From Marine Venoms to Drugs: Efficiently Supported by a Combination of Transcriptomics and Proteomics



Abstract
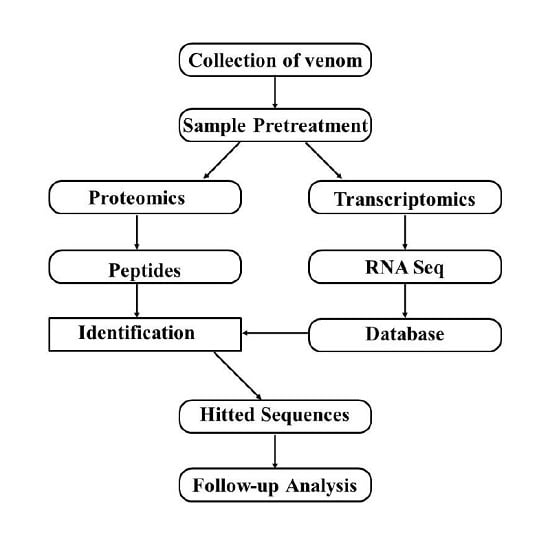
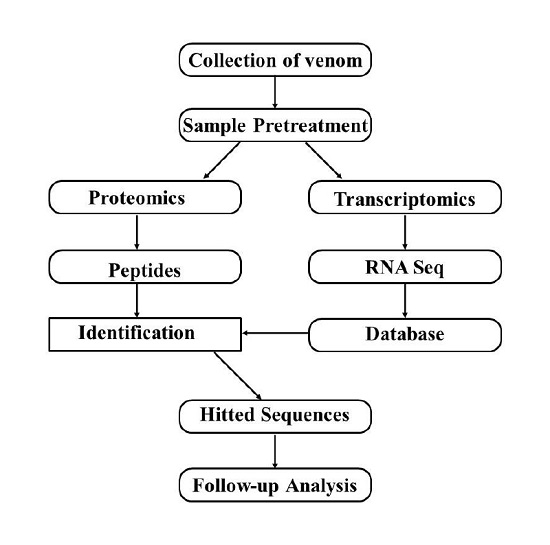
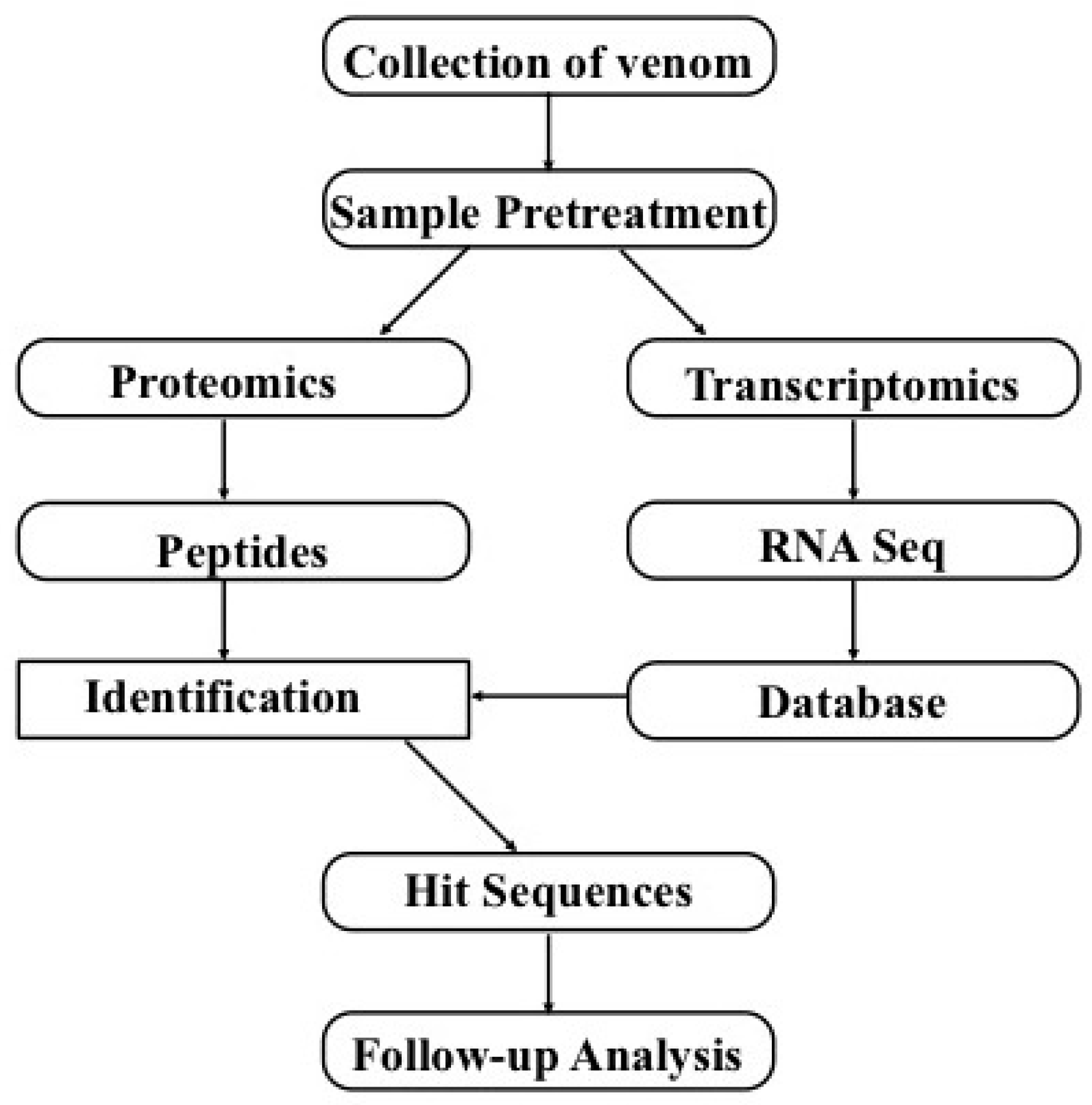
:
1. Introduction
2. Toxin Database
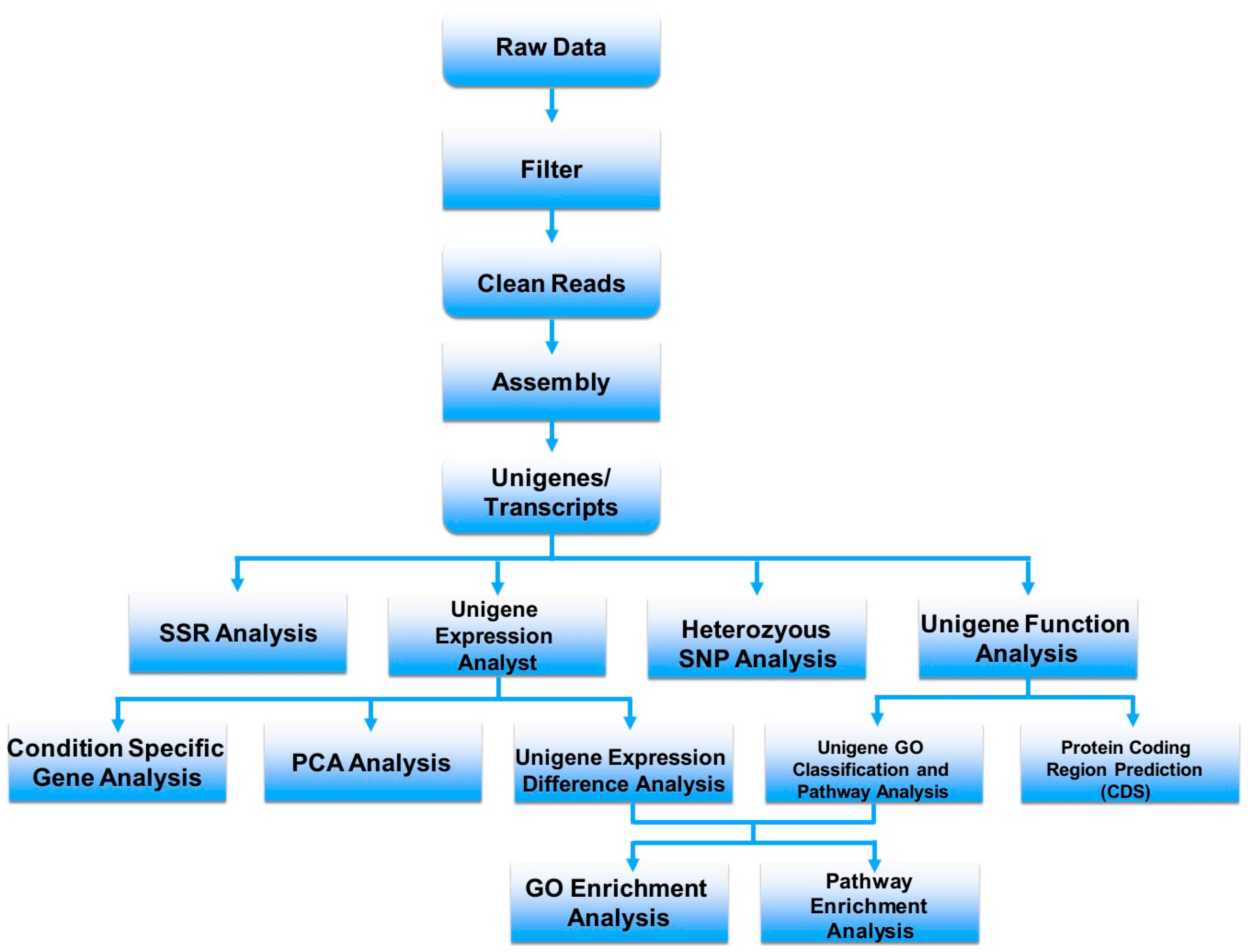
3. Venom-Gland Transcriptomics
4. Venom-Gland Proteomics
5. Combination of Transcriptomics and Proteomics
6. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Calvete, J.J. Venomics, what else? Toxicon 2012, 60, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Durban, J.; Pérez, A.; Sanz, L.; Gómez, A.; Bonilla, F.; Chacón, D.; Sasa, M.; Angulo, Y.; Gutiérrez, J.M.; Calvete, J.J. Integrated “omics” profiling indicates that mirnas are modulators of the ontogenetic venom composition shift in the central american rattlesnake, crotalus simus simus. BMC Genom. 2013, 14, 234. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Jin, A.-H.; Kaas, Q.; Jones, A.; Alewood, P.F.; Lewis, R.J. Deep venomics reveals the mechanism for expanded peptide diversity in cone snail venom. Mol. Cell. Proteom. 2013, 12, 312–329. [Google Scholar] [CrossRef] [PubMed]
- Escoubas, P.; Bosmans, F. Spider peptide toxins as leads for drug development. Expert Opin. Drug Discov. 2007, 2, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Escoubas, P.; King, G.F. Venomics as a drug discovery platform. Expert Rev. Proteom. 2009, 6, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Roelants, K.; Champagne, D.E.; Scheib, H.; Tyndall, J.D.; King, G.F.; Nevalainen, T.J.; Norman, J.A.; Lewis, R.J.; Norton, R.S.; et al. The toxicogenomic multiverse: Convergent recruitment of proteins into animal venoms. Annu. Rev. Genom. Hum. Genet. 2009, 10, 483–511. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Li, X.; Lin, Z.; Ruan, Z.; Wang, M.; Liu, J.; Tong, T.; Li, J.; Huang, Y.; Wen, B.; et al. Prediction of toxin genes from chinese yellow catfish based on transcriptomic and proteomic sequencing. Int. J. Mol. Sci. 2016, 17, 556. [Google Scholar] [CrossRef] [PubMed]
- Bringans, S.; Eriksen, S.; Kendrick, T.; Gopalakrishnakone, P.; Livk, A.; Lock, R.; Lipscombe, R. Proteomic analysis of the venom of heterometrus longimanus (asian black scorpion). Proteomics 2008, 8, 1081–1096. [Google Scholar] [CrossRef] [PubMed]
- Prashanth, J.R.; Lewis, R.J.; Dutertre, S. Towards an integrated venomics approach for accelerated conopeptide discovery. Toxicon 2012, 60, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Roelants, K.; Winter, K.; Hodgson, W.C.; Griesman, L.; Kwok, H.F.; Scanlon, D.; Karas, J.; Shaw, C.; Wong, L.; et al. Novel venom proteins produced by differential domain-expression strategies in beaded lizards and gila monsters (genus heloderma). Mol. Biol. Evol. 2009, 27, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.T.; Khan, A.M.; Brusic, V. Bioinformatics for venom and toxin sciences. Brief. Bioinform. 2003, 4, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Menschaert, G.; Vandekerckhove, T.T.; Baggerman, G.; Schoofs, L.; Luyten, W.; Criekinge, W.V. Peptidomics coming of age: A review of contributions from a bioinformatics angle. J. Proteome Res. 2010, 9, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.; Yiu, S.-M.; Chin, F.Y. Meta-idba: A de novo assembler for metagenomic data. Bioinformatics 2011, 27, i94–i101. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Wheeler, D.L. Genbank. Nucleic Acids Res. 2005, 33, D34–D38. [Google Scholar] [CrossRef] [PubMed]
- Jungo, F.; Bairoch, A. Tox-prot, the toxin protein annotation program of the swiss-prot protein knowledgebase. Toxicon 2005, 45, 293–301. [Google Scholar] [CrossRef] [PubMed]
- He, Q.Y.; He, Q.Z.; Deng, X.C.; Yao, L.; Meng, E.; Liu, Z.H.; Liang, S.P. Atdb: A uni-database platform for animal toxins. Nucleic Acids Res. 2008, 36, D293–D297. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.C.; Halai, R.; Wang, C.K.; Craik, D.J. Conoserver, a database for conopeptide sequences and structures. Bioinformatics 2008, 24, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.L.; Miljenović, T.; Cai, S.; Raven, R.J.; Kaas, Q.; Escoubas, P.; Herzig, V.; Wilson, D.; King, G.F. Arachnoserver: A database of protein toxins from spiders. BMC Genom. 2009, 10, 375. [Google Scholar] [CrossRef] [PubMed]
- Herzig, V.; Wood, D.L.; Newell, F.; Chaumeil, P.A.; Kaas, Q.; Binford, G.J.; Nicholson, G.M.; Gorse, D.; King, G.F. Arachnoserver 2.0, an updated online resource for spider toxin sequences and structures. Nucleic Acids Res. 2010, 39, D653–D657. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Yu, R.; Jin, A.H.; Dutertre, S.; Craik, D.J. Conoserver: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2011, 40, D325–D330. [Google Scholar] [CrossRef] [PubMed]
- Roly, Z.Y.; Hakim, M.A.; Zahan, A.S.; Hossain, M.M.; Reza, M.A. Isob: A database of indigenous snake species of bangladesh with respective known venom composition. Bioinformation 2015, 11, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Jungo, F.; Bougueleret, L.; Xenarios, I.; Poux, S. The uniprotkb/swiss-prot tox-prot program: A central hub of integrated venom protein data. Toxicon 2012, 60, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using conoserver. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef] [PubMed]
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, D.; Arni, R.K.; Betzel, C. Proteome analysis of snake venom toxins: Pharmacological insights. Expert Rev. Proteom. 2008, 5, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Duda, T.F. Extensive and continuous duplication facilitates rapid evolution and diversification of gene families. Mol. Biol. Evol. 2012, 29, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, K.; Undheim, E.A.; Chan, A.H.; Koludarov, I.; Muñoz-Gómez, S.A.; Antunes, A.; Fry, B.G. Evolution stings: The origin and diversification of scorpion toxin peptide scaffolds. Toxins 2013, 5, 2456–2487. [Google Scholar] [CrossRef] [PubMed]
- Duda, T.F., Jr.; Chang, D.; Lewis, B.D.; Lee, T. Geographic variation in venom allelic composition and diets of the widespread predatory marine gastropod conus ebraeus. PLoS ONE 2009, 4, e6245. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.J. Diversity, phylogenetic distribution, and origins of venomous catfishes. BMC Evol. Biol. 2009, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S. Soapdenovo-trans: De novo transcriptome assembly with short rna-seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- NCBI Resource Coordinators. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2017, 45, D12–D17. [Google Scholar]
- Uniprot Consortium. Activities at the universal protein resource (uniprot). Nucleic Acids Res. 2014, 42, D191–D198. [Google Scholar]
- Yin, W.; Wang, Z.; Li, Q.; Lian, J.; Zhou, Y.; Lu, B.; Jin, L.; Qiu, P.; Zhang, P.; Zhu, W.; et al. Evolution trajectories of snake genes and genomes revealed by comparative analyses of five-pacer viper. Nat. Commun. 2016, 7, 13107. [Google Scholar] [CrossRef] [PubMed]
- Castoe, T.A.; De Koning, A.J.; Hall, K.T.; Card, D.C.; Schield, D.R.; Fujita, M.K.; Ruggiero, R.P.; Degner, J.F.; Daza, J.M.; Gu, W.; et al. The burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc. Natl. Acad. Sci. USA 2013, 110, 20645–20650. [Google Scholar] [CrossRef] [PubMed]
- Vonk, F.J.; Casewell, N.R.; Henkel, C.V.; Heimberg, A.M.; Jansen, H.J.; McCleary, R.J.; Kerkkamp, H.M.; Vos, R.A.; Guerreiro, I.; Calvete, J.J.; et al. The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc. Natl. Acad. Sci. USA 2013, 110, 20651–20656. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Bandyopadhyay, P.K.; Olivera, B.M.; Yandell, M. Characterization of the conus bullatus genome and its venom-duct transcriptome. BMC Genom. 2011, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Terrat, Y.; Biass, D.; Dutertre, S.; Favreau, P.; Remm, M.; Stocklin, R. High-resolution picture of a venom gland transcriptome: Case study with the marine snail conus consors. Toxicon 2012, 59, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Yu, Y.; Wu, Y.; Hao, P.; Di, Z.; He, Y.; Chen, Z.; Yang, W.; Shen, Z.; He, X.; et al. The genome of mesobuthus martensii reveals a unique adaptation model of arthropods. Nat. Commun. 2013, 4, 2602. [Google Scholar] [CrossRef] [PubMed]
- Sanggaard, K.W.; Bechsgaard, J.S.; Fang, X.; Duan, J.; Dyrlund, T.F.; Gupta, V.; Jiang, X.; Cheng, L.; Fan, D.; Feng, Y.; et al. Spider genomes provide insight into composition and evolution of venom and silk. Nat. Commun. 2014, 5, 3765. [Google Scholar] [PubMed]
- Consortium, H.G.S. Insights into social insects from the genome of the honeybee apis mellifera. Nature 2006, 443, 931–949. [Google Scholar]
- Werren, J.H.; Richards, S.; Desjardins, C.A.; Niehuis, O.; Gadau, J.; Colbourne, J.K.; Group, N.G.W. Functional and evolutionary insights from the genomes of three parasitoid nasonia species. Science 2010, 327, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, Y.; Hu, W.; Xu, D.; Tao, H.; Yang, X.; Li, Y.; Jiang, L.; Liang, S. Molecular diversification of peptide toxins from the tarantula haplopelma hainanum (ornithoctonus hainana) venom based on transcriptomic, peptidomic, and genomic analyses. J. Proteome Res. 2010, 9, 2550–2564. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, Y.; He, Q.; Liu, J.; Luo, J.; Zhu, L.; Lu, S.; Huang, P.; Chen, X.; Zeng, X.; et al. Toxin diversity revealed by a transcriptomic study of ornithoctonus huwena. PLoS ONE 2014, 9, e100682. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Wüster, W. Assembling an arsenal: Origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences. Mol. Biol. Evol. 2004, 21, 870–883. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Vidal, N.; Van der Weerd, L.; Kochva, E.; Renjifo, C. Evolution and diversification of the toxicofera reptile venom system. J. Proteom. 2009, 72, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Lavergne, V.; Dutertre, S.; Jin, A.-H.; Lewis, R.J.; Taft, R.J.; Alewood, P.F. Systematic interrogation of the conus marmoreus venom duct transcriptome with conosorter reveals 158 novel conotoxins and 13 new gene superfamilies. BMC Genom. 2013, 14, 708. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Safavi-Hemami, H.; McIntosh, L.D.; Purcell, A.W.; Norton, R.S.; Papenfuss, A.T. Diversity of conotoxin gene superfamilies in the venomous snail, conus victoriae. PLoS ONE 2014, 9, e87648. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.F.; Diego-Garcia, E.; de la Vega, R.C.R.; Possani, L.D. Transcriptome analysis of the venom gland of the mexican scorpion hadrurus gertschi (arachnida: Scorpiones). BMC Genom. 2007, 8, 119. [Google Scholar] [CrossRef] [PubMed]
- Koua, D.; Brauer, A.; Laht, S.; Kaplinski, L.; Favreau, P.; Remm, M.; Lisacek, F.; Stöcklin, R. Conodictor: A tool for prediction of conopeptide superfamilies. Nucleic Acids Res. 2012, 40, W238–W241. [Google Scholar] [CrossRef] [PubMed]
- Koua, D.; Laht, S.; Kaplinski, L.; Stöcklin, R.; Remm, M.; Favreau, P.; Lisacek, F. Position-specific scoring matrix and hidden markov model complement each other for the prediction of conopeptide superfamilies. Biochim. Biophys. Acta 2013, 1834, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Yao, G.; Gao, B.-M.; Fan, C.-X.; Bian, C.; Wang, J.; Cao, Y.; Wen, B.; Zhu, Y.; Ruan, Z.; et al. High-throughput identification of novel conotoxins from the chinese tubular cone snail (conus betulinus) by multi-transcriptome sequencing. GigaScience 2016, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.W.; Serrano, S.M. Exploring snake venom proteomes: Multifaceted analyses for complex toxin mixtures. Proteomics 2008, 8, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Jin, A.-H.; Vetter, I.; Hamilton, B.; Sunagar, K.; Lavergne, V.; Dutertre, V.; Fry, B.G.; Antunes, A.; Venter, D.J.; et al. Evolution of separate predation-and defence-evoked venoms in carnivorous cone snails. Nat. Commun. 2014, 5, 3521. [Google Scholar] [CrossRef] [PubMed]
- Carrijo, L.C.; Andrich, F.; De Lima, M.E.; Cordeiro, M.N.; Richardson, M.; Figueiredo, S.G. Biological properties of the venom from the scorpionfish (scorpaena plumieri) and purification of a gelatinolytic protease. Toxicon 2005, 45, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Jones, A.; Lewis, R.J. Remarkable inter-and intra-species complexity of conotoxins revealed by lc/ms. Peptides 2009, 30, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.-H.; Dutertre, S.; Kaas, Q.; Lavergne, V.; Kubala, P.; Lewis, R.J.; Alewood, P.F. Transcriptomic messiness in the venom duct of conus miles contributes to conotoxin diversity. Mol. Cell. Proteom. 2013, 12, 3824–3833. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J.; Ghezellou, P.; Paiva, O.; Matainaho, T.; Ghassempour, A.; Goudarzi, H.; Kraus, F.; Sanz, L.; Williams, D.J. Snake venomics of two poorly known hydrophiinae: Comparative proteomics of the venoms of terrestrial toxicocalamus longissimus and marine hydrophis cyanocinctus. J. Proteom. 2012, 75, 4091–4101. [Google Scholar] [CrossRef] [PubMed]
- Sollod, B.L.; Wilson, D.; Zhaxybayeva, O.; Gogarten, J.P.; Drinkwater, R.; King, G.F. Were arachnids the first to use combinatorial peptide libraries? Peptides 2005, 26, 131–139. [Google Scholar] [CrossRef] [PubMed]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M.; Miljanich, G.P.; Ramachandran, J.; Adams, M.E. Calcium channel diversity and neurotransmitter release: The ω-conotoxins and ω-agatoxins. Annu. Rev. Biochem. 1994, 63, 823–867. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.M.; Olivera, B.M.; Cruz, L.J. Conus peptides as probes for ion channels. Methods Enzymol. 1999, 294, 605–624. [Google Scholar] [PubMed]
- Sun, Y.; Huang, Y.; Li, X.; Baldwin, C.C.; Zhou, Z.; Yan, Z.; Crandall, K.A.; Zhang, Y.; Zhao, X.; Wang, M.; et al. Fish-t1k (transcriptomes of 1,000 fishes) project: Large-scale transcriptome data for fish evolution studies. GigaScience 2016, 5, 18. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Species | Taxonomy Name | Numbers of Sequences |
|---|---|---|
| Snakes | Serpents | 1684 |
| Scorpions | Scorpions | 1510 |
| Spiders | Araneae | 1391 |
| Cone snails | Conus | 3860 |
| Sea anemones | Actiniaria | 308 |
| Insects | Hexapoda | 162 |
| Fish | Teleostei | 44 |
| Mammals | Mammalias | 106 |
| Lizards | Heloderma | 241 |
| Jellyfish | Cubomedusae/Scyphozoa | 175 |
| Sea stars | Asteroidea | 8 |
| Hydra | Hydroida | 14 |
| Worms | Cerebratulus | 5 |
| Forg, Toad | Amphibia | 85 |
| Sea-urchin | Echinoidea | 2 |
| Sea hare | Aplysiomorpha | 44 |
| Scolopendra | Myriapoda | 49 |
| All | Metazoa | 9688 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, B.; Huang, Y.; Baumann, K.; Fry, B.G.; Shi, Q. From Marine Venoms to Drugs: Efficiently Supported by a Combination of Transcriptomics and Proteomics. Mar. Drugs 2017, 15, 103. https://doi.org/10.3390/md15040103
Xie B, Huang Y, Baumann K, Fry BG, Shi Q. From Marine Venoms to Drugs: Efficiently Supported by a Combination of Transcriptomics and Proteomics. Marine Drugs. 2017; 15(4):103. https://doi.org/10.3390/md15040103
Chicago/Turabian StyleXie, Bing, Yu Huang, Kate Baumann, Bryan Grieg Fry, and Qiong Shi. 2017. "From Marine Venoms to Drugs: Efficiently Supported by a Combination of Transcriptomics and Proteomics" Marine Drugs 15, no. 4: 103. https://doi.org/10.3390/md15040103
APA StyleXie, B., Huang, Y., Baumann, K., Fry, B. G., & Shi, Q. (2017). From Marine Venoms to Drugs: Efficiently Supported by a Combination of Transcriptomics and Proteomics. Marine Drugs, 15(4), 103. https://doi.org/10.3390/md15040103