Tricholides A and B and Unnarmicin D: New Hybrid PKS-NRPS Macrocycles Isolated from an Environmental Collection of Trichodesmium thiebautii

Abstract
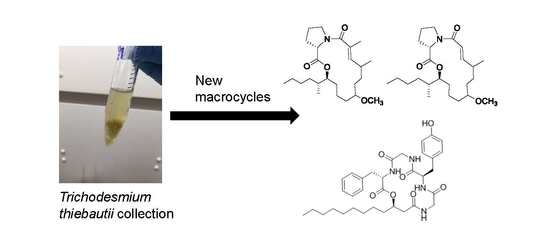
:
1. Introduction
2. Results
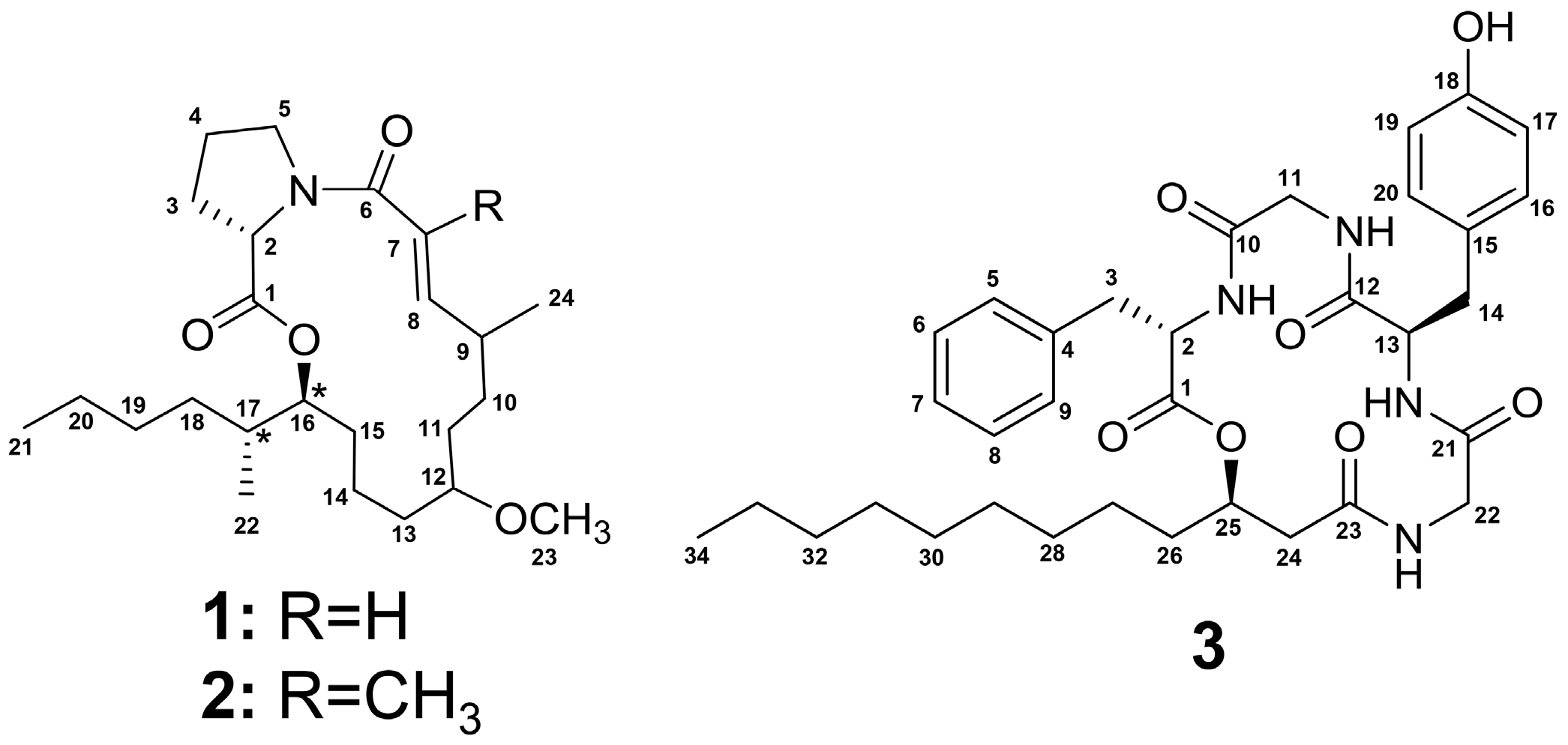
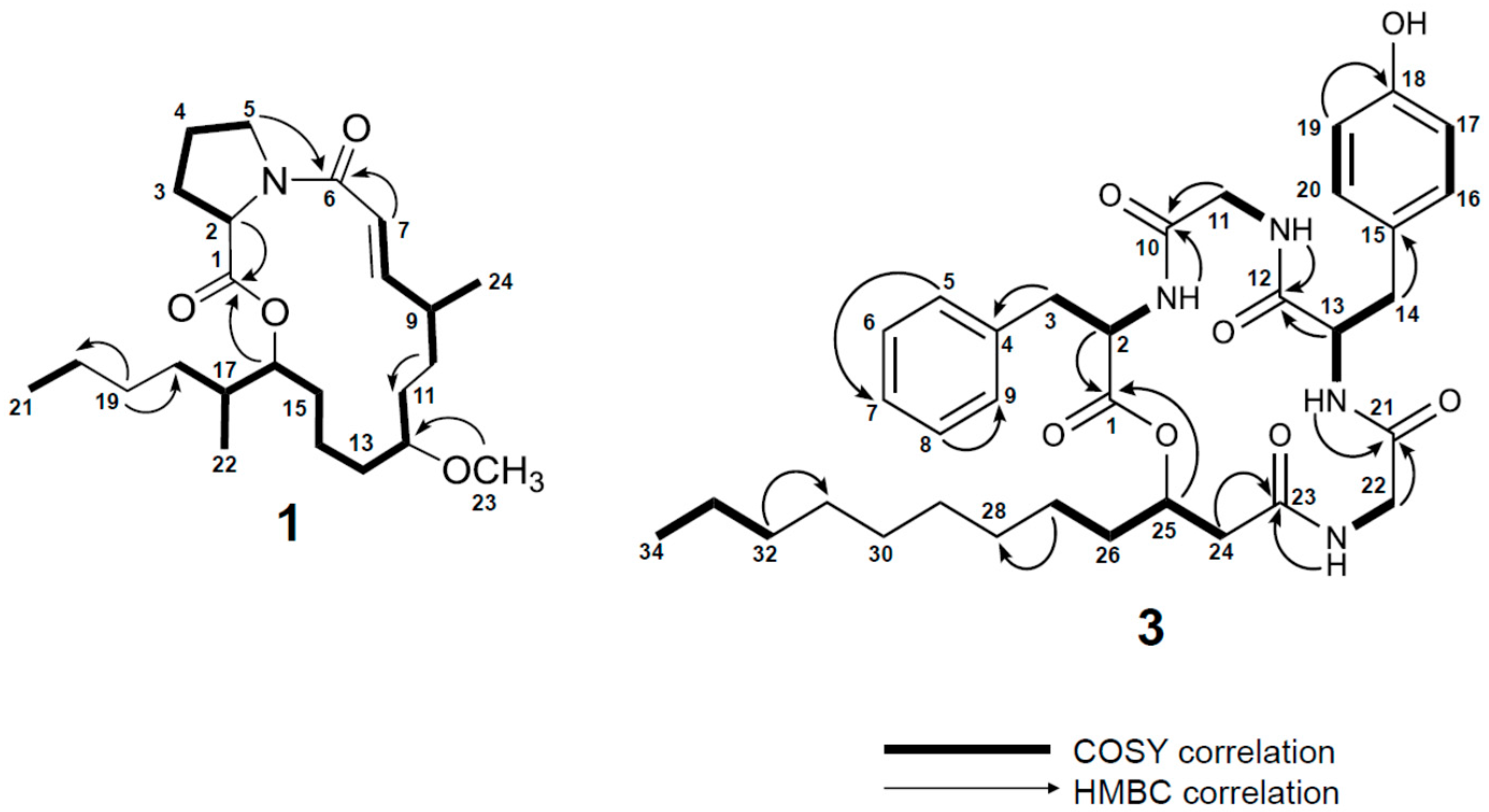
2.1. Isolation and Structure Determination of 1–3
2.2. Biological Evaluation of 1–3
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Collection of Biological Material
4.3. Extraction and Isolation
4.4. Acid Hydrolysis and Marfey’s Protocol
4.5. Hydrolysis of 3
4.6. Preparation of MTPA Esters of 4
4.7. Antimicrobial Assay
4.7.1. Bacterial Strains
4.7.2. Antimicrobial Activity
4.8. Cytotoxicity Assay
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tan, L.T. Pharmaceutical agents from filamentous marine cyanobacteria. Drug Discov. Today 2013, 18, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry 2007, 68, 954–979. [Google Scholar] [CrossRef] [PubMed]
- Nunnery, J.K.; Mevers, E.; Gerwick, W.H. Biologically active secondary metabolites from marine cyanobacteria. Curr. Opin. Biotechnol. 2010, 21, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Mooberry, S.L. Isolation, structure determination, and biological activity of Lyngbyabellin A from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; Krunic, A.; Orjala, J. Sanctolide A, a 14-membered PK-NRP hybrid macrolide from the cultured cyanobacterium Oscillatoria sancta (SAG 74.79). Tetrahedron Lett. 2012, 53, 3563–3567. [Google Scholar] [CrossRef] [PubMed]
- Belakhov, V.V.; Garabadzhiu, A.V. Polyene macrolide antibiotics: Mechanisms of inactivation, ways of stabilization, and methods of disposal of unusable drugs. Russ. J. Gen. Chem. 2015, 85, 2985–3001. [Google Scholar] [CrossRef]
- Kollár, P.; Rajchard, J.; Balounová, Z.; Pazourek, J. Marine natural products: Bryostatins in preclinical and clinical studies. Pharm. Biol. 2014, 52, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.L.; Linington, R.G.; Balunas, M.J.; Centeno, A.; Boudreau, P.; Zhang, C.; Engene, N.; Spadafora, C.; Mutka, T.S.; Kyle, D.E.; et al. Bastimolide A, a Potent Antimalarial Polyhydroxy Macrolide from the Marine Cyanobacterium Okeania hirsuta. J. Org. Chem. 2015, 80, 7849–7855. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.R.; Cao, Z.Y.; Engene, N.; Soria-Mercado, I.E.; Murray, T.F.; Gerwick, W.H. Palmyrolide A, an unusually stabilized neuroactive macrolide from palmyra atoll cyanobacteria. Org. Lett. 2010, 12, 4490–4493. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.E. Cyclic peptides and depsipeptides from cyanobacteria: A review. J. Ind. Microbiol. 1996, 16, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Kehr, J.C.; Picchi, D.; Dittmann, E. Natural product biosyntheses in cyanobacteria: A treasure trove of unique enzymes. Beilstein J. Org. Chem. 2011, 7, 1622–1635. [Google Scholar] [CrossRef] [PubMed]
- Welker, M.; von Döhren, H. Cyanobacterial peptides—Nature’s own combinatorial biosynthesis. FEMS Microbiol. Rev. 2006, 30, 530–563. [Google Scholar] [CrossRef] [PubMed]
- Elsworth, J.F.; Grove, J.F. Cyclodepsipeptides from Beauveria bassiana Bals. Part 1. Beauverolides H and I. J. Chem. Soc. Perkin Trans. 1 1977, 3, 270–273. [Google Scholar] [CrossRef]
- Elsworth, J.F.; Grove, J.F. Cyclodepsipeptides from Beauveria bassiana. Part 2. Beauverolides A to F and their relationship to isarolide. J. Chem. Soc. Perkin Trans. 1 1980, 8, 1795–1799. [Google Scholar] [CrossRef]
- Grove, J.F. Cyclodepsipeptides from Beauveria bassiana. Part 3. The isolation of beauverolides Ba, Ca, Ja, and Ka. J. Chem. Soc. Perkin Trans. 1 1980, 12, 2878–2880. [Google Scholar] [CrossRef]
- Oku, N.; Kawabata, K.; Adachi, K.; Katsuta, A.; Shizuri, Y. Unnarmicins A and C, new antibacterial depsipeptides produced by marine bacterium Photobacterium sp. MBIC06485. J. Antibiot. 2008, 61, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Månsson, M.; Nielsen, A.; Kjaerulff, L.; Gotfredsen, C.H.; Wietz, M.; Ingmer, H.; Gram, L.; Larsen, T.O. Inhibition of virulence gene expression in Staphylococcus aureus by novel depsipeptides from a marine photobacterium. Mar. Drugs 2011, 9, 2537–2552. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Yamamoto, K.; Fukuda, T.; Shojima, A.; Nakayama, J.; Carro, L.; Trujillo, M.E. Arthroamide, a cyclic depsipeptide with quorum sensing inhibitory activity from Arthrobacter sp. J. Nat. Prod. 2015, 78, 2827–2831. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Carr, G.; Zhang, Y.; Williams, D.E.; Amlani, A.; Bottriell, H.; Mui, A.L.-F.; Anderson, R.J. Turnagainolides A and B, cyclic depsipeptides produced in culture by a Bacillus sp.: Isolation, structure elucidation, and synthesis. J. Nat. Prod. 2011, 74, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Kawabata, Y.; Kasai, H.; Katsuta, M.; Shizuri, Y. New Antibiotic. Japanese Patent JP 2007230911, 13 September 2007. [Google Scholar]
- Krunic, A.; Vallat, A.; Mo, S.; Lantvit, D.D.; Swanson, S.M.; Orjala, J. Scytonemides A and B, cyclic peptides with 20S proteasome inhibitory activity from the cultured cyanobacterium Scytonema hofmanii. J. Nat. Prod. 2010, 73, 1927–1932. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, G.G.; Harrigan, B.L.; Davidson, B.S. Kailuins A-D, new cyclic acyldepsipeptides from cultures of a marine-derived bacterium. Tetrahedron 1997, 53, 1577–1582. [Google Scholar] [CrossRef]
- Theodore, C.M.; Lorig-Roach, N.; Still, P.C.; Johnson, T.A.; Drašković, M.; Schwochert, J.A.; Naphen, C.N.; Crews, M.S.; Barker, S.A.; Valeriote, F.A.; et al. Biosynthetic products from a nearshore-derived gram-negative bacterium enable reassessment of the kailuin depsipeptides. J. Nat. Prod. 2015, 78, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Sudek, S.; Haygood, M.G.; Youssef, D.T.A.; Schmidt, E.W. Structure of trichamide, a cyclic peptide from the bloom-forming cyanobacterium Trichodesmium erythraeum, predicted from the genome sequence. Appl. Environ. Microbiol. 2006, 72, 4382–4387. [Google Scholar] [CrossRef] [PubMed]
- Bertin, M.J.; Wahome, P.G.; Zimba, P.V.; He, H.; Moeller, P.D.R. Trichophycin A, a cytotoxic linear polyketide isolated from a Trichodesmium thiebautii bloom. Mar. Drugs 2017, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Bertin, M.J.; Zimba, P.V.; He, H.; Moeller, P.D.R. Structure revision of trichotoxin, a chlorinated polyketide isolated from a Trichodesmium thiebautii bloom. Tetrahedron Lett. 2016, 57, 5864–5867. [Google Scholar] [CrossRef]
- Hove, T.R.; Jeffrey, C.S.; Shao, F. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol. Nat. Protoc. 2007, 2, 2451–2458. [Google Scholar]
- Klein, D.; Braekman, J.C.; Daloze, D.; Hoffman, L.; Castillo, G.; Demoulin, V.V. Madangolide and laingolide A, two novel macrolides from Lyngbya bouillonii (Cyanobacteria). J. Nat. Prod. 1999, 62, 934–936. [Google Scholar] [CrossRef] [PubMed]
- Nakao, Y.; Maki, T.; Matsunaga, S.; van Soest, R.V.M.; Fusetani, N. Penarolide sulfates A1 and A2, new α-glucosidase inhibitors from a marine sponge Penares sp. Tetrahedron 2000, 56, 8977–8987. [Google Scholar] [CrossRef]
- Komárek, J.; Anagnostidis, K. Cyanoprokarota 19 Part 2: Oscillatoriales; Elsevier: München, Germany, 2005; pp. 1–759. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Position | δC | δH (J in Hz) | HMBC | COSY |
|---|---|---|---|---|
| 1 | 172.2, qC | |||
| 2 | 59.1, CH | 4.50, dd (9.0, 2.0) | 1, 3, 4, 5 | 3a, 3b |
| 3a | 31.8, CH2 | 2.29, m | 1, 2, 4 | 2, 3b, 4 |
| 3b | 2.10, m | 1, 4, 5 | 3a, 4 | |
| 4 | 22.3, CH2 | 1.94, m | 2, 3, 5 | 3a, 5a, 5b |
| 5a | 46.7, CH2 | 3.77, m | 2, 3, 4, 6 | 4, 5b |
| 5b | 3.62, m | 2, 3, 4, 6 | 4, 5a | |
| 6 | 165.2, qC | |||
| 7 | 117.1, CH | 5.84, d (15.5) | 6, 8, 9, 24 | 8 |
| 8 | 151.1, CH | 7.16, dd (15.5, 5.8) | 6, 7, 9, 10, 24 | 7, 9 |
| 9 | 35.9, CH | 2.34, m | 7, 8, 10, 24 | 8, 10, 24 |
| 10 | 30.3, CH2 | 1.36, m | 8, 9, 11 | 9 |
| 11a | 30.9, CH2 | 1.62, m | 10, 12 | 11b, 12 |
| 11b | 1.43, ovlp b | 10, 12 | 12 | |
| 12 | 79.9, CH | 3.20, m | 11, 13, 14, 23 | 11b, 13a |
| 13a | 30.0, CH2 | 1.55, m | 12, 14 | 12, 13b, 14b |
| 13b | 1.43, ovlp | 12, 14 | 12, 13a, 14b | |
| 14a | 19.6, CH2 | 1.40, m | 13, 15, 16 | 13b, 14a |
| 14b | 1.16, m | 15, 16 | 13a, 14a | |
| 15a | 32.7, CH2 | 1.57, m | 14, 16 | 14b, 15b, 16 |
| 15b | 1.47, m | 14, 16 | 14b, 15a, 16 | |
| 16 | 78.3, CH | 4.98, ddd (10.6, 4.6) | 1, 14, 15, 17, 22 | 15a, 15b, 17 |
| 17 | 37.5, CH | 1.60, m | 16, 18, 19, 22 | 16, 18b, 22 |
| 18a | 32.9, CH2 | 1.34, ovlp | 17, 19 | 17, 18b |
| 18b | 1.10, m | 17, 19 | 17, 18a | |
| 19a | 29.2, CH2 | 1.30, ovlp | 18, 20 | |
| 19b | 1.26, ovlp | 18, 20 | 18b | |
| 20 | 22.9, CH2 | 1.28, ovlp | 21 | 21 |
| 21 | 14.0, CH3 | 0.89, t (7.1) | 19, 20 | 20 |
| 22 | 14.7, CH3 | 0.91, d (6.8) | 16, 17, 18 | 17 |
| 23 | 56.0, CH3 | 3.27, s | 12 | |
| 24 | 22.5, CH3 | 1.04, d (7.0) | 8, 9, 10 | 9 |
| Residue | Position | δC | Type δH (J in Hz) | HMBC | COSY |
|---|---|---|---|---|---|
| Phe | 1 | 170.1, qC | |||
| 2 | 52.6 CH | 4.58, td (9.5, 5.0) | 1, 3, 4, 10 | 3a, 3b, NH-1 | |
| 3a | 36.5, CH2 | 3.15, dd (13.7, 5.0) | 1, 2, 4, 5, 9 | 2, 3b | |
| 3b | 2.78, ovlp | 1, 2, 4, 5, 9 | 2, 3a | ||
| 4 | 137.6, qC | ||||
| 5/9 | 129.3, CH | 7.20, d (7.2) | 3, 7 | ||
| 6/8 | 128.0, CH | 7.24, t (7.2) | 4, 5, 9 | ||
| 7 | 126.2, CH | 7.17, t (7.2) | 5, 9 | ||
| NH-1 | 7.40, d (9.3) | 2, 10 | 2 | ||
| Gly-1 | 10 | 168.2, qC | |||
| 11a | 42.1, CH2 | 3.90, dd (17.1, 8.3) | 10, 12 | 11b, NH-2 | |
| 11b | 3.37, dd (17.1, 4.6) | 10, 12 | 11a, NH-2 | ||
| NH-2 | 8.02, (8.4, 4.8) | 11, 12 | 11a, 11b | ||
| Tyr | 12 | 171.3, qC | |||
| 13 | 57.2, CH | 4.13, m | 12, 14, 15, 21 | 14a, 14b, NH-3 | |
| 14a | 35.0, CH2 | 3.02, dd (14.3, 3.8) | 12, 13, 15, 16, 20 | 13, 14b | |
| 14b | 2.78, ovlp | 12, 13, 15, 16, 20 | 13, 14a | ||
| 15 | 127.8, qC | ||||
| 16/20 | 129.8, CH | 7.13, d (8.5) | 14, 18 | 17/19 | |
| 17/19 | 115.1, CH | 6.69, d (8.5) | 15, 18 | 16/20 | |
| 18 | 156.0, qC | ||||
| NH-3 | 8.98, d (5.5) | 13, 14, 21 | 13 | ||
| Gly-2 | 21 | 172.4, qC | |||
| 22a | 42.7 CH2 | 3.83, dd (14.7, 3.8) | 21, 23 | 22b, NH-4 | |
| 22b | 3.51, dd (14.7, 6.8) | 21, 23 | 22a, NH-4 | ||
| NH-4 | 8.61, dd (6.8, 3.9) | 22, 23 | 22a, 22b | ||
| Hdda b | 23 | 170.7, qC | |||
| 24a | 40.4, CH2 | 2.60, dd (13.7, 3.6) | 23, 25, 26 | 24b, 25 | |
| 24b | 2.19, dd (13.7, 10.5) | 23, 25, 26 | 24a, 25 | ||
| 25 | 72.5, CH | 5.14, m | 1, 24, 26, 27 | 24b, 26a, 26b | |
| 26a | 33.8, CH2 | 1.54, m | 24, 25, 27, 28 | 25, 26b, 27 | |
| 26b | 1.46, m | 24, 25, 27, 28 | 25, 26a, 27 | ||
| 27 | 24.4, CH2 | 1.16, m | 25, 26, 28 | 26a, 26b | |
| 28 | 28.9, CH2 | 1.22, ovlp c | |||
| 29 | 28.9, CH2 | 1.22, ovlp | |||
| 30 | 28.7, CH2 | 1.25, ovlp | |||
| 31 | 28.7, CH2 | 1.25, ovlp | |||
| 32 | 31.3, CH2 | 1.24, ovlp | 31, 33, 34 | ||
| 33 | 22.1, CH2 | 1.28, m | 32, 34 | 34 | |
| 34 | 14.0, CH3 | 0.87, t (7.2) | 32, 33 | 33 |
| Compound | Residue Sequence | Source |
|---|---|---|
| Unnarmicin D (3) | (R)-Hdda Gly d-Tyr Gly l-Phe | environmental collection of T. thiebautii |
| Unnarmicin A [17] | (R)-Hha a l-Leu d-Phe l-Leu l-Phe | Photobacterium sp. strain MBIC0648517 |
| Unnarmicin C [17] | (R)-Hoa b l-Leu d-Phe l-Leu l-Phe | Photobacterium sp. strain MBIC0648517 |
| Solonamide A [18] | (R)-Hha l-Phe d-Leu d-Ala l-Leu | Photobacterium sp. strain S275318 |
| Solonamide B [18] | (R)-Hoa l-Phe d-Leu d-Ala l-Leu | Photobacterium sp. strain S275318 |
| Arthroamide [19] | (R)-Hppa c l-Val d-Ala l-Val l-Val | Arthrobacter sp. strain PGVB119 |
| Turnagainolide A [20] | (R)-Hppa l-Val d-Ala l-Ile l-Val | Bacillus sp. strain RJA219420 |
| Turnagainolide B [20] | (S)-Hppa l-Val d-Ala l-Ile l-Val | Bacillus sp. strain RJA219420 |
| Ngercheumicin C [21] | Hoa Phe Leu Leu Leu | Photobacterium sp. |
| Ngercheumicin D [21] | Hoa Phe Met Leu Leu | Photobacterium sp. |
| Ngercheumicin E [21] | Hoa Phe Phe Leu Leu | Photobacterium sp. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertin, M.J.; Roduit, A.F.; Sun, J.; Alves, G.E.; Via, C.W.; Gonzalez, M.A.; Zimba, P.V.; Moeller, P.D.R. Tricholides A and B and Unnarmicin D: New Hybrid PKS-NRPS Macrocycles Isolated from an Environmental Collection of Trichodesmium thiebautii. Mar. Drugs 2017, 15, 206. https://doi.org/10.3390/md15070206
Bertin MJ, Roduit AF, Sun J, Alves GE, Via CW, Gonzalez MA, Zimba PV, Moeller PDR. Tricholides A and B and Unnarmicin D: New Hybrid PKS-NRPS Macrocycles Isolated from an Environmental Collection of Trichodesmium thiebautii. Marine Drugs. 2017; 15(7):206. https://doi.org/10.3390/md15070206
Chicago/Turabian StyleBertin, Matthew J., Alexandre F. Roduit, Jiadong Sun, Gabriella E. Alves, Christopher W. Via, Miguel A. Gonzalez, Paul V. Zimba, and Peter D. R. Moeller. 2017. "Tricholides A and B and Unnarmicin D: New Hybrid PKS-NRPS Macrocycles Isolated from an Environmental Collection of Trichodesmium thiebautii" Marine Drugs 15, no. 7: 206. https://doi.org/10.3390/md15070206
APA StyleBertin, M. J., Roduit, A. F., Sun, J., Alves, G. E., Via, C. W., Gonzalez, M. A., Zimba, P. V., & Moeller, P. D. R. (2017). Tricholides A and B and Unnarmicin D: New Hybrid PKS-NRPS Macrocycles Isolated from an Environmental Collection of Trichodesmium thiebautii. Marine Drugs, 15(7), 206. https://doi.org/10.3390/md15070206