,His285,Thr321,Ile323,Asp362,Val380,Phe384,Leu401,Gln413,Phe416,Leu420 |
| UNK1:H67—A:ASP253:OD1 | 2.14352 | |||||
| IL-17A | 5HI4 | −7.43 | 3.60 uM | A:TRP67:HN—:UNK1:O25 | 2.23528 | Tyr62,Pro63,Ile66,Trp67,Ile96,Leu97,Val98,Leu99,Leu112, Val117 |
| UNK1:H67—A:TRP67:O | 1.86307 | |||||
| IL-17D | Modeled from MODBASE server | −8.22 | 947.55 nM | UNK1:H67—A:VAL165:O | 2.01343 | Arg80,Arg81,Phe82,Trp94,Tyr96,Pro106,Tyr108,Pro110,Val165 |
| TNF-α | 1A8M | −7.10 | 6.20 uM | UNK1:H67—A:GLN67:OE1 | 2.0408 | Pro20,Lys65,Gly66,Gln67,Asp140,Tyr141,Leu142,Asp143,Phe144,Ala145 |
| IL-1β | 6Y8M | −6.19 | 29.24 uM | UNK1:H67—A:MET148:O | 2.23593 | Leu6,Met44,Phe46,Lys103,Glu105,Asn108,Leu110,Thr147,Met148,Gln149,Phe150 |
| Prostaglandin E2 | 4YHL | −8.58 | 514.41 nM | NA | NA | Ile23,Pro24,Met27,Thr69,Val72,Ser73,Thr76,Tyr80,Arg316,Ser319,Val320 |
| Prostaglandin F synthase | 1RY0 | −7.39 | 3.83 uM | NA | NA | Arg223,Leu236,Gly220,Leu219,Ala218,Ala269,Tyr55,Ser217,Tyr55,Tyr24,Tyr216,Gly22,Asp50,Leu268,Thr23,Lys270,Gln222,Ser221 |
| S.N0. | Ligands | Van der Wall Energy (kJ/mol) | Electrostatic Energy (kJ/mol) | Polar Salvation Energy (kJ/mol) | SASA Energy (kJ/mol) | Binding Energy (kJ/mol) |
|---|---|---|---|---|---|---|
| 1. | Arb_E | −300.730 +/−13.113 | −22.633 +/−9.119 | 47.260 +/−188.830 | −28.499 +/−0.977 | −277.602 +/−39.964 |
| 2. | Beta_sito | −232.379 +/−11.525 | −0.160 +/−2.069 | 39.170 +/−34.210 | −21.016 +/−1.008 | −214.385 +/−36.906 |
| 3. | Celecoxib | −220.267 +/−0.184 | −59.621 +/−0.124 | 103.2250 +/−0.94 | −16.972 +/−0.011 | −193.635 +/−0.573 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, V.; Khan, M.I.; Jamal, Q.M.S.; Alzahrani, F.A.; Albiheyri, R. Computational Molecular Docking and Simulation-Based Assessment of Anti-Inflammatory Properties of Nyctanthes arbor-tristis Linn Phytochemicals. Pharmaceuticals 2024, 17, 18. https://doi.org/10.3390/ph17010018
Ahmad V, Khan MI, Jamal QMS, Alzahrani FA, Albiheyri R. Computational Molecular Docking and Simulation-Based Assessment of Anti-Inflammatory Properties of Nyctanthes arbor-tristis Linn Phytochemicals. Pharmaceuticals. 2024; 17(1):18. https://doi.org/10.3390/ph17010018
Chicago/Turabian StyleAhmad, Varish, Mohammad Imran Khan, Qazi Mohammad Sajid Jamal, Faisal A. Alzahrani, and Raed Albiheyri. 2024. "Computational Molecular Docking and Simulation-Based Assessment of Anti-Inflammatory Properties of Nyctanthes arbor-tristis Linn Phytochemicals" Pharmaceuticals 17, no. 1: 18. https://doi.org/10.3390/ph17010018