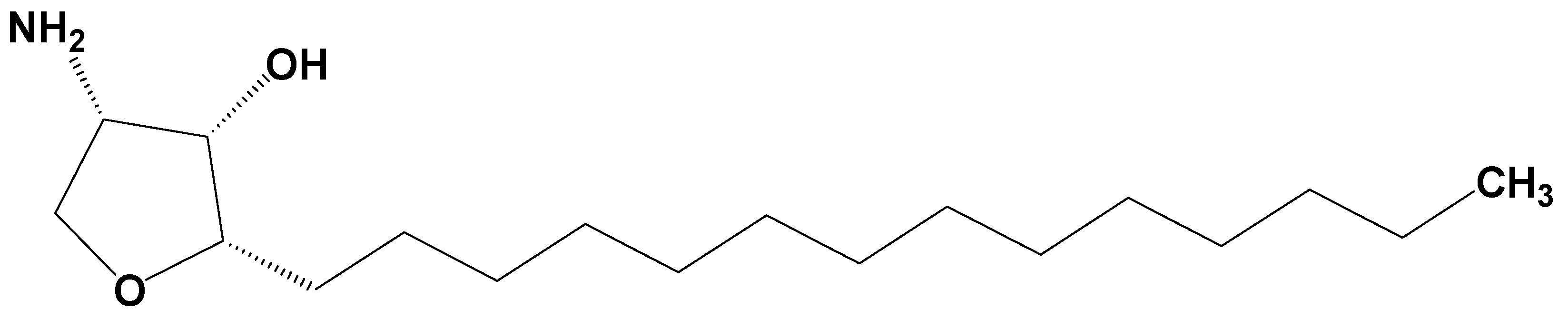
Pharmacokinetics of Jaspine B and Enhancement of Intestinal Absorption of Jaspine B in the Presence of Bile Acid in Rats


Abstract
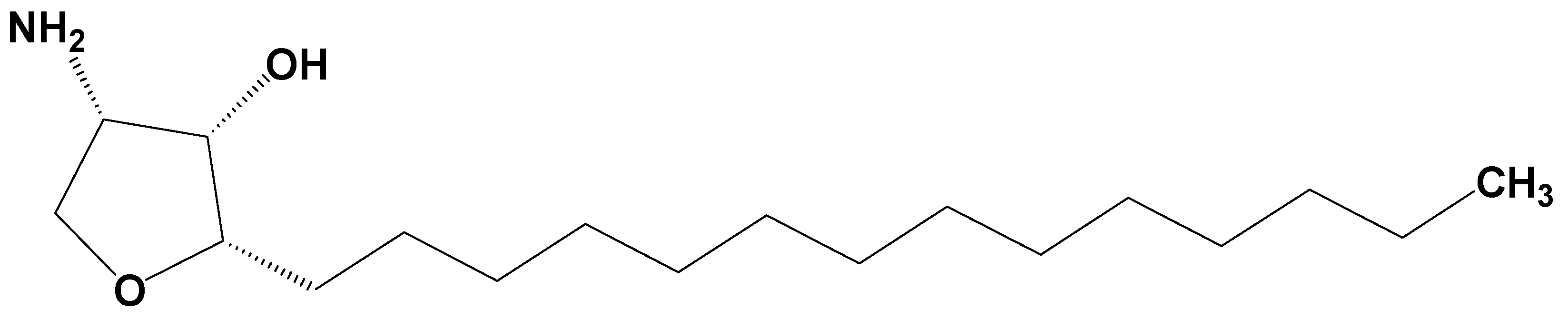
:
1. Introduction
2. Results
2.1. LC/MS-MS Analysis of Jaspine B in the Biological Samples
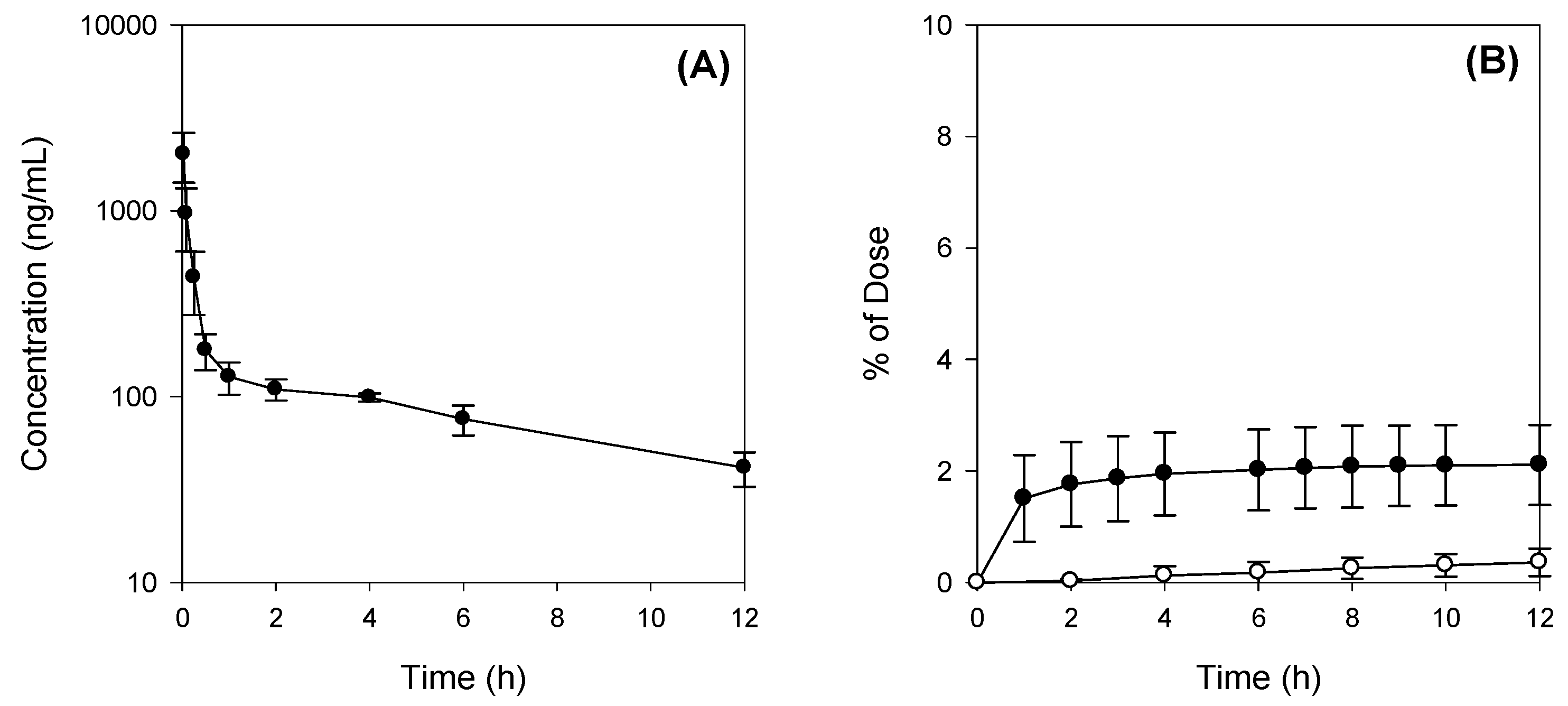
2.2. Pharmacokinetics of Jaspine B Following Intravenous Injection of 10 mg/kg Jaspine B
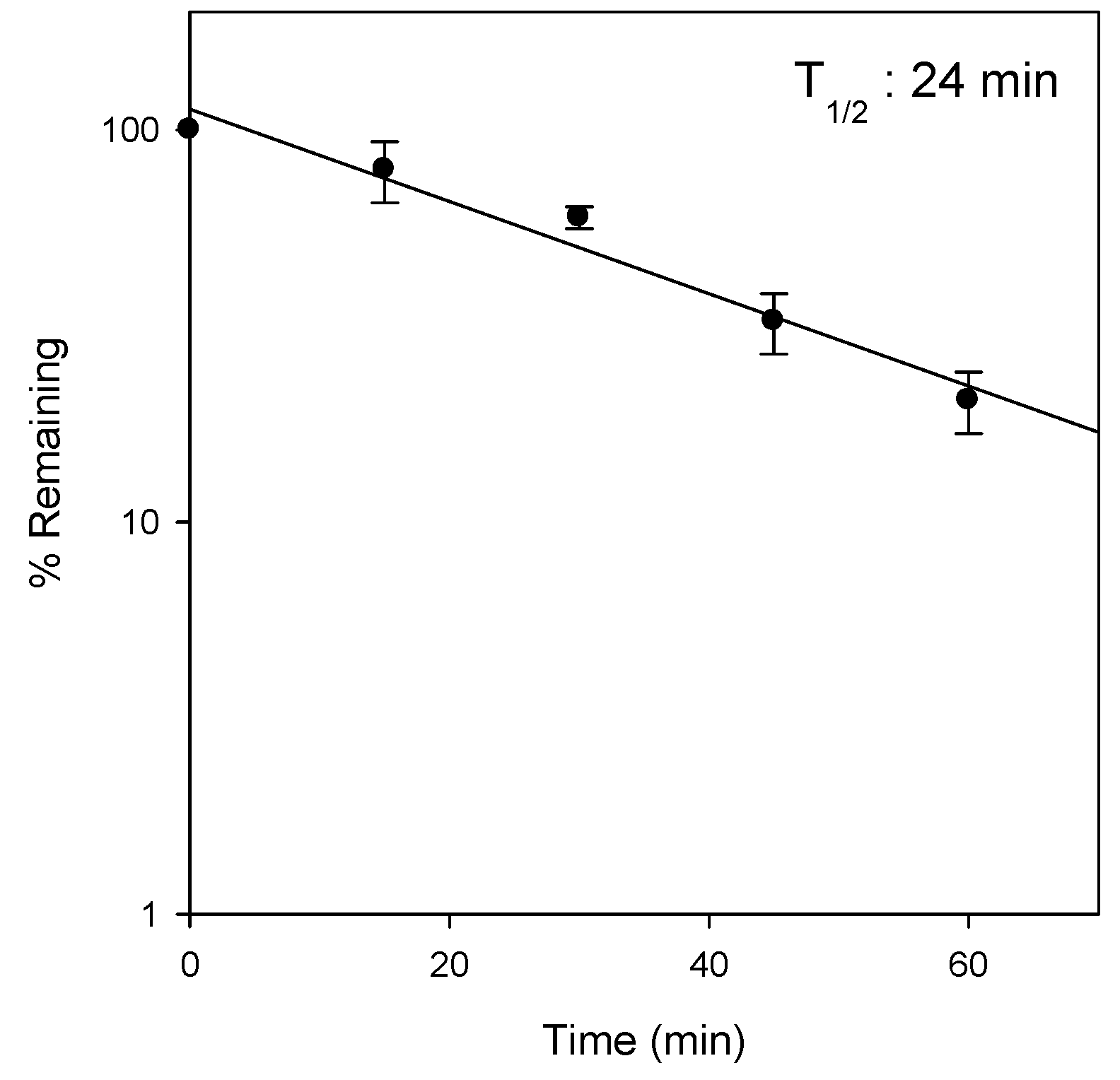
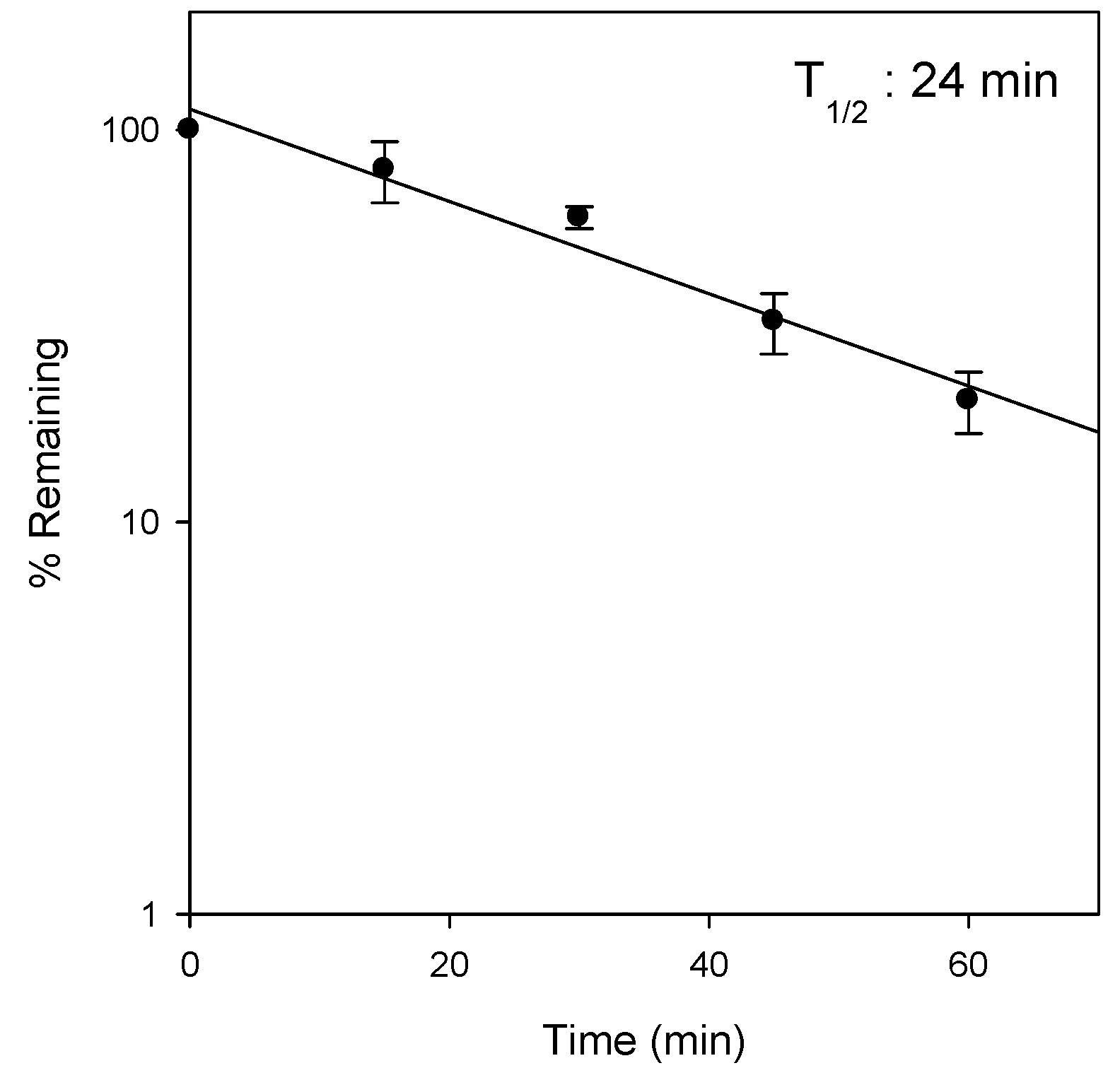
2.3. Metabolic Instability of Jaspine B
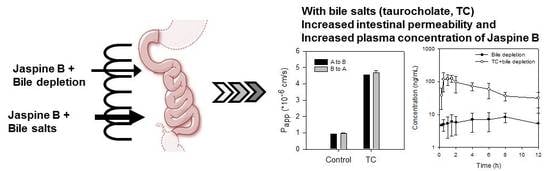
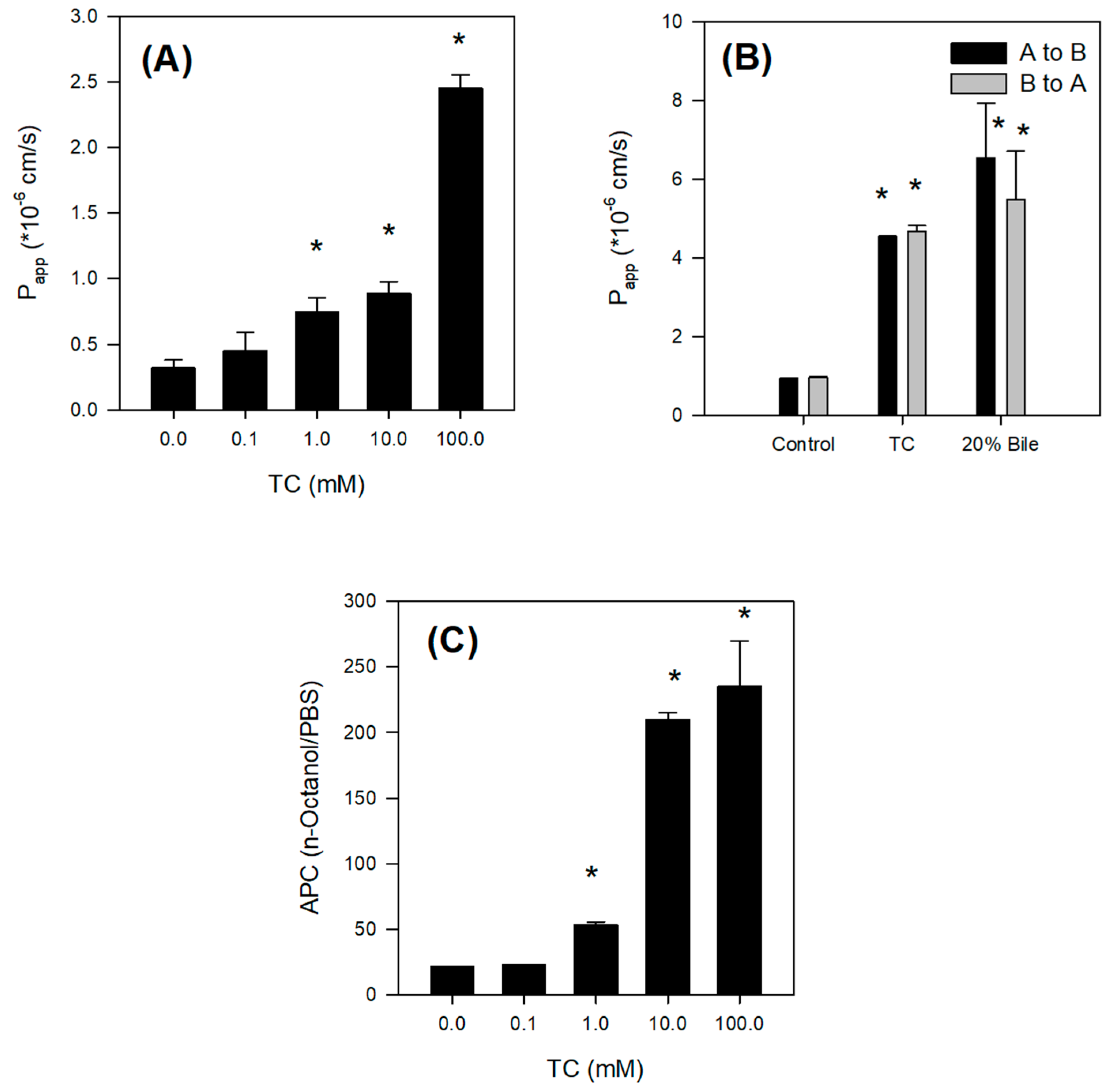
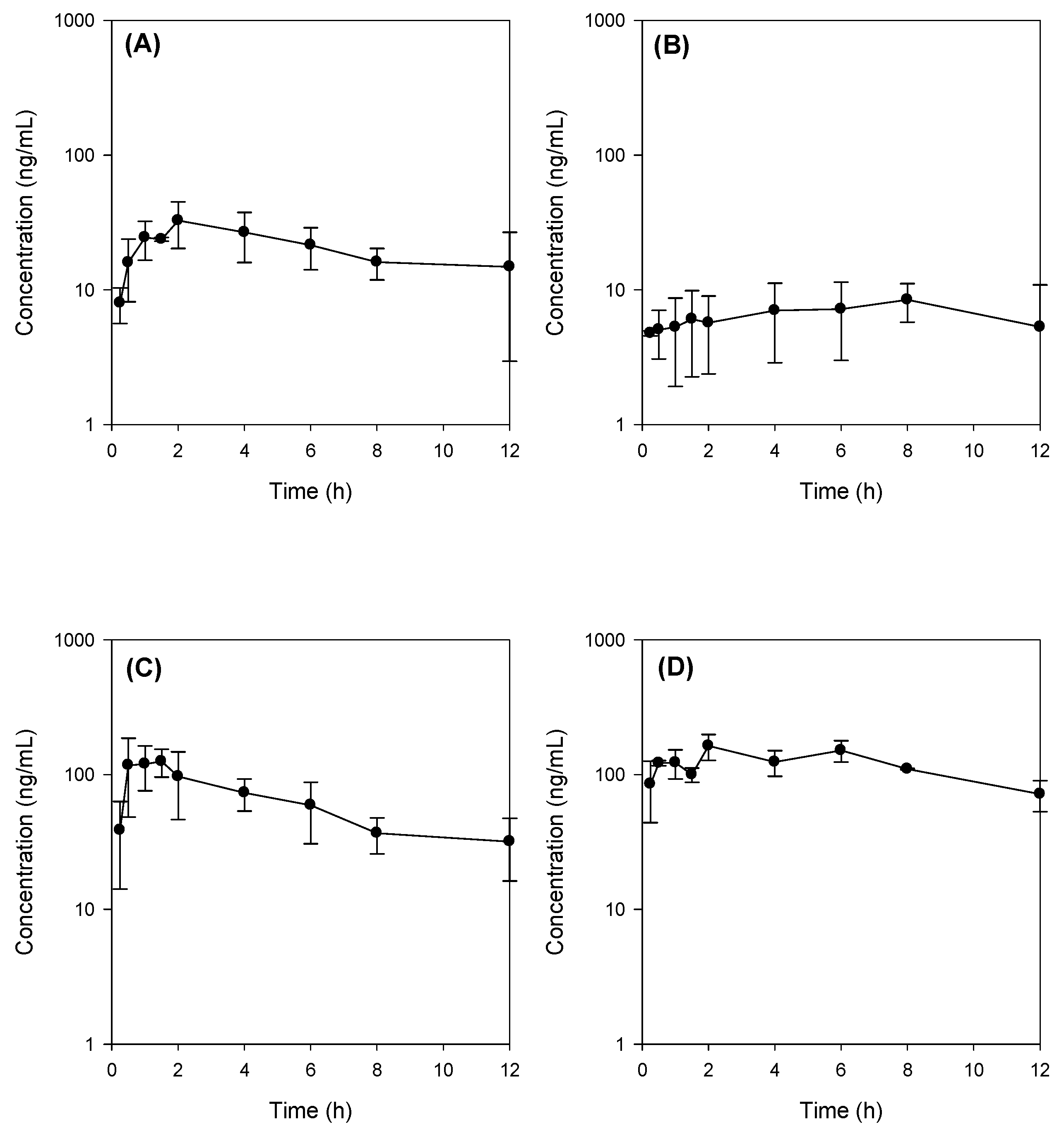
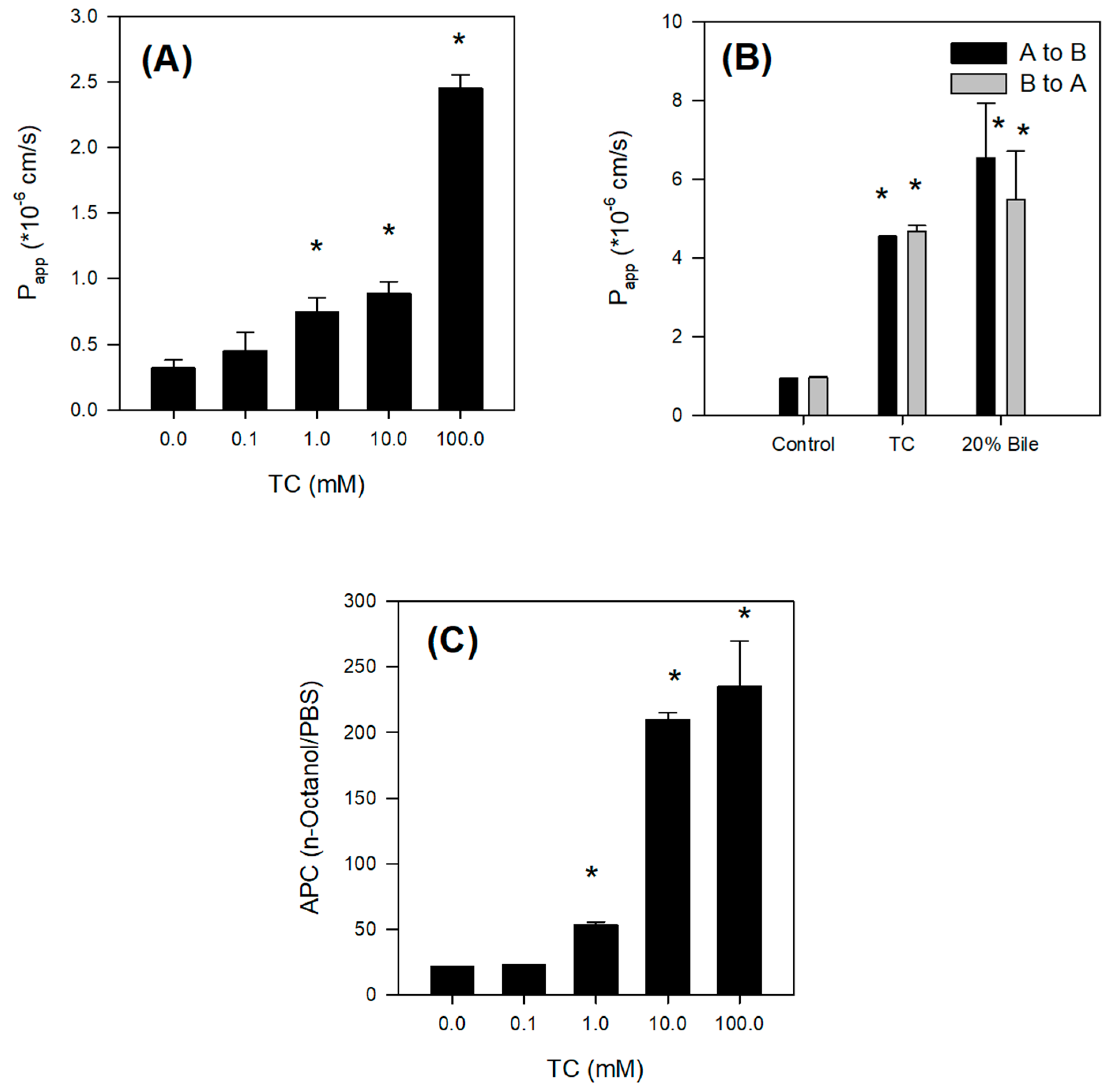
2.4. Bile Acid Facilitates the Intestinal Absorption of Jaspine B
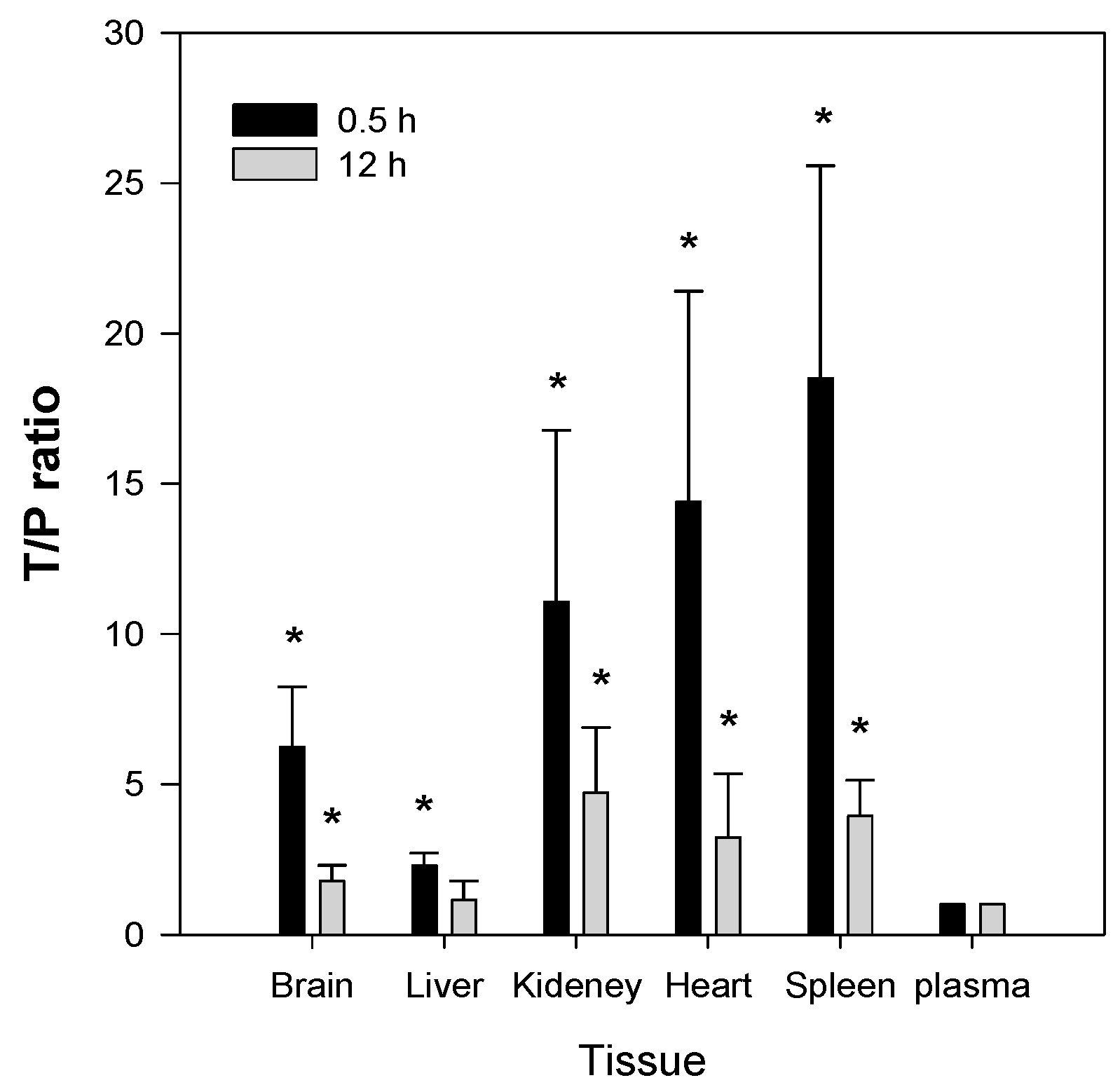
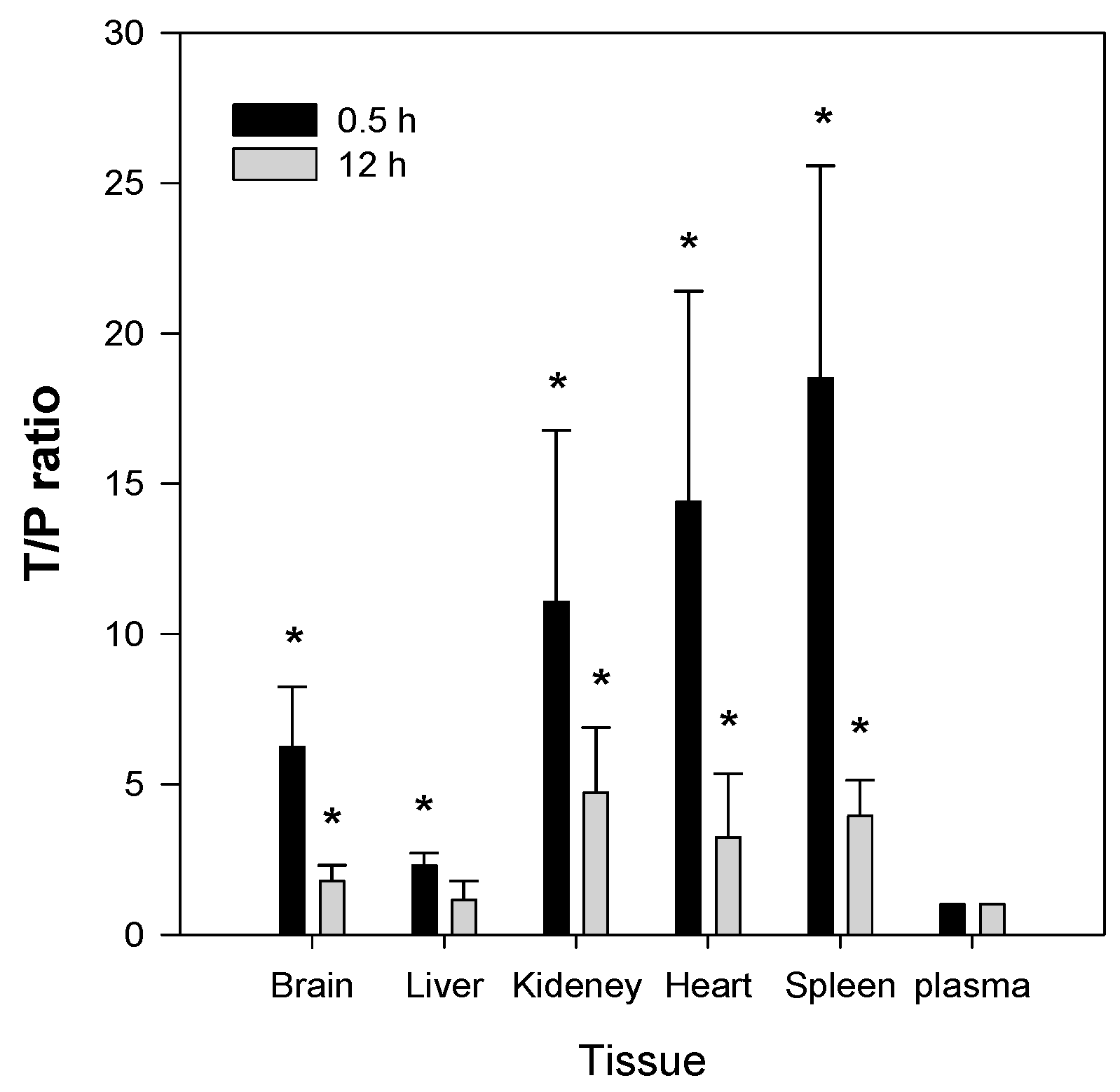
2.5. Tissue Distribution of Jaspine B
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. LC-MS/MS Analysis of Jaspine B
4.3. Pharmacokinetics of Jaspine B
4.4. Tissue Distribution of Jaspine B
4.5. Metabolic Stability of Jaspine B in Rat Liver Microsomes (RLMs)
4.6. Determination of Bile Salts Concentration in Bile
4.7. Determination of the Intestinal Permeability of Jaspine B
4.8. Effect of TC on the Lipophilicity of Jaspine B
4.9. Data Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Indumathy, S.; Dass, C.R. Finding chemo: The search for marine-based pharmaceutical drugs active against cancer. J. Pharm. Pharmacol. 2013, 65, 1280–1301. [Google Scholar] [CrossRef] [PubMed]
- Haefner, B. Drugs from the deep: Marine natural products as drug candidates. Drug Discov. Today 2003, 8, 536–544. [Google Scholar] [CrossRef]
- Sima, P.; Vetvicka, V. Bioactive substances with anti-neoplastic efficacy from marine invertebrates: Porifera and coelenterata. World J. Clin. Oncol. 2011, 2, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Sharma, M.; Joshi, P.; Rawat, D.S. Clinical status of anti-cancer agents derived from marine sources. Anticancer Agents Med. Chem. 2008, 8, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Nastrucci, C.; Cesario, A. From the sea to anticancer therapy. Curr. Med. Chem. 2011, 18, 3551–3562. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, G.; Towle, M.J.; Cheng, H.; Kawamura, T.; TenDyke, K.; Liu, D.; Kishi, Y.; Yu, M.J.; Littlefield, B.A. Induction of morphological and biochemical apoptosis following prolonged mitotic blockage by halichondrin b macrocyclic ketone analog e7389. Cancer Res. 2004, 64, 5760–5766. [Google Scholar] [CrossRef] [PubMed]
- Shida, D.; Takabe, K.; Kapitonov, D.; Milstien, S.; Spiegel, S. Targeting SphK1 as a new strategy against cancer. Curr. Drug Targets 2008, 9, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, G.T.; Maceyka, M.; Milstien, S.; Spiegel, S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discov. 2013, 12, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.G.; Gray, A.I.; Pyne, S.; Pyne, N.J. Resveratrol dimers are novel sphingosine kinase 1 inhibitors and affect sphingosine kinase 1 expression and cancer cell growth and survival. Br. J. Pharmacol. 2012, 166, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.G.; Tonelli, F.; Li, Z.; Lu, X.; Bittman, R.; Pyne, S.; Pyne, N.J. Fty720 analogues as sphingosine kinase 1 inhibitors: Enzyme inhibition kinetics, allosterism, proteasomal degradation, and actin rearrangement in mcf-7 breast cancer cells. J. Biol. Chem. 2011, 286, 18633–18640. [Google Scholar] [CrossRef] [PubMed]
- Pyne, S.; Bittman, R.; Pyne, N.J. Sphingosine kinase inhibitors and cancer: Seeking the golden sword of hercules. Cancer Res. 2011, 71, 6576–6582. [Google Scholar] [CrossRef] [PubMed]
- French, K.J.; Schrecengost, R.S.; Lee, B.D.; Zhuang, Y.; Smith, S.N.; Eberly, J.L.; Yun, J.K.; Smith, C.D. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003, 63, 5962–5969. [Google Scholar] [PubMed]
- Johnson, K.R.; Johnson, K.Y.; Crellin, H.G.; Ogretmen, B.; Boylan, A.M.; Harley, R.A.; Obeid, L.M. Immunohistochemical distribution of sphingosine kinase 1 in normal and tumor lung tissue. J. Histochem. Cytochem. 2005, 53, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Sobue, S.; Iwasaki, T.; Sugisaki, C.; Nagata, K.; Kikuchi, R.; Murakami, M.; Takagi, A.; Kojima, T.; Banno, Y.; Akao, Y.; et al. Quantitative rt-pcr analysis of sphingolipid metabolic enzymes in acute leukemia and myelodysplastic syndromes. Leukemia 2006, 20, 2042–2046. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, I.; Musman, M.; Ohtani, I.I.; Ichiba, T.; Tanaka, J.; Gravalos, D.G.; Higa, T. Pachastrissamine, a cytotoxic anhydrophytosphingosine from a marine sponge, Pachastrissa sp. J. Nat. Prod. 2002, 65, 1505–1506. [Google Scholar] [CrossRef] [PubMed]
- Salma, Y.; Lafont, E.; Therville, N.; Carpentier, S.; Bonnafe, M.J.; Levade, T.; Genisson, Y.; Andrieu-Abadie, N. The natural marine anhydrophytosphingosine, Jaspine B, induces apoptosis in melanoma cells by interfering with ceramide metabolism. Biochem. Pharmacol. 2009, 78, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Stoica, B.A.; Movsesyan, V.A.; Lea, P.M.; Faden, A.I. Ceramide-induced neuronal apoptosis is associated with dephosphorylation of AKT, BAD, FKHR, GSK-3beta, and induction of the mitochondrial-dependent intrinsic caspase pathway. Mol. Cell. Neurosci. 2003, 22, 365–382. [Google Scholar] [CrossRef]
- Yoo, H.; Lee, Y.S.; Lee, S.; Kim, S.; Kim, T.Y. Pachastrissamine from pachastrissa sp. Inhibits melanoma cell growth by dual inhibition of CDK2 and ERK-mediated FOXO3 downregulation. Phytother. Res. 2012, 26, 1927–1933. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, K.; Kwon, M.; Lee, D.; Choi, M.K.; Song, I.S. Differential cytotoxic effects of Jaspine B in various cancer cells. J. Life Sci. 2016, 26, 101–109. [Google Scholar] [CrossRef]
- Choi, M.K.; Song, I.S.; Park, S.R.; Hong, S.S.; Kim, D.D.; Chung, S.J.; Shim, C.K. Mechanism of the stationary canalicular excretion of tributylmethyl ammonium in rats with a ccl4-induced acute hepatic injury. J. Pharm. Sci. 2005, 94, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Parniak, M.A.; Sarafianos, S.G.; Empey, P.E.; Rohan, L.C. In vitro transport characteristics of efda, a novel nucleoside reverse transcriptase inhibitor using caco-2 and mdckii cell monolayers. Eur. J. Pharmacol. 2014, 732, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.A.; Yoon, Y.H.; Choi, K.; Kwon, M.; Goo, S.H.; Cha, J.S.; Choi, M.K.; Lee, H.S.; Song, I.S. Enhanced oral bioavailability of morin administered in mixed micelle formulation with pluronicf127 and tween80 in rats. Biol. Pharm. Bull. 2015, 38, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Oude Elferink, R.P.; Meijer, D.K.; Kuipers, F.; Jansen, P.L.; Groen, A.K.; Groothuis, G.M. Hepatobiliary secretion of organic compounds; molecular mechanisms of membrane transport. Biochim. Biophys. Acta 1995, 1241, 215–268. [Google Scholar] [CrossRef]
- Martinez-Augustin, O.; Sanchez de Medina, F. Intestinal bile acid physiology and pathophysiology. World J. Gastroenterol. 2008, 14, 5630–5640. [Google Scholar] [CrossRef] [PubMed]
- Song, I.S.; Choi, M.K.; Shim, W.S.; Shim, C.K. Transport of organic cationic drugs: Effect of ion-pair formation with bile salts on the biliary excretion and pharmacokinetics. Pharmacol. Ther. 2013, 138, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Song, I.S.; Chung, S.J.; Shim, C.K. Contribution of ion pair complexation with bile salts to biliary excretion of organic cations in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, 515–525. [Google Scholar]
- Song, I.S.; Han, Y.H.; Chung, S.J.; Shim, C.K. Contribution of ion-pair complexation with bile salts to the transport of organic cations across llc-pk1 cell monolayers. Pharm. Res. 2003, 20, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Yoshimitsu, Y.; Oishi, S.; Miyagaki, J.; Inuki, S.; Ohno, H.; Fujii, N. Pachastrissamine (Jaspine B) and its stereoisomers inhibit sphingosine kinases and atypical protein kinase c. Bioorg. Med. Chem. 2011, 19, 5402–5408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, J.L.; Scheig, R.L.; Klatskin, G. The effect of sodium taurocholate on the hepatic metabolism of sulfobromophthalein sodium (bsp). The role of bile flow. J. Clin. Investig. 1970, 49, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jamei, M.; Yeo, K.R.; Tucker, G.T.; Rostami-Hodjegan, A. Prediction of intestinal first-pass drug metabolism. Curr. Drug Metab. 2007, 8, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A. Role of the intestinal bile acid transporters in bile acid and drug disposition. Handb. Exp. Pharmacol. 2011, 201, 169–203. [Google Scholar]
- Prabagar, B.; Yoo, B.K.; Woo, J.S.; Kim, J.A.; Rhee, J.D.; Piao, M.G.; Choi, H.G.; Yong, C.S. Enhanced bioavailability of poorly water-soluble clotrimazole by inclusion with beta-cyclodextrin. Arch. Pharm. Res. 2007, 30, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.W.; Armstrong, A.; Newman, C.; Alakhov, V.; Pietrzynski, G.; Brewer, J.; Campbell, S.; Corrie, P.; Rowinsky, E.K.; Ranson, M. A phase 2 study of sp1049c, doxorubicin in p-glycoprotein-targeting pluronics, in patients with advanced adenocarcinoma of the esophagus and gastroesophageal junction. Investig. New Drugs 2011, 29, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Yang, Q.; Deng, L.; Lu, J.; Chen, J. Phospholipid-tween 80 mixed micelles as an intravenous delivery carrier for paclitaxel. Drug Dev. Ind. Pharm. 2011, 37, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Adedokun, O.J.; Sandborn, W.J.; Feagan, B.G.; Rutgeerts, P.; Xu, Z.; Marano, C.W.; Johanns, J.; Zhou, H.; Davis, H.M.; Cornillie, F.; et al. Association between serum concentration of infliximab and efficacy in adult patients with ulcerative colitis. Gastroenterology 2014, 147, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.T.; Chen, H.H.; Song, I.S.; Savaraj, N.; Ishikawa, T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007, 26, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Song, J.; Bae, H.; Kim, W.J.; Lee, J.Y.; Han, G.H.; Lee, S.K.; Kim, S. Synthesis and biological evaluation of carbocyclic analogues of pachastrissamine. Mar. Drugs 2015, 13, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.K.; Kwon, M.; Ahn, J.H.; Kim, N.J.; Bae, M.A.; Song, I.S. Transport characteristics and transporter-based drug-drug interactions of tm-25659, a novel taz modulator. Biopharm. Drug Dispos. 2014, 35, 183–194. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | IV (10 mg/kg) | |
|---|---|---|
| Plasma | T1/2 (h) | 6.7 ± 1.6 |
| MRT (h) | 3.6 ± 0.5 | |
| AUC12h (ng∙h/mL) | 1286.9 ± 165.6 | |
| AUC∞ (ng∙h/mL) | 1701.9 ± 137.1 | |
| Vss (L/kg) | 21.2 ± 2.6 | |
| CLtotal (mL/min/kg) | 98.3 ± 7.7 | |
| Bile | Xbile (% of dose) | 2.4 ± 0.8 |
| CLbile (mL/min/kg) | 3.1 ± 0.6 | |
| Urine | Xurine (% of dose) | 0.4 ± 0.2 |
| CLurine (mL/min/kg) | 0.5 ± 0.3 | |
| Parameters | Control (n = 3) | Bile Depletion (n = 3) | Bile Depletion + TC (n = 5) | Control + TC (n = 3) |
|---|---|---|---|---|
| Cmax (ng/mL) | 36.3 ± 11.3 | 8.5 ± 2.8 * | 163.4 ± 28.4 * | 174.1 ± 13.0 * |
| Tmax (h) | 2.5 ± 1.3 | 9.3 ± 2.3 * | 1.0 ± 0.5 | 3.3 ± 2.3 |
| T1/2 (h) | 5.5 ± 1.1 | − | 7.2 ± 1.5 | 6.4 ± 2.8 |
| MRT (h) | 4.9 ± 0.4 | 6.1 ± 0.3 * | 4.9 ± 0.5 | 5.5 ± 0.03 |
| AUC12h (ng∙h/mL) | 240.6 ± 57.8 | 80.3 ± 42.7 * | 828.3 ± 180 * | 1406.3 ± 4.3 *,# |
| AUC∞ (ng∙h/mL) | 314.4 ± 76.1 | − | 1201.7 ± 254 * | 2100.9 ± 419 *,# |
| BA (%) | 6.2 | 1.6 | 23.5 | 41.2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, M.-K.; Lee, J.; Nam, S.J.; Kang, Y.J.; Han, Y.; Choi, K.; Choi, Y.A.; Kwon, M.; Lee, D.; Song, I.-S. Pharmacokinetics of Jaspine B and Enhancement of Intestinal Absorption of Jaspine B in the Presence of Bile Acid in Rats. Mar. Drugs 2017, 15, 279. https://doi.org/10.3390/md15090279
Choi M-K, Lee J, Nam SJ, Kang YJ, Han Y, Choi K, Choi YA, Kwon M, Lee D, Song I-S. Pharmacokinetics of Jaspine B and Enhancement of Intestinal Absorption of Jaspine B in the Presence of Bile Acid in Rats. Marine Drugs. 2017; 15(9):279. https://doi.org/10.3390/md15090279
Chicago/Turabian StyleChoi, Min-Koo, Jihoon Lee, So Jeong Nam, Yun Ju Kang, Youjin Han, Kwangik Choi, Young A. Choi, Mihwa Kwon, Dongjoo Lee, and Im-Sook Song. 2017. "Pharmacokinetics of Jaspine B and Enhancement of Intestinal Absorption of Jaspine B in the Presence of Bile Acid in Rats" Marine Drugs 15, no. 9: 279. https://doi.org/10.3390/md15090279
APA StyleChoi, M. -K., Lee, J., Nam, S. J., Kang, Y. J., Han, Y., Choi, K., Choi, Y. A., Kwon, M., Lee, D., & Song, I. -S. (2017). Pharmacokinetics of Jaspine B and Enhancement of Intestinal Absorption of Jaspine B in the Presence of Bile Acid in Rats. Marine Drugs, 15(9), 279. https://doi.org/10.3390/md15090279