
Comparing Low-Dose Carvedilol Continuous Manufacturing by Solid and Liquid Feeding in Self-Emulsifying Delivery Systems via Hot Melt EXtrusion (SEDEX)

 , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
2.1. CAV Solubility, Liquid SEDDS Preparation and CAV Loading in the SEDDS
2.2. Hot Melt Extrusion (HME)
2.2.1. CAV in Preblend
2.2.2. Carvedilol in the SEDDS
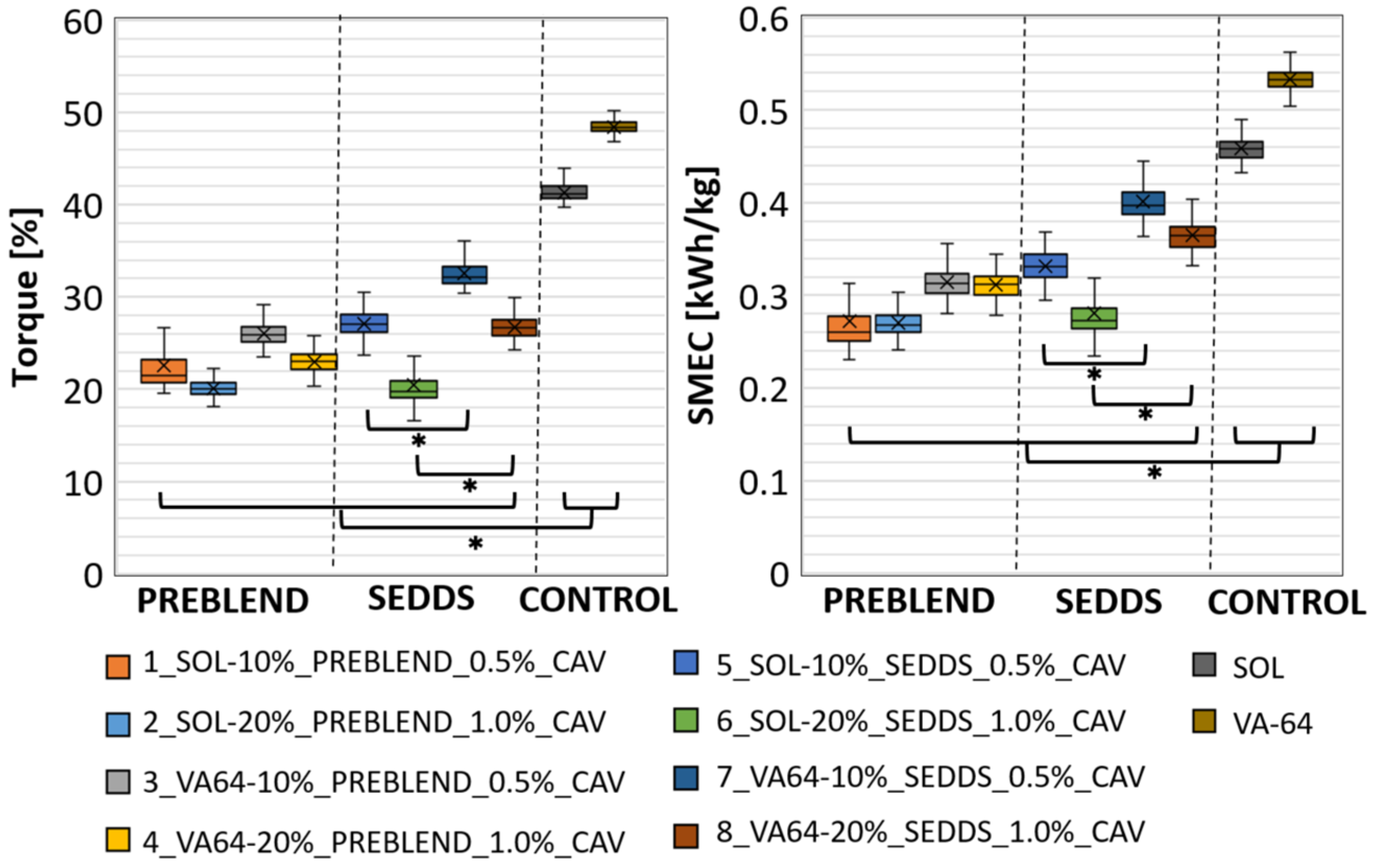
2.2.3. Comparing CAV Introduced to HME via Solid and Liquid Feeding
2.3. HME-SEDDS Solid-State Characterization
2.3.1. Organoleptic Evaluation
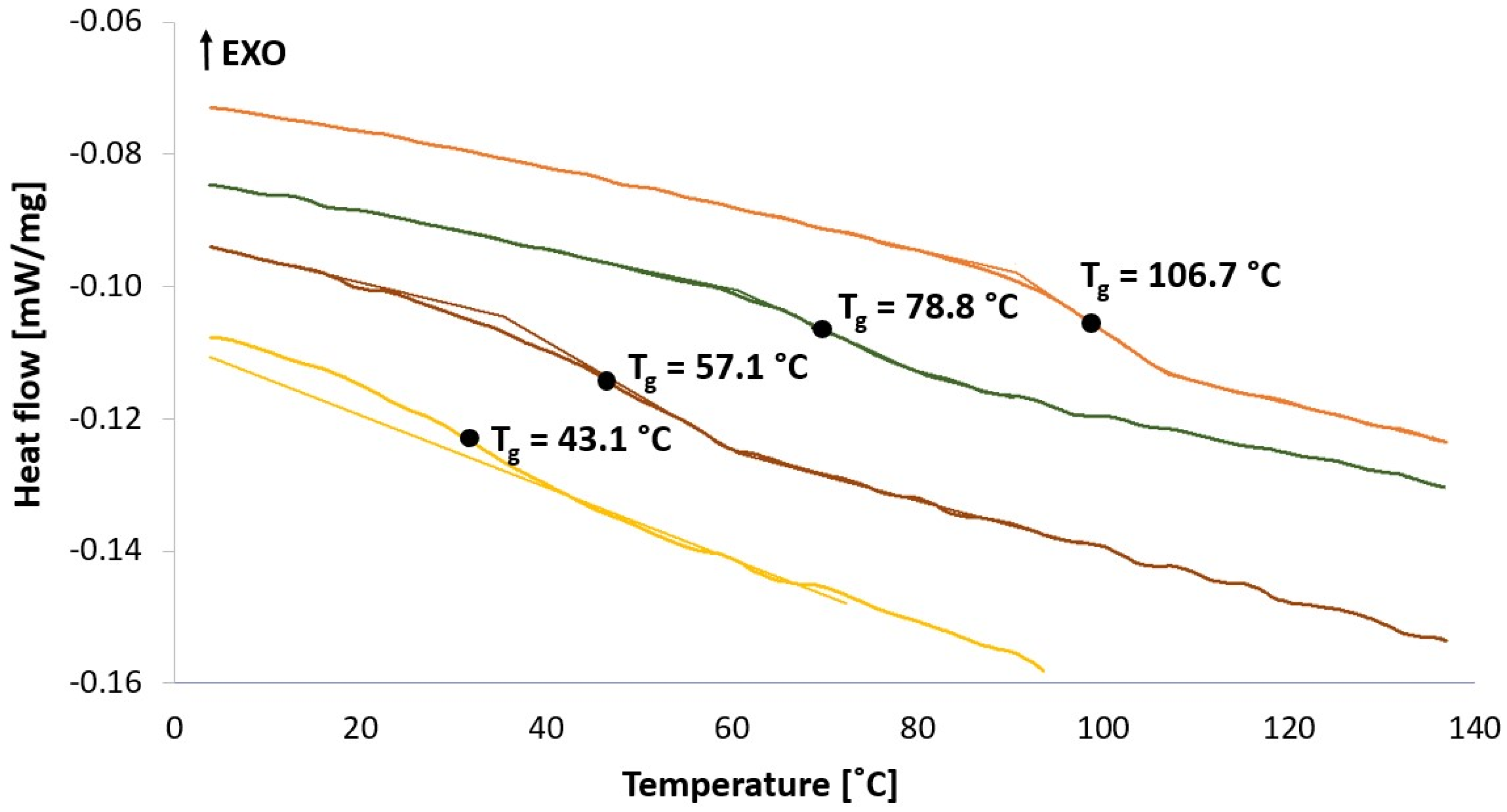
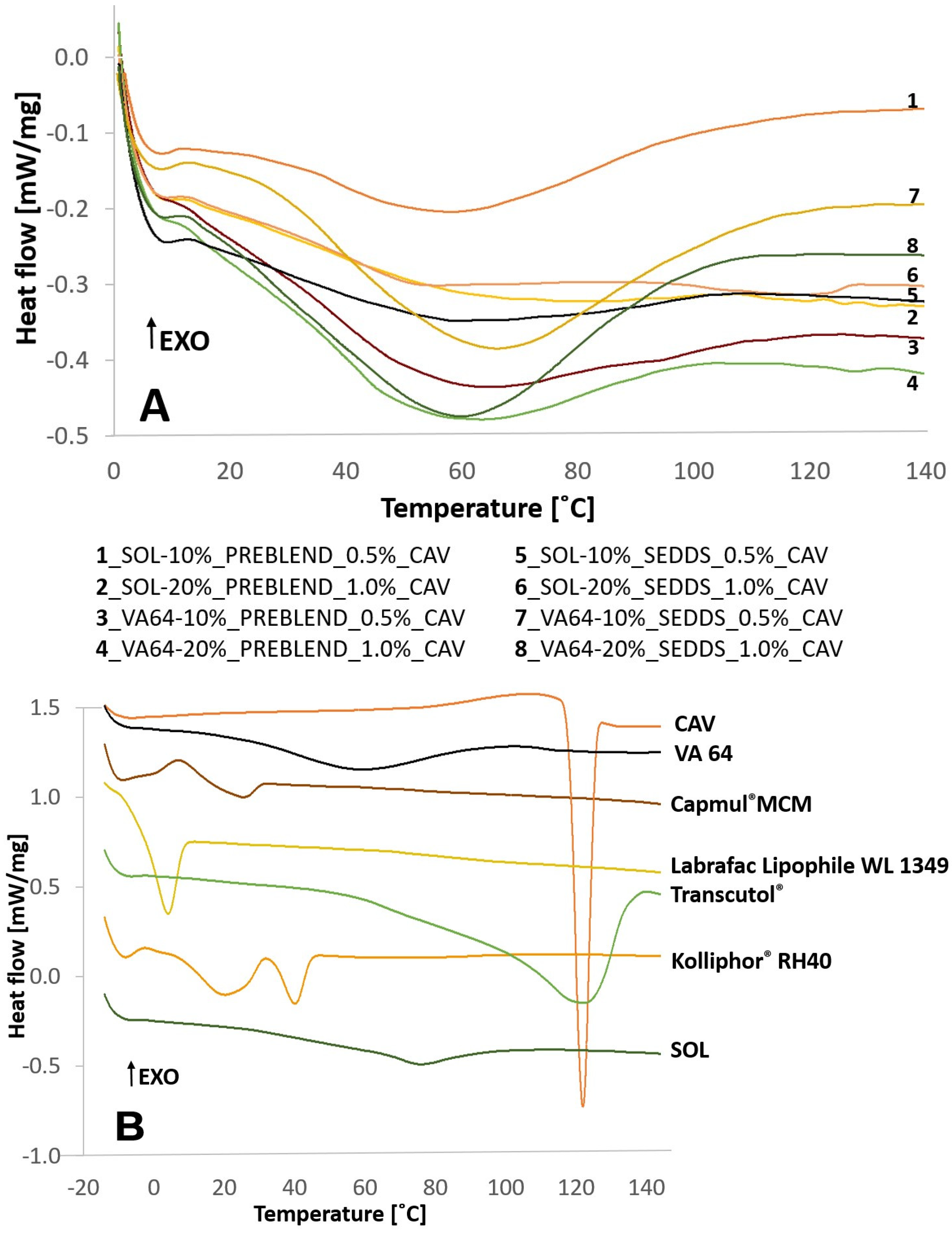
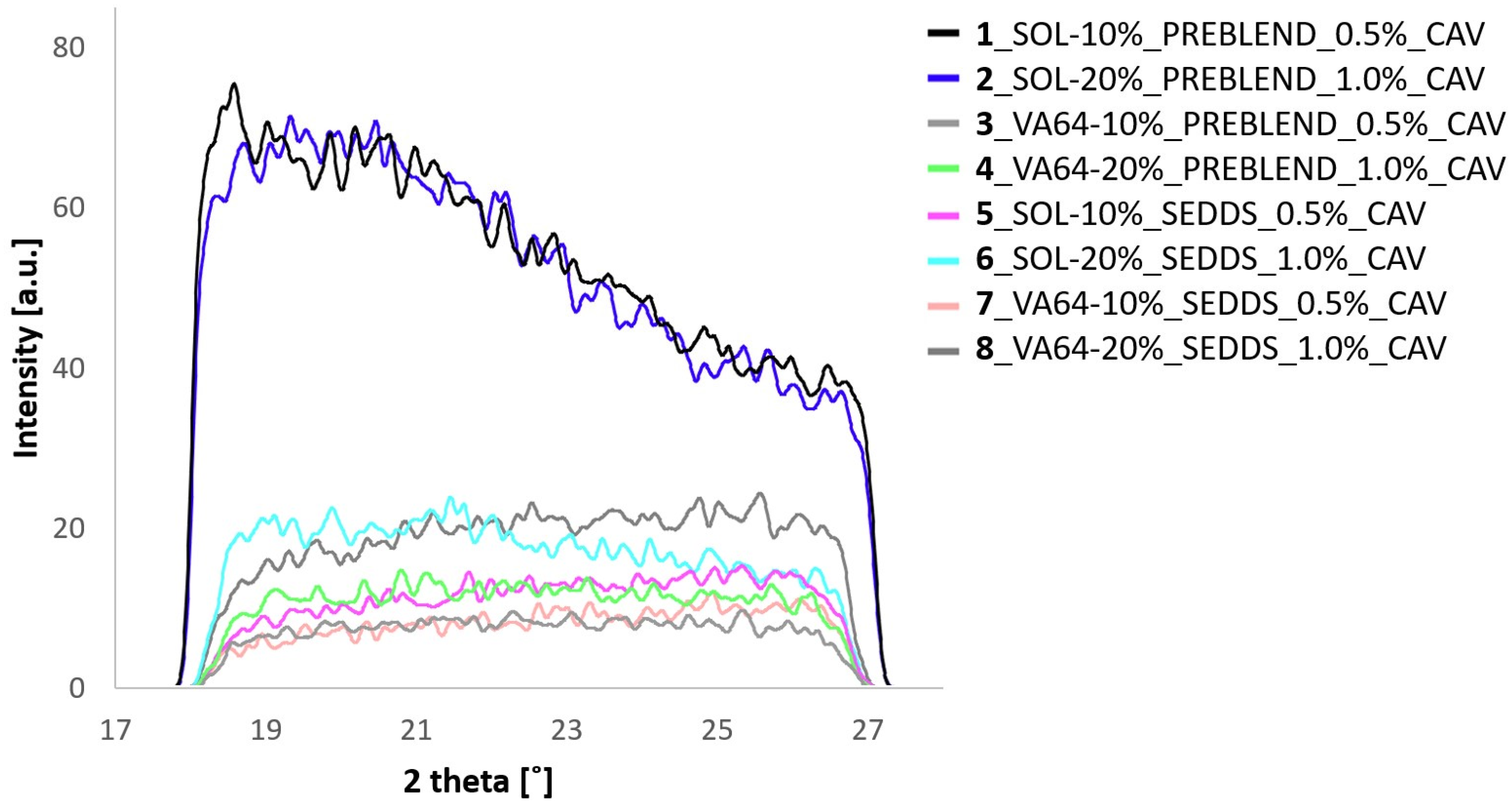
2.3.2. Differential Scanning Calorimetry (DSC) and Wide-Angle X-ray Scattering (WAXS)
2.4. In Vitro Characterization
2.4.1. Reconstitution Time
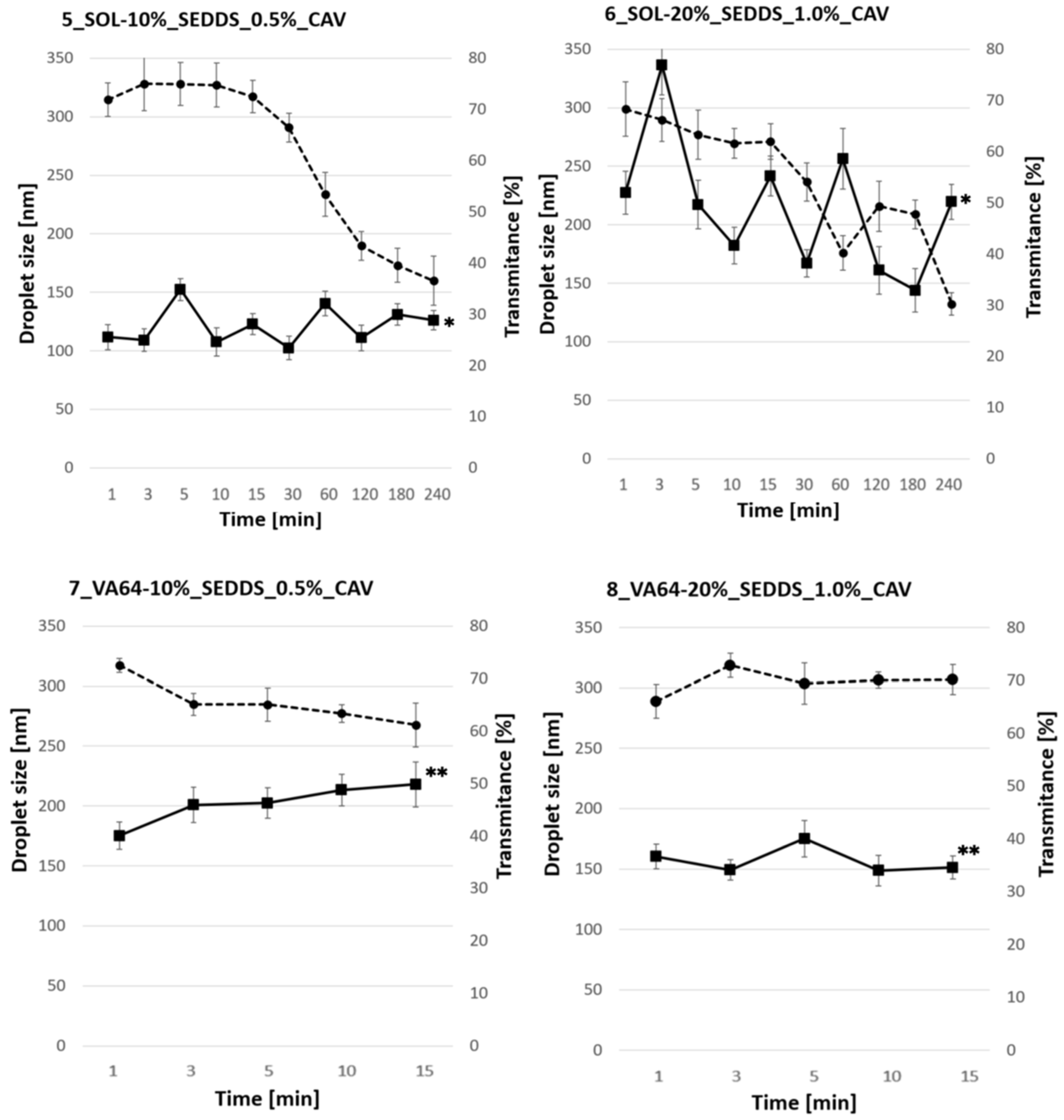
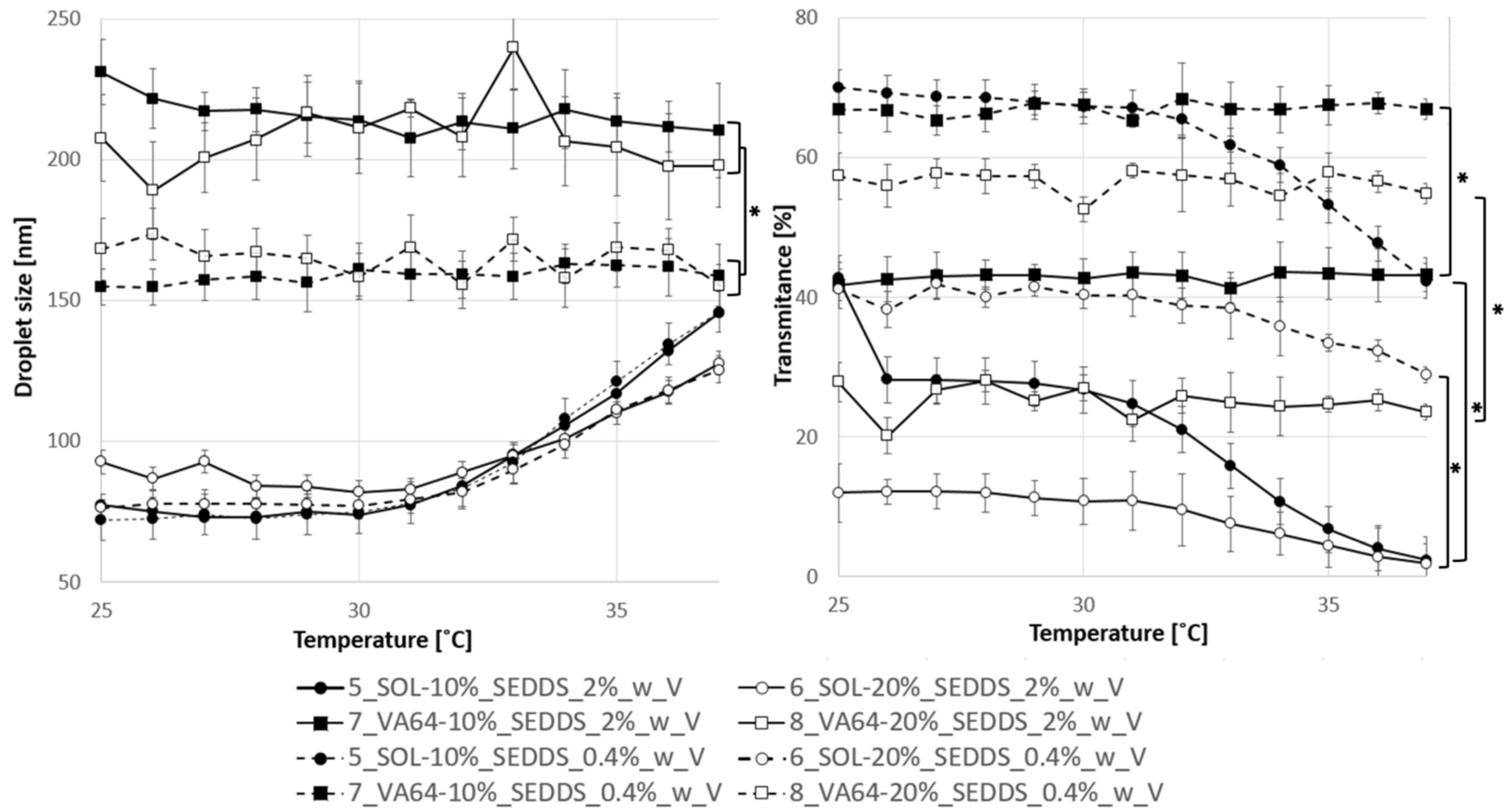
2.4.2. Emulsification Properties of HME-SEDDS
3. Materials and Methods
3.1. Materials
3.2. Carvedilol Solubility, Liquid SEDDS Preparation and Carvedilol Loading in SEDDS
3.3. Preblend Preparation
3.4. Particle Size Distribution (PSD)
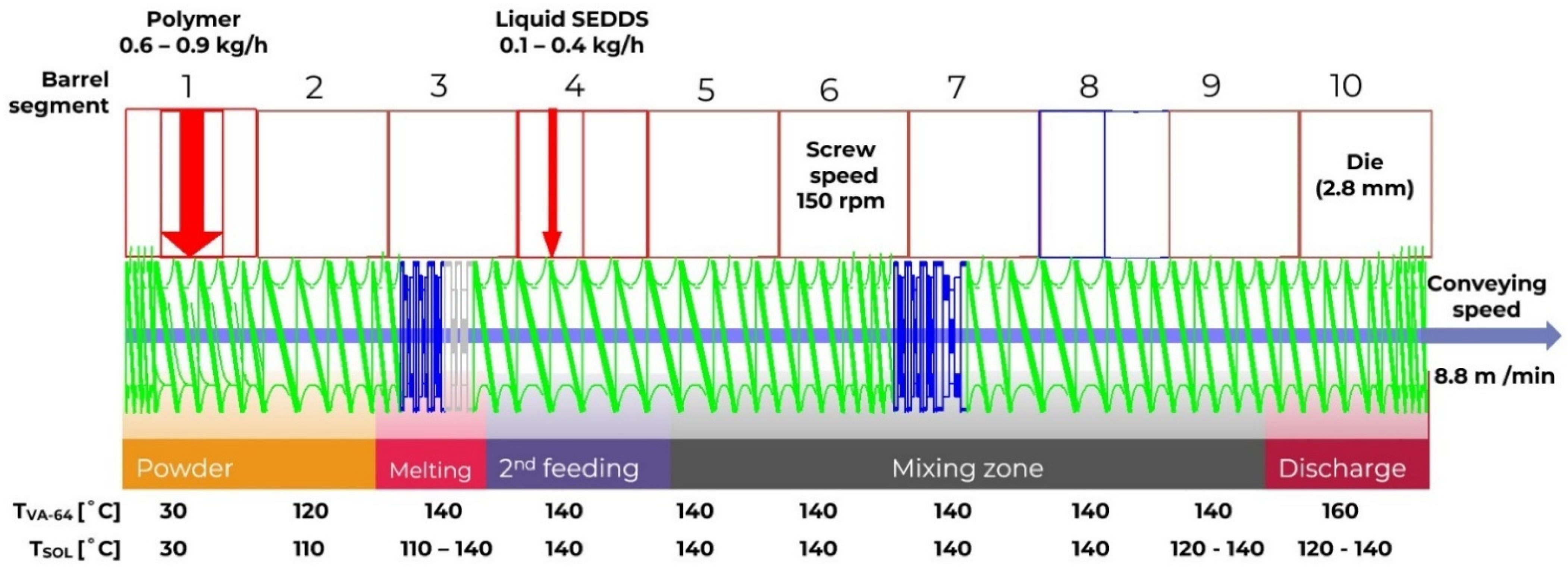
3.5. Hot Melt Extrusion
3.6. Cryomilling
3.7. Solid-State Characterization
3.7.1. Differential Scanning Calorimetry (DSC)
3.7.2. Wide-Angle X-ray Scattering (WAXS)
3.8. In Vitro Characterization
3.8.1. Reconstitution Time
3.8.2. Droplet Size, Polydispersity Index (PDI) and Transmittance
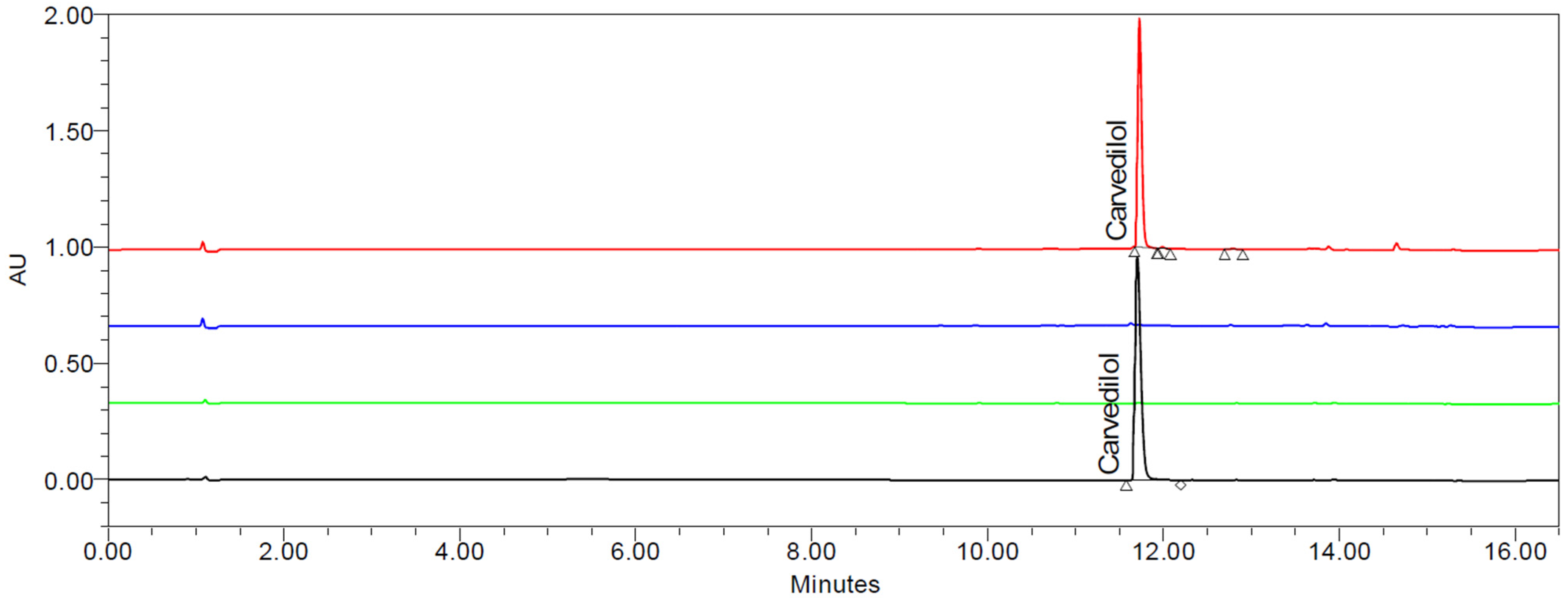
3.9. UPLC Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ASD | Amorphous solid dispersion |
| CAV | Carvedilol |
| DSC | Differential scanning calorimetry |
| HME | Hot melt extrusion |
| HME-SEDDS | Solid SEDDS prepared by hot melt extrusion |
| HPC | Hydroxy propyl cellulose |
| HPMC | Hydroxypropyl methylcellulose |
| HPMCAS | Hydroxy propyl methylcellulose-acetate/succinate |
| LBDS | Lipid-based delivery systems |
| MCC | Microcrystalline cellulose |
| PDI | Polydispersity index |
| PSD | Particle size distribution |
| RSD | Relative standard deviation |
| SEDDS | Self-emulsifying drug delivery systems |
| SMEC | Specific mechanical energy consumption |
| SOL | Soluplus® |
| SOL-HME-SEDDS | Solid SEDDS prepared by hot melt extrusion with Soluplus® |
| UPLC | Ultra-High-performance liquid chromatography |
| VA-64 | Kollidon® VA-64, copovidone |
| VA-64-HME-SEDDS | Solid SEDDS prepared by hot melt extrusion with Kollidon® VA-64 |
| WAXS | Wide-angle X-ray scattering |
References
- Kumari, L.; Choudhari, Y.; Patel, P.; Gupta, G.D.; Singh, D.; Rosenholm, J.M.; Bansal, K.K.; Kurmi, B. Das Advancement in Solubilization Approaches: A Step towards Bioavailability Enhancement of Poorly Soluble Drugs. Life 2023, 13, 1099. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Huckle, J.; Newman, J.; Reid, D.L. Trends in small molecule drug properties: A developability molecule assessment perspective. Drug Discov. Today 2022, 27, 103366. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef] [PubMed]
- Zupančič, O.; Spoerk, M.; Paudel, A. Lipid-based solubilization technology via hot melt extrusion: Promises and challenges. Expert Opin. Drug Deliv. 2022, 19, 1013–1032. [Google Scholar] [CrossRef]
- Nardin, I.; Köllner, S. Successful development of oral SEDDS: Screening of excipients from the industrial point of view. Adv. Drug Deliv. Rev. 2019, 142, 128–140. [Google Scholar] [CrossRef]
- Zupančič, O.; Partenhauser, A.; Lam, H.T.; Rohrer, J.; Bernkop-Schnürch, A. Development and in vitro characterisation of an oral self-emulsifying delivery system for daptomycin. Eur. J. Pharm. Sci. 2016, 81, 129–136. [Google Scholar] [CrossRef]
- Matić, J.; Alva, C.; Eder, S.; Reusch, K.; Paudel, A.; Khinast, J. Towards predicting the product quality in hot-melt extrusion: Pilot plant scale extrusion. Int. J. Pharm. X 2021, 3, 100084. [Google Scholar] [CrossRef]
- Silva, L.A.D.; Almeida, S.L.; Alonso, E.C.P.; Rocha, P.B.R.; Martins, F.T.; Freitas, L.A.P.; Taveira, S.F.; Cunha-Filho, M.S.S.; Marreto, R.N. Preparation of a solid self-microemulsifying drug delivery system by hot-melt extrusion. Int. J. Pharm. 2018, 541, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schmied, F.P.; Bernhardt, A.; Klein, S. Preparation of Solid Self-Nanoemulsifying Drug Delivery Systems (S-SNEDDS) by Co-Extrusion of Liquid SNEDDS and Polymeric Carriers—A New and Promising Formulation Approach to Improve the Solubility of Poorly Water-Soluble Drugs. Pharmaceuticals 2022, 15, 1135. [Google Scholar] [CrossRef]
- Raman Kallakunta, V.; Dudhipala, N.; Nyavanandi, D.; Sarabu, S.; Yadav Janga, K.; Ajjarapu, S.; Bandari, S.; Repka, M.A. Formulation and processing of solid self-emulsifying drug delivery systems (HME S-SEDDS): A single-step manufacturing process via hot-melt extrusion technology through response surface methodology. Int. J. Pharm. 2023, 641, 123055. [Google Scholar] [CrossRef]
- Zupančič, O.; Doğan, A.; Matić, J.; Kushwah, V.; Alva, C.; Spoerk, M.; Paudel, A. SEDEX—Self-Emulsifying Delivery Via Hot Melt Extrusion: A Continuous Pilot-Scale Feasibility Study. Pharmaceutics 2022, 14, 2617. [Google Scholar] [CrossRef] [PubMed]
- Llusa, M.; Mohr, S.; Baumgartner, R.; Paudel, A.; Koscher, G.; Khinast, J. Continuous low-dose feeding of highly active pharmaceutical ingredients in hot-melt extrusion. Drug Dev. Ind. Pharm. 2016, 42, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Vasoya, J.M.; Desai, H.H.; Gumaste, S.G.; Tillotson, J.; Kelemen, D.; Dalrymple, D.M.; Serajuddin, A.T.M. Development of Solid Dispersion by Hot Melt Extrusion Using Mixtures of Polyoxylglycerides With Polymers as Carriers for Increasing Dissolution Rate of a Poorly Soluble Drug Model. J. Pharm. Sci. 2019, 108, 888–896. [Google Scholar] [CrossRef]
- Singh, S.K.; Verma, P.R.P.; Razdan, B. Development and characterization of a carvedilol-loaded self-microemulsifying delivery system. Clin. Res. Regul. Aff. 2009, 26, 50–64. [Google Scholar] [CrossRef]
- Krstić, M.; Manić, L.; Martić, N.; Vasiljević, D.; Mračević, S.Đ.; Vukmirović, S.; Rašković, A. Binary polymeric amorphous carvedilol solid dispersions: In vitro and in vivo characterization. Eur. J. Pharm. Sci. 2020, 150, 105343. [Google Scholar] [CrossRef]
- Silva, L.A.D.; Cintra, E.R.; Alonso, E.C.P.; Alves, G.L.; Lima, E.M.; Taveira, S.F.; da Cunha-Filho, M.S.S.; Marreto, R.N. Selection of excipients for the development of carvedilol loaded lipid-based drug delivery systems. J. Therm. Anal. Calorim. 2017, 130, 1593–1604. [Google Scholar] [CrossRef]
- Singh, B.; Singh, R.; Bandyopadhyay, S.; Kapil, R.; Garg, B. Optimized nanoemulsifying systems with enhanced bioavailability of carvedilol. Colloids Surf. B Biointerfaces 2013, 101, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Mandić, J.; Luštrik, M.; Vrečer, F.; Gašperlin, M.; Zvonar Pobirk, A. Solidification of carvedilol loaded SMEDDS by swirling fluidized bed pellet coating. Int. J. Pharm. 2019, 566, 89–100. [Google Scholar] [CrossRef]
- Czajkowska-Kośnik, A.; Szekalska, M.; Amelian, A.; Szymańska, E.; Winnicka, K. Development and evaluation of liquid and solid self-emulsifying drug delivery systems for atorvastatin. Molecules 2015, 20, 21010–21022. [Google Scholar] [CrossRef]
- Becker, K.; Salar-Behzadi, S.; Zimmer, A. Solvent-Free Melting Techniques for the Preparation of Lipid-Based Solid Oral Formulations. Pharm. Res. 2015, 32, 1519–1545. [Google Scholar] [CrossRef]
- Guns, S.; Mathot, V.; Martens, J.A.; Van Den Mooter, G. Upscaling of the hot-melt extrusion process: Comparison between laboratory scale and pilot scale production of solid dispersions with miconazole and Kollicoat® IR. Eur. J. Pharm. Biopharm. 2012, 81, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Joyce, P.; Dening, T.J.; Meola, T.R.; Schultz, H.B.; Holm, R.; Thomas, N.; Prestidge, C.A. Solidification to improve the biopharmaceutical performance of SEDDS: Opportunities and challenges. Adv. Drug Deliv. Rev. 2019, 142, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Gumaste, S.G.; Gupta, S.S.; Serajuddin, A.T.M. Investigation of Polymer-Surfactant and Polymer-Drug-Surfactant Miscibility for Solid Dispersion. AAPS J. 2016, 18, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Thakore, S.D.; Sirvi, A.; Joshi, V.C.; Panigrahi, S.S.; Manna, A.; Singh, R.; Sangamwar, A.T.; Bansal, A.K. Biorelevant dissolution testing and physiologically based absorption modeling to predict in vivo performance of supersaturating drug delivery systems. Int. J. Pharm. 2021, 607, 120958. [Google Scholar] [CrossRef]
- Gallo, R.C.; Ferreira, A.P.G.; Castro, R.E.A.; Cavalheiro, E.T.G. Studying the thermal decomposition of carvedilol by coupled TG-FTIR. J. Therm. Anal. Calorim. 2016, 123, 2307–2312. [Google Scholar] [CrossRef]
- Thakkar, R.; Thakkar, R.; Pillai, A.; Ashour, E.A.; Repka, M.A. Systematic screening of pharmaceutical polymers for hot melt extrusion processing: A comprehensive review. Int. J. Pharm. 2020, 576, 118989. [Google Scholar] [CrossRef] [PubMed]
- Mandić, J.; Pirnat, V.; Luštrik, M.; German Ilić, I.; Vrečer, F.; Gašperlin, M.; Zvonar Pobirk, A. Solidification of SMEDDS by fluid bed granulation and manufacturing of fast drug release tablets. Int. J. Pharm. 2020, 583, 119377. [Google Scholar] [CrossRef]
- Afonso Urich, J.A.; Marko, V.; Boehm, K.; Lara Garcia, R.A.; Fedorko, A.; Salar-Behzadi, S.; Jeremic, D. Stability-Indicating UPLC-PDA-QDa Methodology for Carvedilol and Felodipine in Fixed-Dose Combinations Using AQbD Principles. Sci. Pharm. 2024, 92, 22. [Google Scholar] [CrossRef]
- Stefanini-Oresic, L. Validation of analytical procedures. Farm. Glas. 1994, 50, 97–105. [Google Scholar] [CrossRef]
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use ICH Q14: Analytical procedure development. ICH Harmon. Tripart. Guidel. 2022, 31, 65. Available online: https://database.ich.org/sites/default/files/ICH_Q14_Document_Step2_Guideline_2022_0324.pdf (accessed on 17 September 2024).
- Committee for Medicinal Products for Human Use. ICH Harmonised Guideline: Validation of analytical procedures—Q2(R2). ICH Eur. Med. Agency 2022, Step 2b, 1–39. [Google Scholar]
- Waters 996 Photodiode. Detector: Peak Purity I. What Is Peak Purity Analysis? pp. 16–17. Available online: https://www.waters.com/webassets/cms/library/docs/wpp16.pdf (accessed on 17 September 2024).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Blank SEDDS | CAR-SEDDS (5% w/w) | ||
|---|---|---|---|---|
| Concentration [w/v] | 0.4% | 2.0% | 0.4% | 2.0% |
| Droplet size [nm] | 35.7 * ± 3.1 | 36.9 * ± 4.2 | 44.7 * ± 5.4 | 49.0 * ± 4.8 |
| Polydispersity index | 0.048 * ± 0.03 | 0.052 * ± 0.04 | 0.088 * ± 0.02 | 0.101 * ± 0.05 |
| Zeta potential [mV] | −3.48 ± 0.98 | 2.59 ± 0.92 | 1.12 ± 0.96 | 0.92 ± 0.90 |
| Transmittance [%] | 73.6 ± 4.2 | 74.5 ± 2.8 | 77.3 ± 3.5 | 63.5 * ± 3.3 |
| Emulsification time [s] | 12 ± 3 | 15 ± 2 | 16 ± 3 | 15 ± 4 |
| Appearance | Bluish clear | Bluish clear | Bluish clear | Bluish clear |
| Sample | D10 | D50 | D90 | Span |
|---|---|---|---|---|
| Carvedilol | 2.9 ± 0.03 | 11.3 * ± 0.1 | 27.6 ± 0.3 | 2.2 |
| Kollidon® VA64 | 38.8 ± 0.6 | 98.9 * ± 1.5 | 171.9 ± 2.8 | 1.3 |
| Soluplus® | 216.8 ± 12.6 | 337.9 * ± 12.4 | 481.9 ± 10.0 | 0.8 |
| Sample | CAV in Preblend [%] | CAV in HME-SEDDS [%] |
|---|---|---|
| 1_SOL-10%_PREBLEND_0.5%_CAV | 96.75 ± 2.61 | 79.35 ± 0.24 |
| 2_SOL-20%_PREBLEND_1.0%_CAV | 37.04 ± 0.50 | 53.29 ± 0.11 |
| 3_VA64-10%_PREBLEND_0.5%_CAV | 76.44 ± 0.63 | 66.16 ± 0.15 |
| 4_VA64-20%_PREBLEND_1.0%_CAV | 81.04 ± 0.79 | 110.07 ± 0.48 |
| 5_SOL-10%_SEDDS_0.5%_CAV | N/A | 76.08 ± 10.47 |
| 6_SOL-20%_SEDDS_1.0%_CAV | N/A | 96.98 ± 0.70 |
| 7_VA64-10%_SEDDS_0.5%_CAV | N/A | 98.18 ± 1.85 |
| 8_VA64-20%_SEDDS_1.0%_CAV | N/A | 97.21 ± 8.83 |
| Raw Material | Description | Tg/Tm [°C] | Td [°C] | SEDDS [% w/w] | HME-SEDDS [% w/w] |
|---|---|---|---|---|---|
| Carvedilol | poorly water-soluble BCS class II drug substance | 115 [13] | 248 [25] | 5 | 0.5–1.0 |
| Labrafac® lipophile | Medium-chain Triglycerides Liquid lipid for CAV solubilization | <−5 * | >250 * | 30 | 3–6 |
| Kolliphor® RH40 | PEG-40 hydrogenated castor oil Surfactant for CAV solubilization | 16–26 * | 300 * | 30 | 3–6 |
| Capmul® MCM | Glyceryl caprylate/caprate Type I Liquid lipid for CAV solubilization | 25 [5] | 148.9 * | 30 | 3–6 |
| Transcutol® | Diethylene Glycol Monoethyl Ether Co-solvent for CAV solubilization | −80 * | 196–200 * | 10 | 1–2 |
| Kollidon® VA 64 | Vinyl pyrrolidone: mvinyl acetate 6:4 Polymer matrix for liquids SEDDS adsorbtion | 105 [26] | 270 [26] | N/A | 80–90 |
| Soluplus® | Poly(vinylcaprolactam-covinylacetate ethylene glycol)graft polymer Polymer matrix for liquids SEDDS adsorbtion | 72 °C [26] | 278 °C [26] | N/A | 80–90 |
| CAV Introduction | Sample | Polymer [%] | SEDDS * [%] | CAV Total [%] |
|---|---|---|---|---|
| PREBLEND | 1_SOL-10%_PREBLEND_0.5%_CAV | 90% Soluplus® | 10 | 0.5 |
| 2_SOL-20%_PREBLEND_1.0%_CAV | 80% Soluplus® | 20 | 1.0 | |
| 3_VA64-10%_PREBLEND_0.5%_CAV | 90% Kollidon® VA-64® | 10 | 0.5 | |
| 4_VA64-20%_PREBLEND_1.0%_CAV | 80% Kollidon® VA-64® | 20 | 1.0 | |
| SEDDS | 5_SOL-10%_SEDDS_0.5%_CAV | 90% Soluplus® | 10 | 0.5 |
| 6_SOL-20%_SEDDS_1.0%_CAV | 80% Soluplus® | 20 | 1.0 | |
| 7_VA64-10%_SEDDS_0.5%_CAV | 90% Kollidon® VA-64® | 10 | 0.5 | |
| 8_VA64-20%_SEDDS_1.0%_CAV | 80% Kollidon® VA-64® | 20 | 1.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zupančič, O.; Matić, J.; Doğan, A.; Gaggero, A.; Khinast, J.; Paudel, A. Comparing Low-Dose Carvedilol Continuous Manufacturing by Solid and Liquid Feeding in Self-Emulsifying Delivery Systems via Hot Melt EXtrusion (SEDEX). Pharmaceuticals 2024, 17, 1290. https://doi.org/10.3390/ph17101290
Zupančič O, Matić J, Doğan A, Gaggero A, Khinast J, Paudel A. Comparing Low-Dose Carvedilol Continuous Manufacturing by Solid and Liquid Feeding in Self-Emulsifying Delivery Systems via Hot Melt EXtrusion (SEDEX). Pharmaceuticals. 2024; 17(10):1290. https://doi.org/10.3390/ph17101290
Chicago/Turabian StyleZupančič, Ožbej, Josip Matić, Aygün Doğan, Alessio Gaggero, Johannes Khinast, and Amrit Paudel. 2024. "Comparing Low-Dose Carvedilol Continuous Manufacturing by Solid and Liquid Feeding in Self-Emulsifying Delivery Systems via Hot Melt EXtrusion (SEDEX)" Pharmaceuticals 17, no. 10: 1290. https://doi.org/10.3390/ph17101290