2. Results and Discussion
Compound
1 was isolated as a yellowish amorphous solid. The molecular formula of
1 was deduced to be C
22H
17N
3O
4 from the [M + H]
+ peak at
m/
z 388.1296 (calcd. for 388.1297) in the HRESIMS, which required 16 degrees of unsaturation. The
1H and HSQC spectra of
1 showed ten aromatic proton signals at δ
H 8.90 (1H, d,
J = 7.4 Hz), 8.70 (1H, d,
J = 8.6 Hz), 8.23 (1H, d,
J = 7.7 Hz), 8.15 (1H, t,
J = 7.4 Hz), 7.99 (1H, overlapped), 7.98 (1H, overlapped), 7.89 (1H, d,
J = 7.6 Hz), 7.03 (1H, t,
J = 8.0 Hz), 6.49 (1H, t,
J = 7.6 Hz) and 6.42 (1H, d,
J = 8.0 Hz), one methine signal at δ
H 6.06 (1H, q,
J = 6.4 Hz) and one methyl signal at δ
H 1.81 (3H, d,
J = 6.4 Hz) (
Figure S1). Moreover, an exchangeable proton signal of a carboxyl group at δ
H 15.60 (1H, s) was detected on the
1H NMR spectrum using CDCl
3 (
Table 1) (
Figure S2). The
13C (
Figure S3) and HSQC (
Figure S4) spectra displayed ten tertiary sp
2 carbons
(δ
C 172.4, 168.6, 151.5, 145.6, 144.8, 143.9, 143.7, 141.0, 126.6 and 112.2), ten aromatic sp
2 carbons (δ
C 137.8, 136.6, 135.3, 134.7, 133.4, 131.5, 128.7, 128.3, 116.1 and 113.3), an oxygenated methine carbon
(δ
C 49.5) and a methyl carbon
(δ
C 24.3) (
Table 2). The characteristic UV spectrum (λ
max 256 and 371 nm), distinct deshielded aromatic proton signals at δ
H 8.90–7.98, an exchangeable proton signal for a carboxyl group at δ
H 15.60, a downshifted methine proton signal at δ
H 6.06 and four nitrogen-bearing quaternary carbons at δ
C 144.8, 143.9, 143.7 and 141.0 revealed the presence of a saphenic acid bearing phenazine moiety [
7,
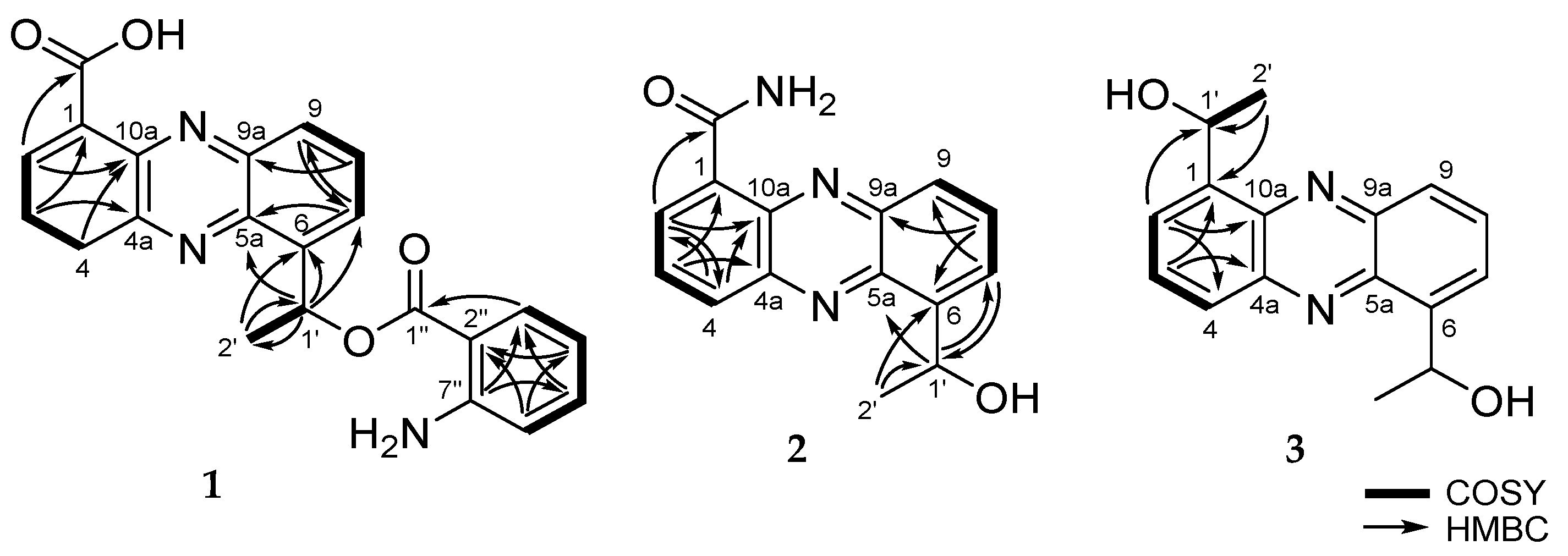
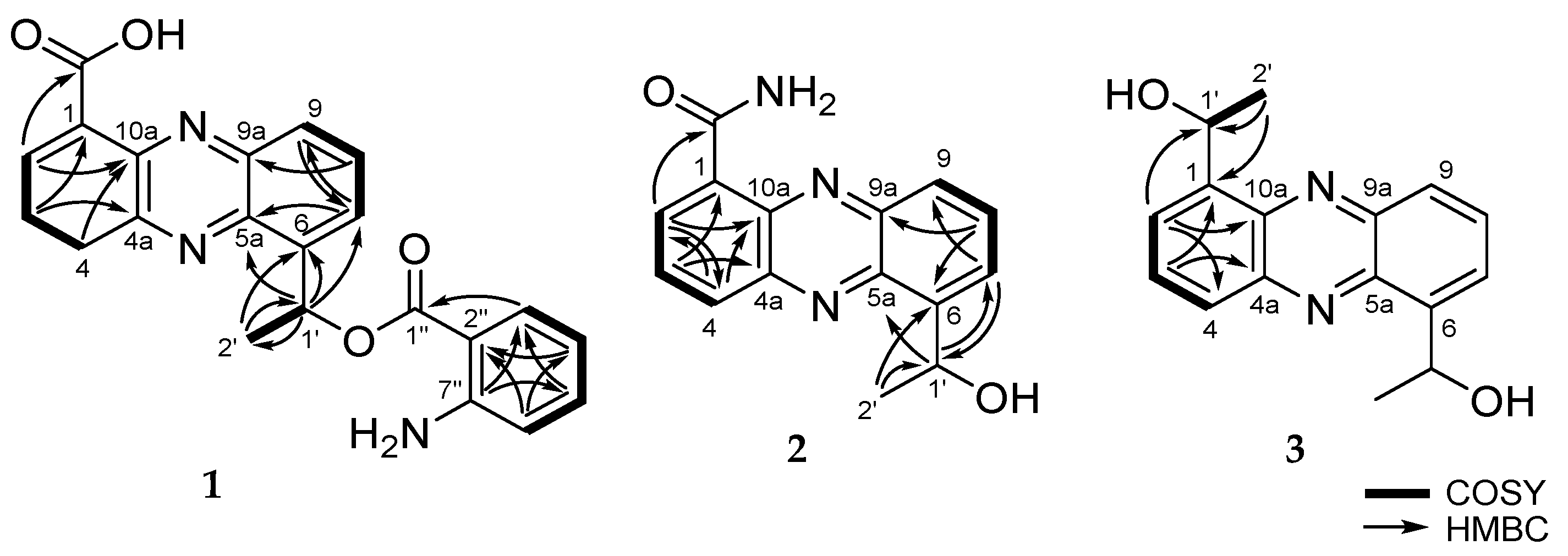
8]. Analysis of the COSY spectrum (
Figure S5) suggested four spin systems: From H-2 (δ
H 8.90) to H-4 (δ
H 8.70), from H-7 (δ
H 7.98) to H-9 (δ
H 8.23), from H-1’ at (δ
H 6.06) to H-2’ (δ
H 1.81) and from H-3” (δ
H 7.89) to H-6” (δ
H 6.42) (
Figure 2). These evidences revealed that
1 had 1,6- or 1,9-disubstituted phenazine ring. Since
1H and
13C NMR chemical shift values of
1 were very similar to
4, as well as already reported saphenic acid [
9], the structure of
1 was concluded to be a 1,6-disubstituted phenazine derivative (
Table 1 and
Table 2). The molecular formula, based on the HR-MS result, revealed that
1 has an amino group. The amino group was linked to a remaining unconnected quaternary carbon C-7” by its chemical shift (δ
C 151.5). These data clearly indicated that
1 belongs to the saphenic acid family with 2-aminobenzoic acid moiety. The position of the carboxylic acid (1-COOH) was determined by the HMBC correlation between H-2 (δ
H 8.90) and the carbonyl carbon
(δ
C 168.6) (
Figure S6). The oxygenated methine proton H-1’ (δ
H 6.06) showed HMBC cross-peaks with C-5a (δ
C 143.7), C-6 (δ
C 145.6) and C-7 (δ
C 128.7). The HMBC correlation between the aromatic methine H-3” (δ
H 7.89) and C-1” (δ
C 172.4) established the presence of 2-amino benzoic ester (
Figure 2). Though we could not directly ascertain the HMBC correlation between the saphenic acid moiety and 2-amino benzoic ester, the ROESY correlation between H-1’ (δ
H 6.06) and H-6” (δ
H 6.42) established their connection (
Figure S7). Furthermore, saphenic acid and 2-amino benzoic ester satisfied the unsaturation number and molecular formula. Thereby, the oxygenated methine C-1’ was found to be connected with C-1” by an ester linkage. Thus, the structure of
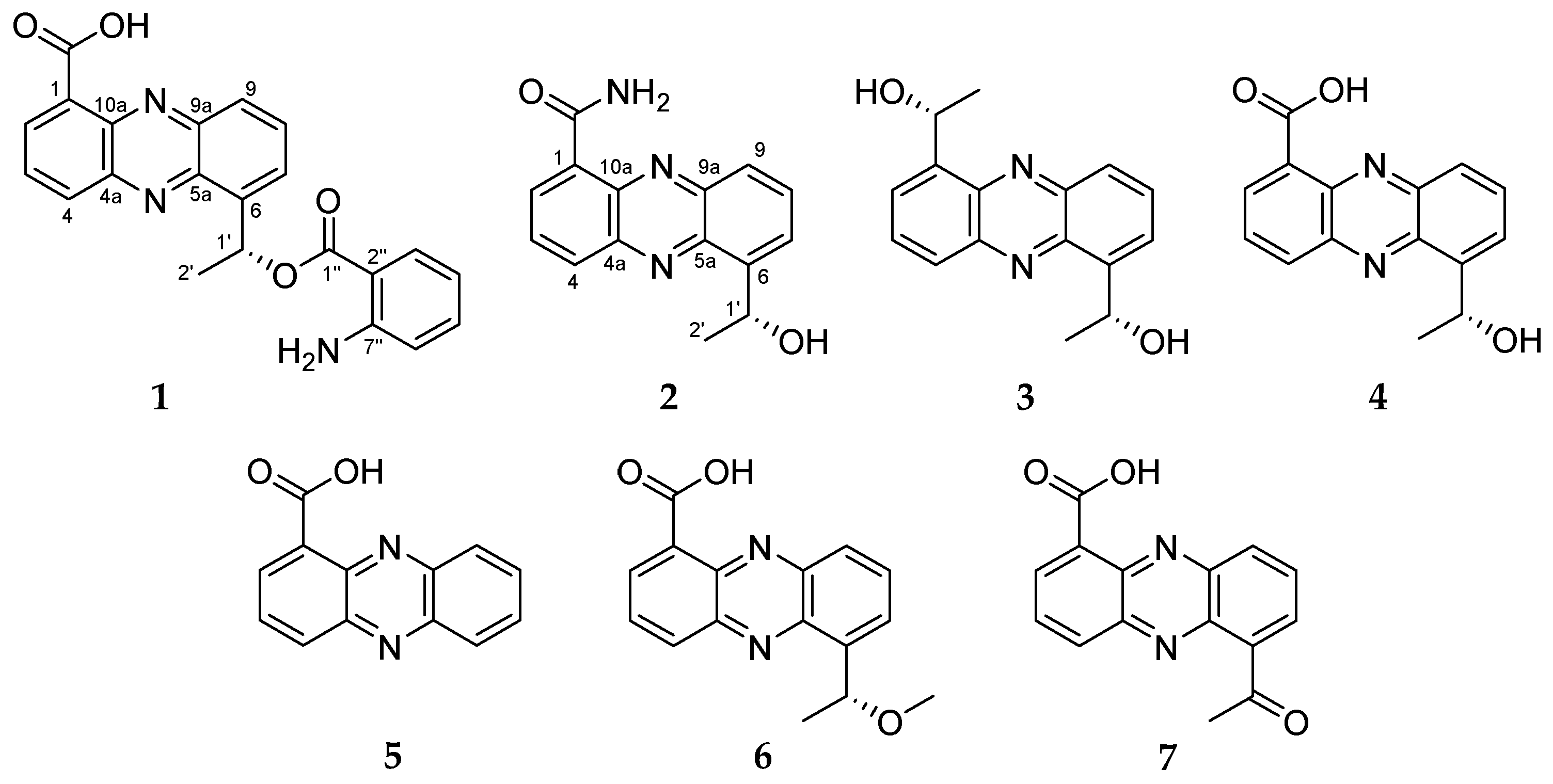
1 was determined as 6-[1-(2-aminobenzoyloxy)ethyl]-1-phenazinecarboxylic acid.
It was supposed that this strain produces phenazine derivatives through the same biosynthesis pathway [
6]. So, we assumed that the stereocenter of C-1’ has the same configuration with (
R)-saphenic acid (
4). To determine the stereochemistry of C-1’ in
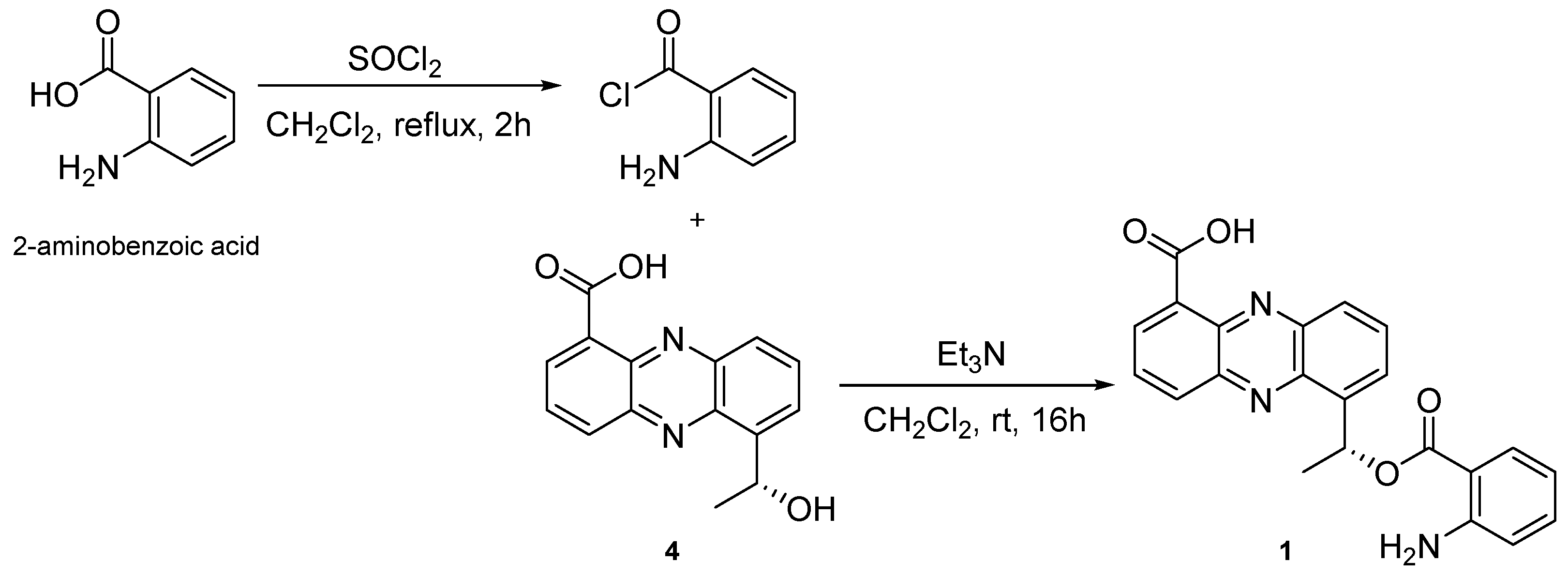
1,
1 was semi-synthesized with
4 and 2-aminobenzoic acid (
Figure 3). Semi-synthesized
1 showed identical
1H NMR and MS spectra with natural
1 (
Figures S26–S28). In addition, natural and semi-synthesized
1 had the same negative optical rotation values (
R-saphenic acid: [α]
−73, (
c 0.05, CHCl
3); natural
1: [α]
−80, (
c 0.05, CHCl
3); semi-synthetic
1: [α]
−40, (
c 0.05, CHCl
3)). Thus, the stereochemistry of C-1’ was determined to be
R-configuration. Peculiarly,
1 was easily changed under the light. However, it could be changelessly stored in the dark and fridge for several weeks.
Compound
2 was purified as a yellowish needle. The molecular formula of
2 was deduced to be C
15H
13N
3O
2 with 11 degrees of unsaturation from the [M + H]
+ peak at
m/
z 268.1088 (calcd. for 268.1086) in the HRESIMS. Based on the HRMS data (
Figure S8),
2 had one more nitrogen and one less oxygen than saphenic acid (
4). Chemical shifts and splitting patterns of
2 (
Figures S9 and S10) were closely similar to those of
4. The main difference was the appearance of two exchangeable proton signals at δ
H 10.69 and δ
H 6.29 for amide protons in
2 instead of the exchangeable proton signal at δ
H 15.44 of a carboxyl group in
1. The amide protons showed correlations each other in the COSY spectrum (
Figure S12). Therefore, the planer structure of
2 was elucidated as a new derivative of saphenic acid and named as saphenic amide.
2 also had a negative optical rotation value [α]
−13, (
c 0.1, CHCl
3), which was in agreement with that of
4 [α]
−13 (
c 0.1, CHCl
3), suggesting that the stereochemistry of C-1’ was
R-form.
Compound
3 was obtained as a yellowish amorphous solid. Based on the HRESIMS spectrum (
Figure S20), the molecular formula was established as C
16H
16N
2O
2 with 10 degrees of unsaturation from the [M + H]
+ peak at
m/
z 269.1290 (calcd. for 269.1290). The
1H NMR (
Figure S15) and HSQC (
Figure S17) spectra of
3 showed three aromatic proton signals at δ
H 8.19 (1H, d,
J = 8.2 Hz), 7.83 (1H, t,
J = 6.9 Hz) and 7.78 (1H, d,
J = 6.9 Hz), an oxygenated methine proton signal at δ
H 5.71 (1H, q,
J = 6.5 Hz) and a methyl signal at δ
H 1.81 (1H, d,
J = 6.5 Hz).
13C NMR and HSQC spectra displayed three quaternary carbons (δ
C 142.8, 141.4 and 141.0), three aromatic sp
2 carbons (δ
C 131.4, 128.6 and 127.7), an oxygenated methine carbon (δ
C 68.9) and a methyl carbon (δ
C 23.8). The pattern of
1H NMR spectrum and UV maximum at 255 and 366 nm revealed the presence of a typical phenazine moiety. The main difference between
3 and the other isolated compounds was the disappearance of a carboxyl group in
3.
1H and
13C NMR data of
3 showed only a half set of signals and
3 had no optical activity, indicating that
3 consisted of two equivalent molecular portions and had a symmetric structure of 1-(2,3-diaminophenyl)ethanol. By considering NMR chemical shifts and biosynthetic pathway of
3 and
4, the stereochemistry of C-1’ in
3 was determined to be the same as that in
4. Thus, the structure of
3 was determined to be a new saphenic acid derivative and named as saphenol.
The structures of the known compounds
4–
7 were identified as the previously reported (
R)-saphenic acid (
4), phenazine-1-carboxylic acid (
5), 6-(1-hydroxyehtyl)phenazine-1-carboxylic acid (
6) and 6-acetyl-phenazine-1-carboxylic acid (
7) by the comparison of NMR, MS and optical rotation values with the literature [
9,
10]. When comparing with saphenic acid (
4),
5 was a lack of 1-hydroxy-ethyl group,
6 was methylated form of saphenic acid (
4), and
7 was oxidized saphenic acid. As previously reported [
9], saphenic acid (
4) of yellow needle form turned brown after several hours in day light as well as fluorescent light. Moreover,
4 was quickly turned from yellow into orange in hydrophilic solvents like methanol and dimethyl sulfoxide under the light within a few minutes.
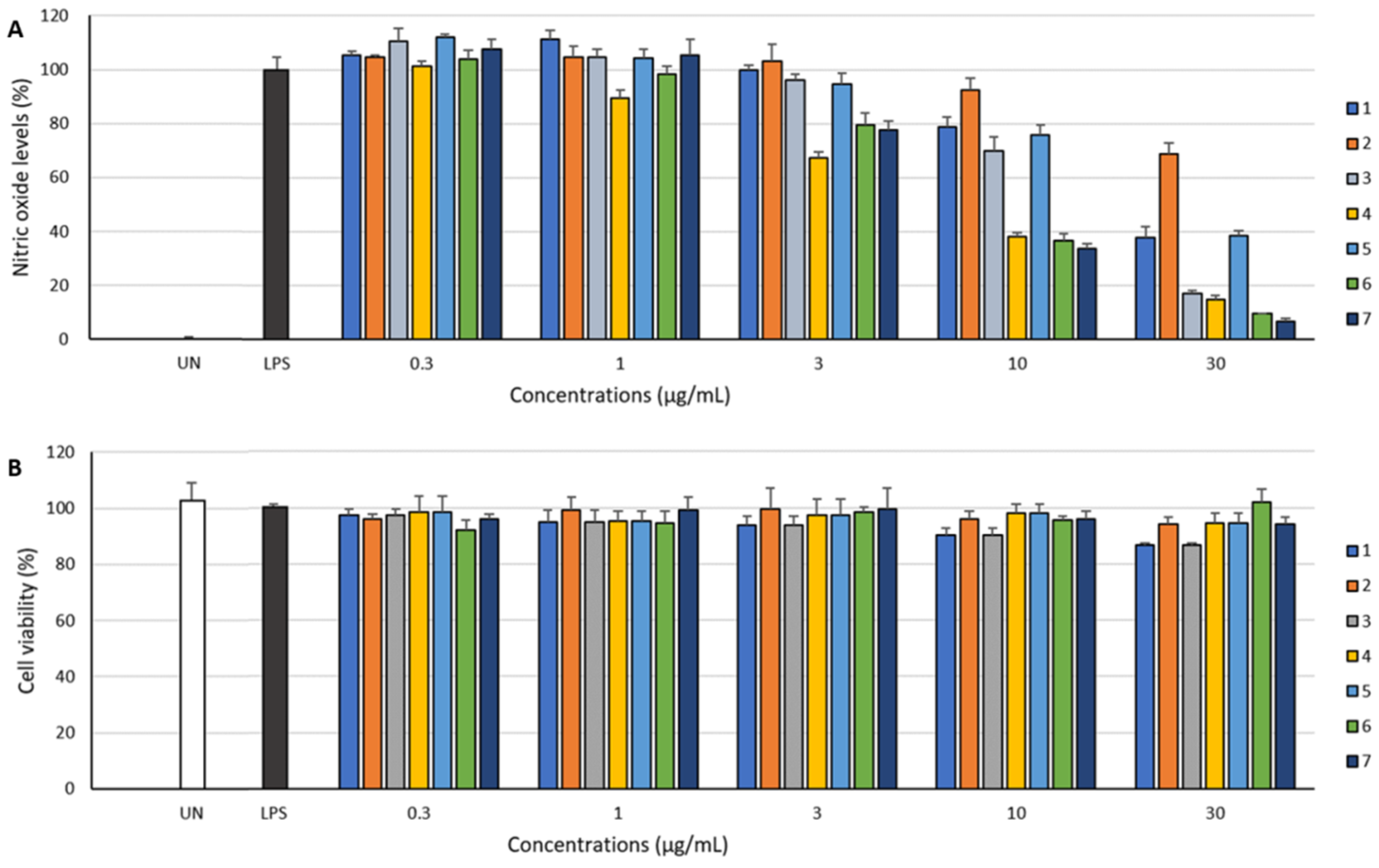
The anti-inflammatory activity of
1–
7 was assessed by NO assay, and their cytotoxicity on RAW 264.7 cells was evaluated by 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) assay. The biological activities of the previously reported compounds
4–
7 have never been reported [
9]. Even though saphenic acid is a common pharmacophore for antibiotics and antitumor reagents, its biological activity has never been reported [
11].
1–
7, except for
2, exhibited inhibitory effect against LPS-induced NO production in RAW 264.7 cells (
Figure 4). Moreover, these compounds did not affect the viability of RAW 264.7 cells at concentrations up to 30 μg/mL (
Figure 4). Among the isolated compounds,
6 inhibited NO production with the highest EC
50 value of 19.6 μM. Saphenic acid (
4) and their derivatives (
6 and
7) showed a similar inhibition tendency (
Table 3). Methylated (
6) and oxidized (
7) saphenic acids showed no significant difference in the activity. However, these compounds were about twice stronger than saphenic acid substituted with 2-amino benzoic acid (
1, EC
50 = 46.8 μM). Furthermore, when there is no functional group at C-6 of phenazine (
5, EC
50 = 76.1 μM), the activity was lowest. We could not carry out further biological activity tests due to insufficient amount. However, our results suggest that NO inhibitory activity is influenced by the carboxylic acid at C-1 as well as the presence of simple functional group at C-6 of phenazine ring.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotation was measured on a Rudolph research analytical Autopol III S2 polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA). UV spectra were obtained on a Shimadzu UV-1650PC spectrophotometer (Shimadzu Corporation, Kyoto, Japan). IR spectra were recorded on a JASCO FT/IR-4100 spectrophotometer (JASCO Corporation, Tokyo, Japan). NMR spectra were collected on a Bruker AVANCE III 600 spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) operating at 600 MHz (1H) and 150 MHz (13C). LRMS data were acquired on an Agilent 6100 single quadrupole mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) with electron ionization (EI). HRESIMS was obtained with a Waters SYNPT G2 Q-TOF mass spectrometer (Waters Corporation, Milford, CT, USA) at Korea Basic Science Institute (KBSI) in Cheongju, Republic of Korea. HPLC was performed using a PrimeLine binary pump (Analytical Scientific Instruments, Inc., El Sobrante, CA, USA) with Shodex RI-101 refractive index detector (Shoko Scientific Co. Ltd., Yokohama, Japan) and S3210 variable UV detector (Schambeck SFD GmbH, Bad Honnef, Germany). Columns used for HPLC were YMC-Pack Silica (250 mm × 10 mm i.d., 5 µm), YMC ODS-A (250 mm × 10 mm i.d., 5 μm) and YMC-Triart C18 (250 mm × 10 mm i.d., 5 µm and 250 mm × 4.6 mm i.d., 5 µm). Silica gel 60 (Merck, 230–400 mesh) and RP-C18 silica gel (YMC-Gel ODS-A, 12 nm S-75 μm) were used for open column chromatography. Mass culture was carried out in a 100 L fermenter (Fermentec Corporation, Cheongju, Republic of Korea). All solvents used were either HPLC grade or distilled prior to use.
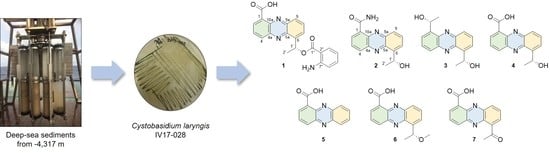
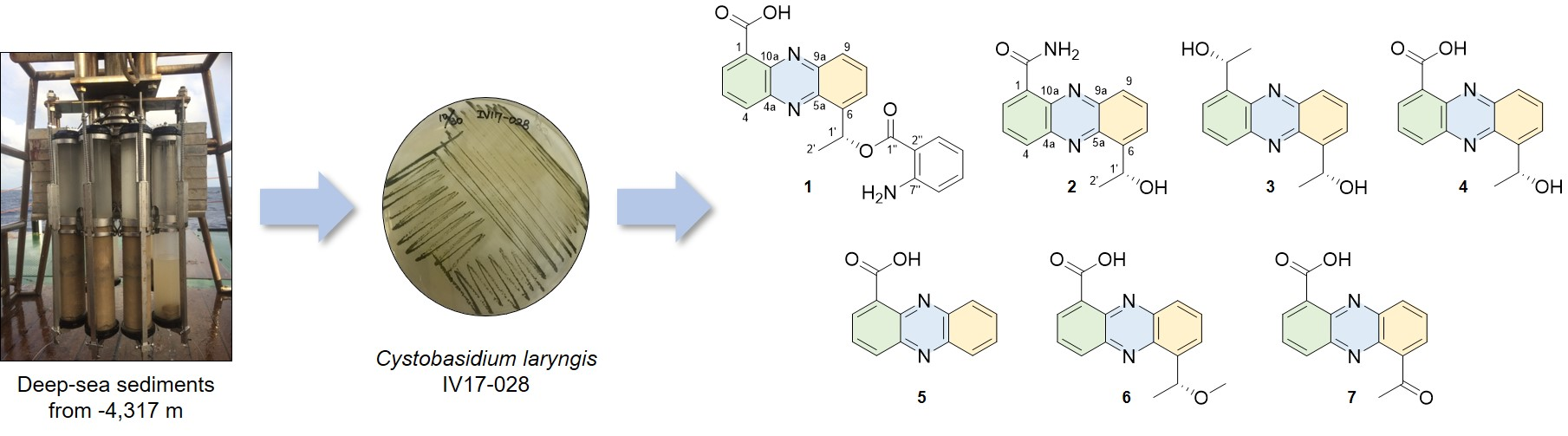
3.2. Microbial Material
The strain IV17-028 was isolated from a deep-sea sediment sample collected using a multi-corer (MC) mounted on the R/V ISABU from the Indian Ocean (date: 9 August 2017, latitude: 008° 07.5121′ S, longtitude: 068° 06.6033′ E, depth: 4317 m). A total of 1 g of the heated sediment sample in the dry oven at 60 °C for 30 min, was spread onto the surface of Bennett (BN)’s agar plates (1% glucose, 0.2% tryptone, 0.1% yeast extract, 0.1% beef extract, 0.5% glycerol, 3.2% artificial sea salt and 1.8% agar, pH 7.02 before sterilization). The plates were incubated for 5 days at room temperature and then 20 days at 4 °C fridge. The resulting colony of dark green color was transferred and maintained on the BN agar plate. The colors of the colonies formed were beige to green on the BN agar medium. The strain was identified as Cystobasidium laryngis on the basis of 26S rRNA sequence analysis. The sequence of IV17-028 was submitted to GenBank under accession number MK131277.
3.3. Fermentation and Isolation of Metabolites
The seed and production cultures were carried out in the BN broth medium (1% glucose, 0.2% tryptone, 0.1% yeast extract, 0.1% beef extract, 0.5% glycerol, 3.2% artificial sea salt, pH 7.02 before sterilization). The seed culture was performed in a 2 L baffled Erlenmeyer flask containing 600 mL of autoclaved medium at 28 °C for 5 days on a rotary shaker at 140 rpm. The seed culture broth was inoculated into a 100 L fermenter containing 60 L of the broth medium under the aseptic condition. The fermenter was operated at 28 °C, 50 rpm and airflow rate of 20 liter per minute (LPM) in the dark for 10 days. After cultivation, the broth was extracted with ethyl acetate (EtOAc, 60 L) twice. The EtOAc extract was evaporated to obtain crude extract (46.5 g). A portion of crude extract (12.8 g) was partitioned with 85% methanol (MeOH) in H2O and hexane (Hex). 85% MeOH layer (1.72 g) was subjected to ODS vacuum column chromatography followed by stepwise gradient elution with MeOH/H2O (1:4, 2:3, 3:2, 4:1 and 5:0, v/v) as eluent. Insoluble part (23.1 mg) containing 4 obtained from the 60% MeOH fraction was recrystallized from acetone to give a pure 4 (9.9 mg) as yellow needles. The 60% MeOH fraction was again applied to an ODS flash column chromatography with a MeOH/H2O solvent system (4:6, 5:5, 6:4, 7:3 and 8:2, v/v). The subfraction eluted with MeOH/H2O (6:4) was purified by a reversed-phase HPLC (YMC ODA-S, 250 × 10 mm i.d., 5 μm; 48% MeOH in H2O; flow rate: 2.0 mL/min; detector: RI) to yield crude 2 (2.3 mg, tR 50 min). The crude 2 was recrystallized in MeOH to afford a pure 2 (1.1 mg) as yellow crystals. The 80% MeOH fraction was purified by a RP-HPLC (YMC ODS-A, 250 × 10 mm i.d., 5 μm; 28% MeCN in H2O containing 0.01% TFA; flow rate: 3.5 mL/min; detector: UV; wavelength: 254 nm) to give 3 (0.7 mg, tR 30 min). The remaining crude extract (33.7 g) was suspended in H2O and sequentially partitioned with Hex, CHCl3, EtOAc and BuOH. The CHCl3 soluble fraction (5.94 g) was separated on silica gel flash column chromatography with MC/MeOH (10:0, 20:1, 10:1 and 5:1, v/v). The MC:MeOH (20:1) fraction was collected into nine fractions. Subfractions 3–5 (34.1 mg) from the MC:MeOH (20:1) fraction was purified by a normal phase HPLC (YMC-Pack SIL; 250 × 10 mm i.d., 5 μm; EtOAc:Hex (2:8); flow rate: 2.0 mL/min; detector: RI) to yield 5 (0.7 mg, tR 35 min) and 6 (0.5 mg, tR 25 min). Subfractions 6 and 7 (4.8 g) were again applied to an ODS-A vacuum column chromatography by stepwise gradient elution with MeOH/H2O (1:4, 2:3, 3:2, 4:1 and 5:0, v/v). Each fraction was divided into three subfractions. The third 80%MeOH fraction (468.5 mg) was separated with a normal phase HPLC (YMC-Pack SIL; 250 × 10 mm i.d., 5 μm; EtOAc:Hex:MeOH (1:1:0.02); flow rate: 2.0 mL/min; detector: RI) to afford 1 (1.7 mg, tR 8 min), 4 (2.1 mg, tR 14 min) and 7 (1.3 mg, tR 12 min).
Compound
1: Yellowish amorphous; [α]
−13 (
c 0.1, CHCl
3); UV (CHCl
3) λ
max (log ε) 371 (1.25), 256 (1.83) nm; IR (CHCl
3)
νmax 3727, 3625, 2883, 1222, 770 cm
−1;
1H and
13C NMR data (CD
3OD), see
Table 1 and
Table 2;
1H NMR (CDCl
3, 600 MHz) δ
H 15.60 (1H, s), 8.99 (1H, d,
J = 6.7 Hz), 8.62 (1H, d,
J = 8.6, 6.7 Hz), 8.15 (1H, d,
J = 8.3 Hz), 8.05 (1H, t,
J = 7.6 Hz), 7.98 (1H, d,
J = 7.8 Hz), 7.95 (1H, d,
J = 6.8 Hz), 7.91 (1H, t,
J = 8.5 Hz), 7.10 (1H, t,
J = 8.1 Hz), 6.56 (1H, t,
J = 7.4 Hz), 6.39 (1H, d,
J = 8.6 Hz), 6.03 (1H, q,
J = 14.2, 7.6 Hz), 1.82 (3H, d,
J = 6.7 Hz); HRESIMS
m/
z 388.1296 [M + H]
+ (calcd. for C
22H
18N
3O
4, 388.1297).
Saphenic amide (
2): Yellowish needle; [α]
−13 (
c 0.1, CHCl
3); UV (CHCl
3) λ
max (log ε) 370 (0.87), 255 (1.52) nm; IR (CHCl
3)
νmax 3328, 2922, 1664, 1218, 770 cm
−1;
1H and
13C NMR data (CDCl
3), see
Table 1 and
Table 2; HRESIMS
m/
z 268.1088 [M + H]
+ (calcd. for C
15H
14N
3O
2, 268.1086).
Saphenol (
3): Yellowish amorphous; [α]
0 (
c 0.1, CHCl
3); UV (CHCl
3) λ
max (log ε) 366 (0.61), 255 (1.41) nm; IR (CHCl
3)
νmax 3413, 2957, 1218, 770 cm
−1;
1H and
13C NMR data (CDCl
3), see
Table 1 and
Table 2; HRESIMS
m/
z 269.1290 [M + H]
+ (calcd. for C
16H
17N
2O
2, 269.1290).
Saphenic acid (
4): Yellowish needle; [α]
−13 (
c 0.1, CHCl
3);
1H and
13C NMR data (CDCl
3), see
Table 1 and
Table 2; LREIMS
m/
z 269.1 [M + H]
+.
Phenazine-1-carboxylic acid (5): Yellowish amorphous; 1H NMR (CDCl3, 600 MHz) δH 15.58 (1H, s), 8.97 (1H, d, J = 7.0 Hz), 8.52 (1H, d, J = 8.7 Hz), 8.34 (1H, d, J = 8.3 Hz), 8.28 (1H, d, J = 8.64 Hz), 8.04–7.96. (1H × 3, overlapped); LREIMS m/z 225.1 [M + H]+.
6-(1-hydroxyehtyl)phenazine-1-carboxylic acid (6): Yellowish amorphous; [α] −60° (c 0.05, CHCl3); 1H NMR (CDCl3, 600 MHz) δH 15.62 (1H, s), 8.95 (1H, d, J = 7.0 Hz), 8.53 (1H, d, J = 8.7 Hz), 8.17 (1H, d, J = 8.3 Hz) 8.05 (1H, br d), 8.03–8.00 (1H × 2, overlapped), 5.76 (1H, q, J = 12.9, 6.5 Hz), 3.42 (3H, s), 1.62 (3H, d, J = 6.4 Hz); LREIMS m/z 283.1 [M + H]+.
6-acetylphenazine-1-carboxylic acid (7): Yellowish amorphous; 1H NMR (CDCl3, 600 MHz) δH 15.26 (1H, s), 9.01 (1H, dd, J = 7.0, 1.3 Hz), 8.55 (1H, dd, J = 8.7, 1.3 Hz), 8.41 (1H, dd, J = 8.7, 1.3 Hz), 8.29 (1H, dd, J = 6.9, 1.3 Hz), 8.08 (1H, dd, J = 8.6, 7.1 Hz), 8.05 (1H, dd, J = 8.6, 6.9 Hz), 3.07 (3H, s); LREIMS m/z 267.1 [M + H]+.
3.4. Semi-Synthesis of 1
To a solution of 2-aminobenzoic acid (50.0 mg, 0.36 mM) in toluene (1.2 mL) was added thionyl chloride solution (1.8 mL, 1.8 mM) at room temperature and the mixture was refluxed for 3 h. After completion of the reaction was confirmed by TLC, the solvent was removed under vacuum to obtain the crude acid chloride as yellow oil, which was used for further reaction without purification [
12].
To a solution of (
R)-saphenic acid (
4) (5.4 mg, 0.02 mM) in DCM (200 μL) was added triethylamine (5 μL) at 0 °C and stirred for 10 min. To this mixture, the solution of acid chloride (150 μL, 0.02 mM) in DCM (2.7 mL) was added drop wise at 0 °C and stirred at room temperature for 16 h. The reaction solution was evaporated in vacuo and separated on PTLC (Hex-EtOAc, 4:6). The fractions containing a yellowish spot (Rf 0.1 ~ 0.4) were combined and further purified by RP-HPLC (YMC Triart-C
18, 250 × 4.6 mm i.d., 5 μm; 45% MeCN in H
2O containing 0.01% TFA; flow rate: 1.0 mL/min; detector: RI) to give semi-synthesized
1 (0.2 mg, t
R 32 min).
1H NMR (CDCl
3, 600 MHz) δ
H 15.61 (1H, s), 8.99 (1H, d,
J = 5.8 Hz), 8.61 (1H, d,
J = 7.5 Hz), 8.14 (1H, d,
J = 8.6 Hz), 8.05 (1H, t,
J = 7.26 Hz), 7.95 (1H, d,
J = 7.8 Hz), 7.94 (1H, d,
J = 5.5 Hz), 7.89 (1H, t,
J = 8.5 Hz), 7.08 (1H, d,
J = 7.1 Hz), 6.54 (1H, t,
J = 7.4 Hz), 6.35 (1H, d,
J = 8.6 Hz), 6.03 (1H, q,
J = 12.7, 5.9 Hz), 1.80 (3H, d,
J = 6.7 Hz) (
Figure S26).
3.5. Nitrite Quantification
NO
2− accumulation was used as an indicator of NO production as described previously [
13]. RAW 264.7 cells were plated at 5 × 10
5 cells/mL, pre-treated with various concentrations (0.3, 1, 3, 10 or 30 μg/mL) of
1–
7 and stimulated with LPS (200 ng/mL) for 24 h. The supernatants were mixed with an equal volume of Griess reagent (1% sulfanilamide, 0.1% naphthylethylenediamine dihydrochloride and 2% phosphoric acid) and incubated at room temperature for 10 min. NaNO
2 was used to generate a standard curve, and the concentration of nitrite in the medium was determined by measuring optical density at 540 nm [
13]. Cell viability was determined by XTT assay as described previously [
14]. And it was confirmed that the concentration of compounds used in this study has no significant effect on cell viability.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}