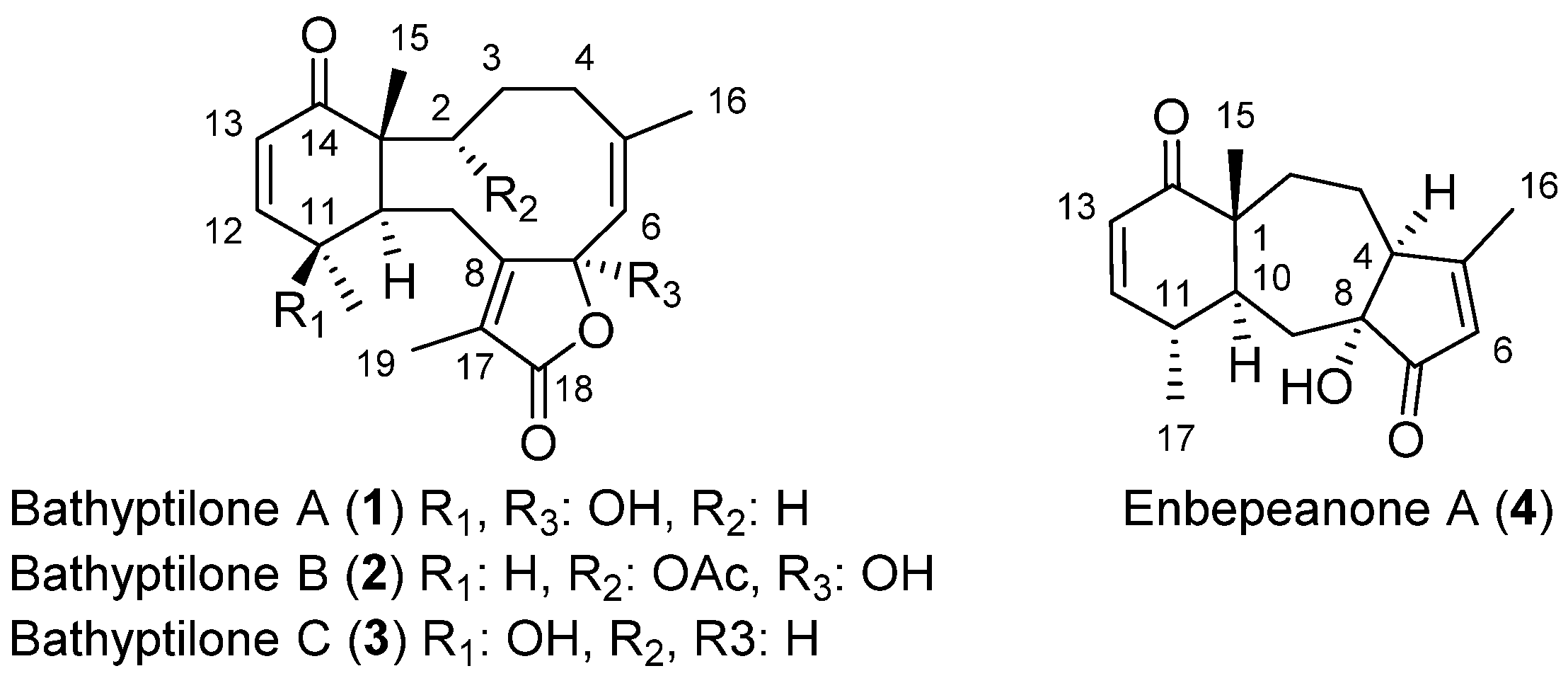
2. Results and Discussion
The coral Anthoptilum grandiflorum was collected by trawl at a depth of between 662 and 944 m during an austral autumn cruise aboard the R/V Nathaniel B. Palmer (NBP) in the vicinity of the Scotia Arc. The sea pens were frozen and freeze-dried, followed by an exhaustive extraction in refluxing DCM using a Soxhlet extractor. Normal-phase MPLC of the 10.6 g Soxhlet extract was conducted for initial fractionation. Normal- and reverse-phase HPLC yielded four new terpenoid metabolites (1–4).
Bathyptilone A (
1) was isolated as a crystalline solid with a molecular formula of C
20H
26O
5, based on HRESIMS with supporting
1H and
13C NMR data (
Table 1). The
13C NMR spectrum indicated the presence of a ketone, an ester-equivalent, four quaternary, four methine, four methylene and four methyl groups, accounting for all carbons and twenty-four of the twenty-six protons. Further, one quaternary carbon had a shift (δ
c 69.3) indicative of an oxygen-bearing sp
3 carbon, while a second quaternary carbon (δ
c 106.2) appeared as a ketal-type carbon. A tricyclic scaffold was derived by the degree of unsaturation, as five of the eight unsaturations could be assigned to double bond functions (ketone, δ
C 203.7; ester-equivalent, δ
C 171.7; and six olefinic carbons, δ
C 160.7, 151.7, 145.3, 127.4, 125.8, 124.1).
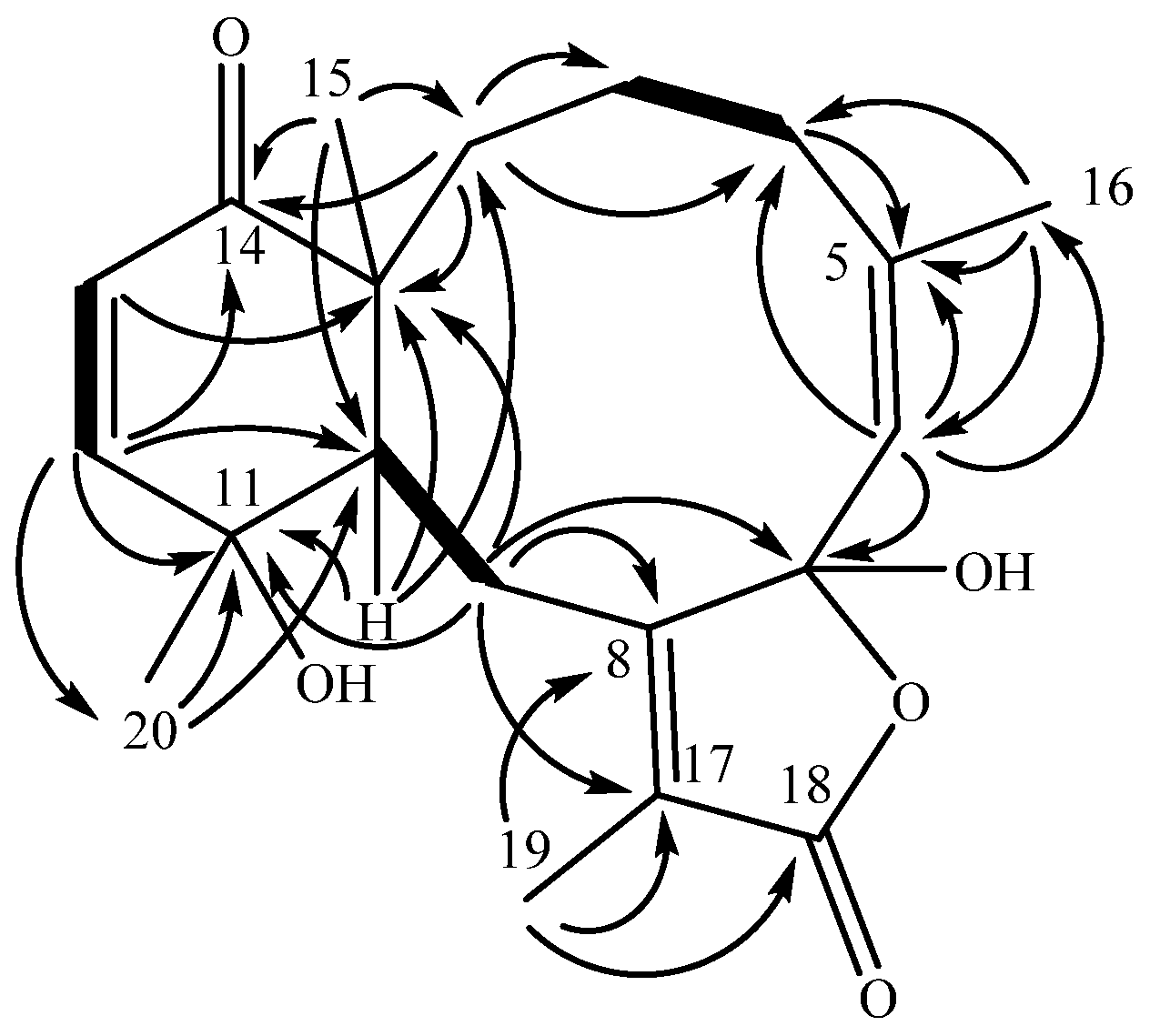
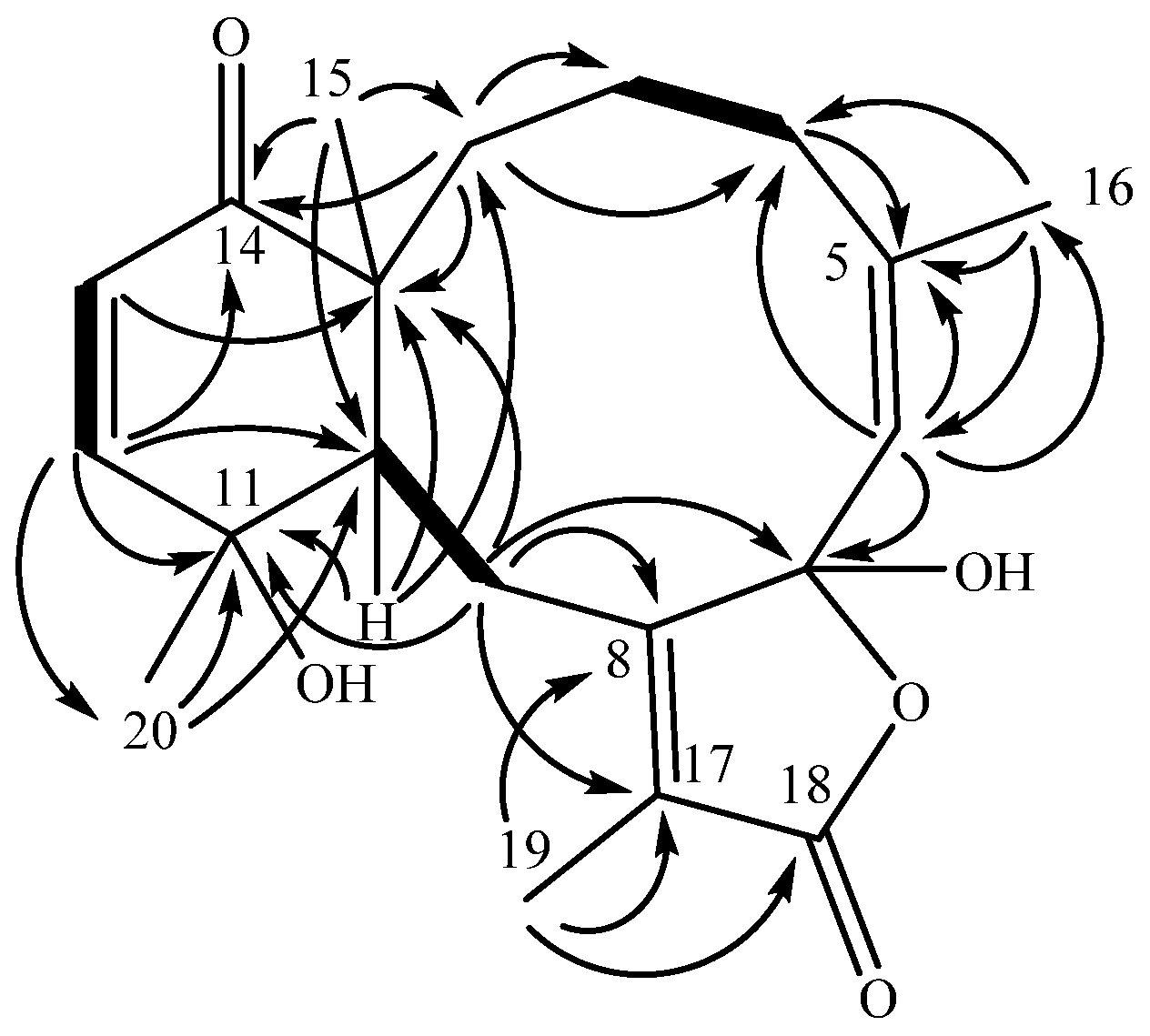
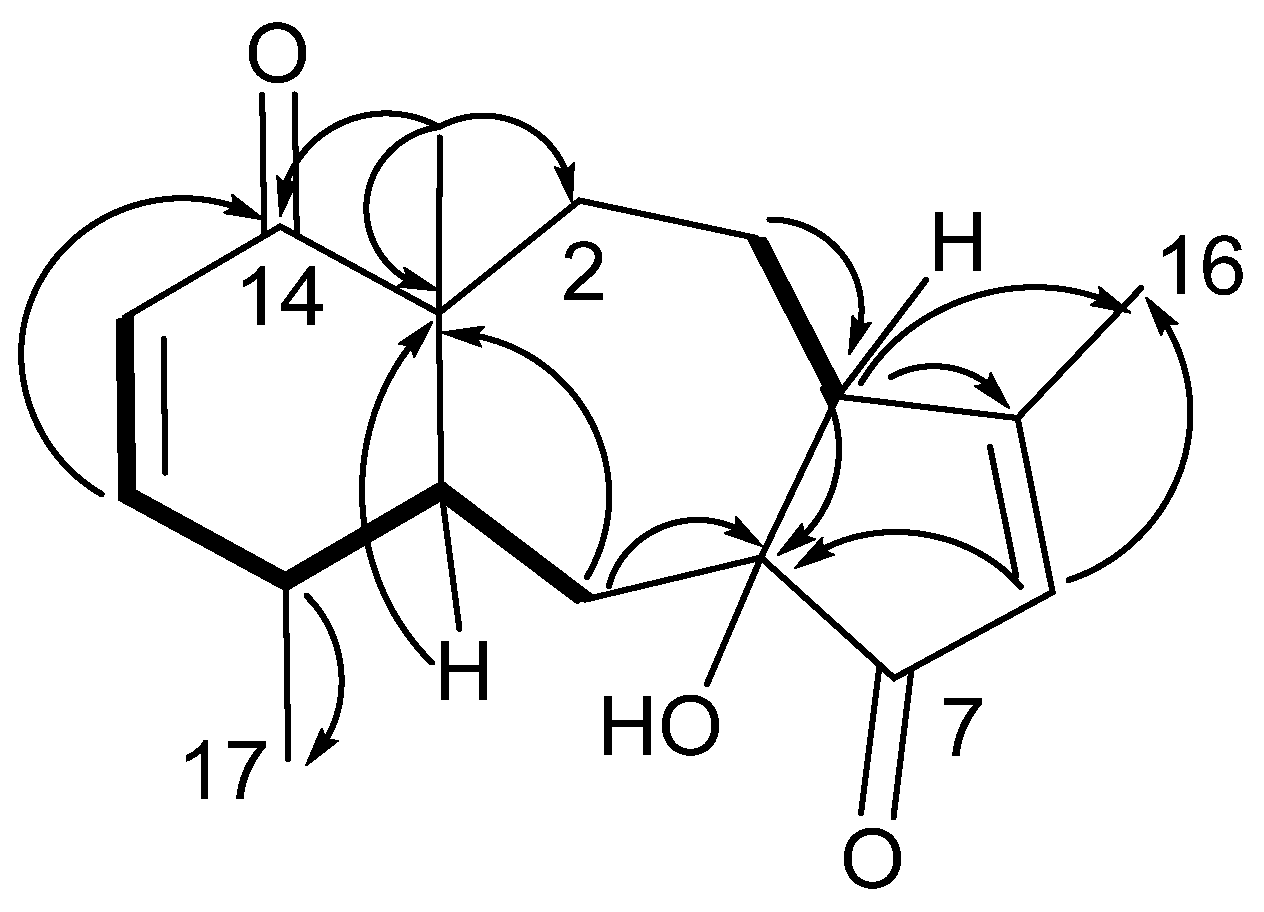
A cyclohexenone ring was constructed from COSY and HMBC data. Two
3JHH coupled olefinic protons (
Figure 1), δ
H 6.63 (H-12) and δ
H 5.97 (H-13), displayed HMBC correlations to δ
C 203.7 (C-14), establishing an α,β-unsaturated ketone. Proton H-12 displayed further HMBC connectivity to an aliphatic methine δ
C 41.4 (C-10), as well as to a quaternary, oxygen-bearing carbon (δ
C 69.3, C-11). Proton H-13 similarly extended the cyclohexenone substructure with HMBC correlation to an aliphatic quaternary carbon (δ
C 50.2, C-1), which, by virtue of correlations from a methyl singlet that networked C-1, -10, -14, into the HMBC spin system, completed the cyclohexenone substructure (
Figure 1). The cyclohexenone portion could be decorated at C-11 with the C-20 methyl group (δ
C 30.4), which displayed HMBC correlations to C-10, -11 and -12 (δ
C 151.7). Additional corroboration of the six-membered ring was obtained from H-10, with HMBC correlations to C-1 and C-11. This cyclohexenone partial structure has two open valences, at C-1 and C-10.
The elaboration of the cyclohexenone resulted in a fused cyclodecene ring. Besides previously described correlations into the cyclohexenone ring, H3-15 (δH 1.16) displayed a further correlation to a methylene carbon at δC 21.8 (C-2). Although proton H-2a (δH 1.54) and H-2b (δH 1.84) overlapped protons of H-3a (δH 1.52) and H-4a (δH 1.85), respectively, preventing clear assignment of COSY correlations, it was observed that H-2b and C-4 (δC 29.5) were correlated in the HMBC spectrum. Both H-3a and H-3b displayed HMBC correlations to C-2, and H-3b further correlated to C-1 (δC 50.2), establishing an alkyl substituent at C-1. Proton H-4a was found to extend the alkyl group through HMBC correlations to two olefinic carbons, C-5 (δC 145.3) and C-6 (δC 124.1). The HMBC also established that quaternary C-5 bears a vinyl methyl δH 1.78 (H3-16) based on HMBC correlations of H-6 (δC 5.35) to C-16 (δC 23.5) and reciprocal correlations from H3-16 to C-6, as well as C-5 (δC 145.3). Further development of this partial structure ended at a quaternary, acetal-type carbon at δC 106.2 (C-7) based on an HMBC correlation from H-6.
An isolated spin system was observed in the COSY spectrum between the broad doublet δH 3.14 (H-10) and the doublet of doublets δH 3.25 (H-9b), the latter of which also correlated to H-9a (δH 2.47). HMBC extended that system further based on H2-9 correlations to two quaternary olefinic carbons, C-8 (δC 160.7) and C-17 (δC 125.8). A key correlation between H-9a and the acetal-like carbon C-7 (δc 106.2) completed a C10 macrocycle.
Remaining to be accounted were an ester-type carbon (δC 171.7, C-18) and a vinyl methyl group (δH 1.95, H3-19), and two open valencies on C-7 (δc 106.2) and C-17 (δC 125.8). The vinyl methyl group correlated in the HMBC spectrum to quaternary olefins C-8 and C-17, locating the methyl group on one of the open valencies on C-17. Methyl protons H3-19 also displayed an HMBC correlation to C-18, accounting for all the carbon atoms in the molecular formula and requiring that the remaining two oxygen atoms occupy the open valencies on C-7 and C-18, resulting in a γ-lactone bearing a C-7 hemiacetal. The planar structure that results from these connections identifies bathyptilone A (1) as a member of the briarane family of octocoral metabolites.
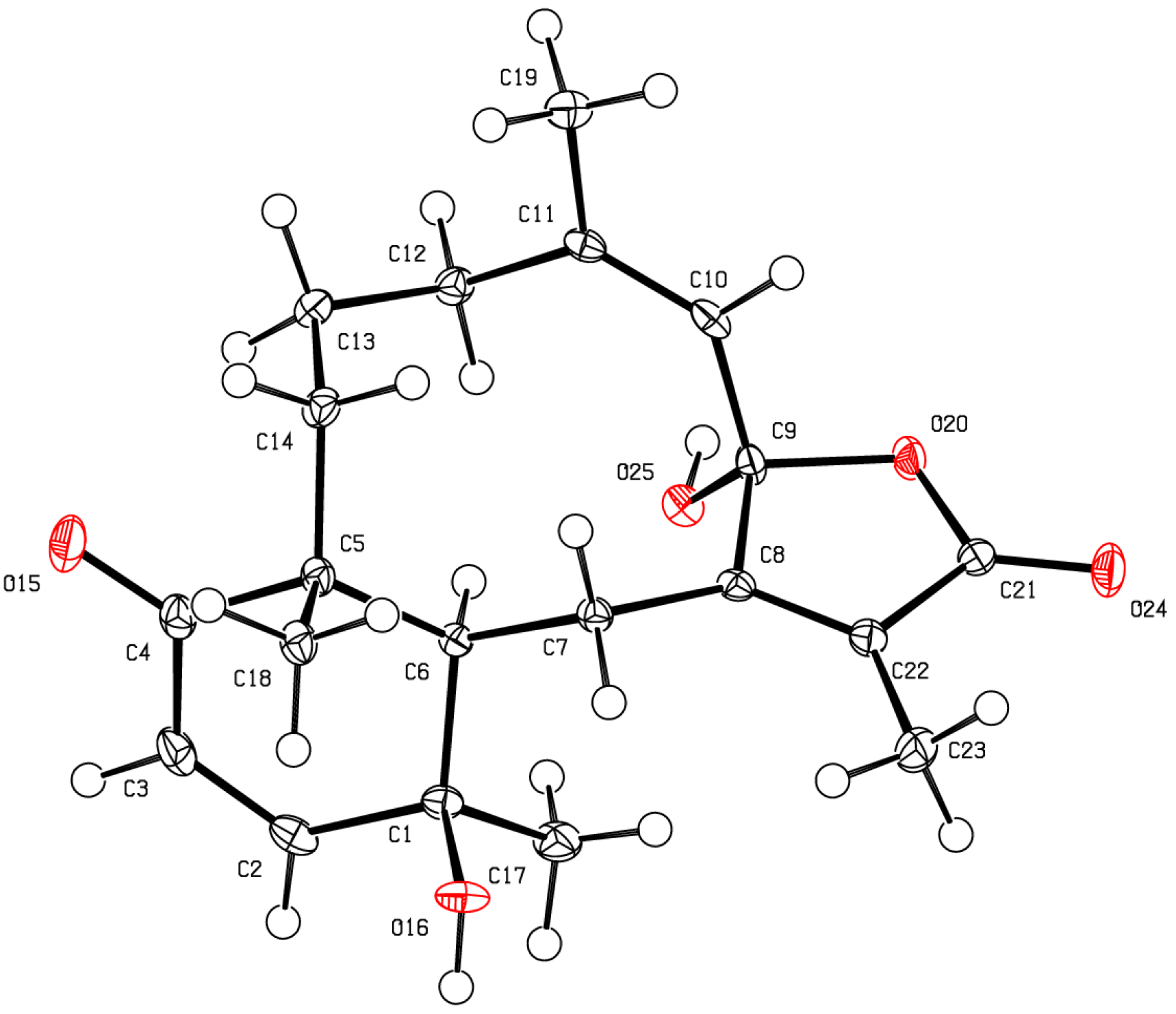
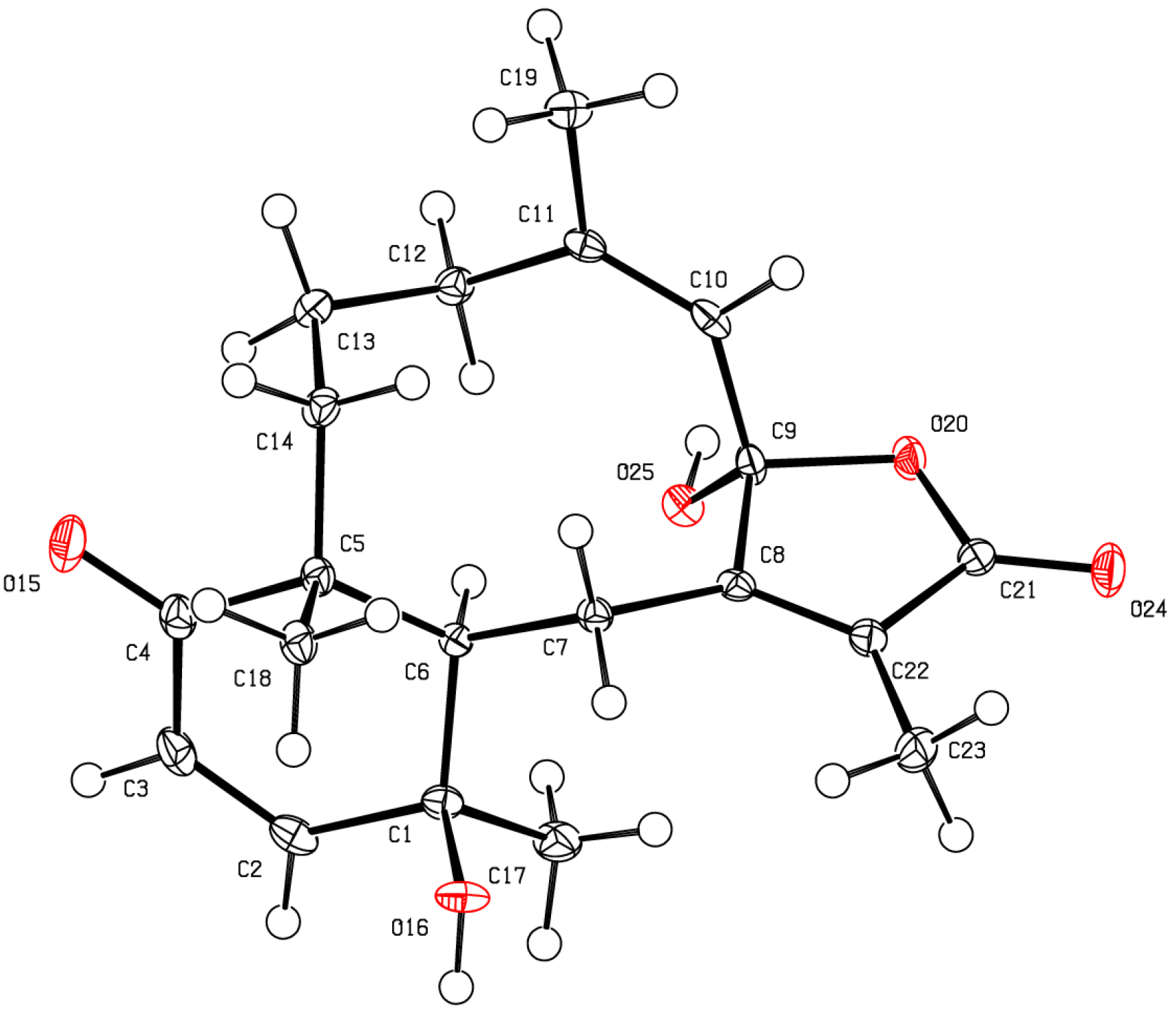
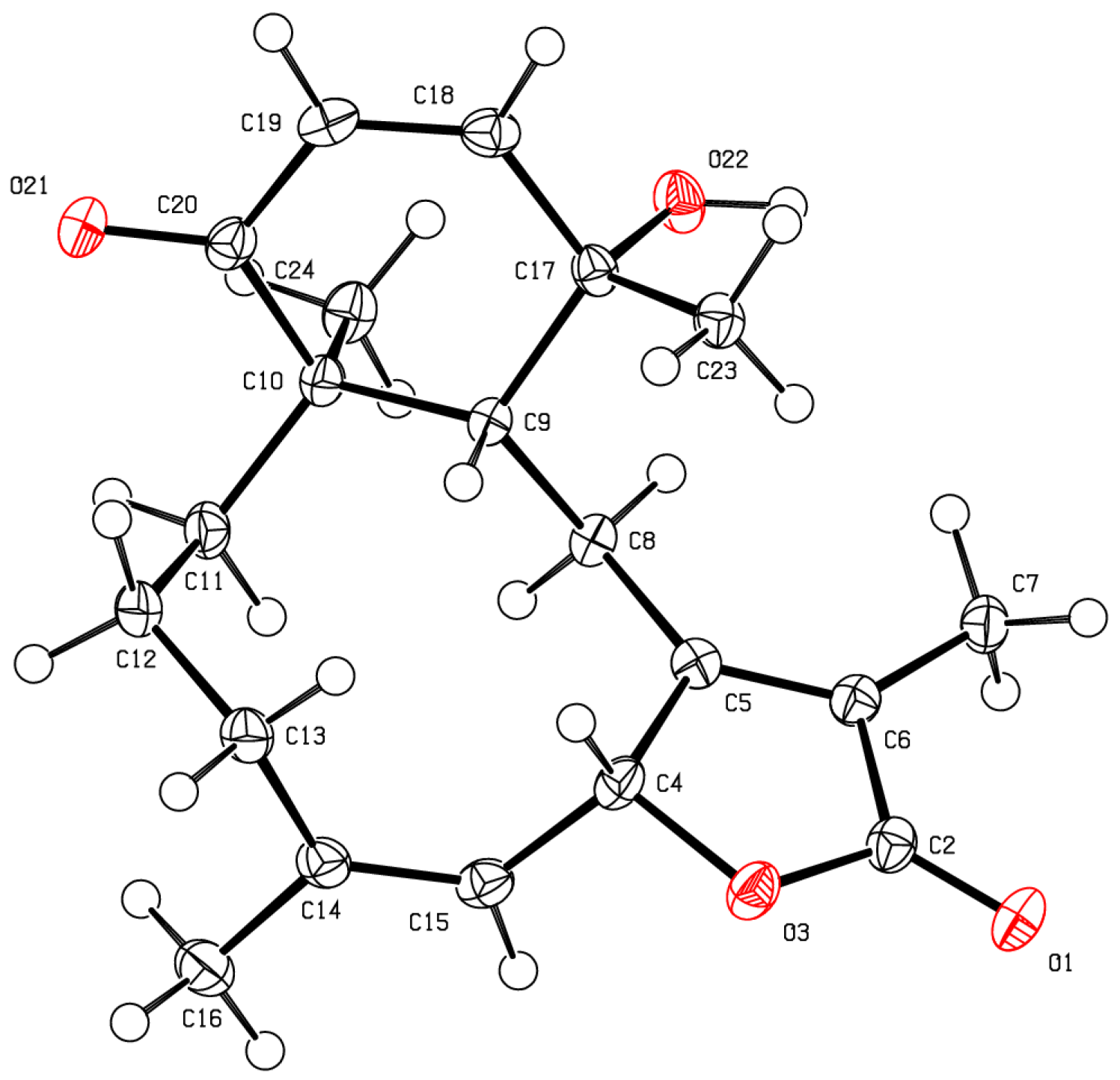
The stereochemical evaluation of bathyptilone A (
1) was approached using NOESY spectroscopy. Proton H-10 (δ
H 3.14) showed NOESY correlations to δ
H 1.35 (H
3-20) and δ
H 3.52 (H-4b), while H
2-9 (δ
H 2.47, H-9a; 3.25, H-9b) displayed proximity to H
3-15 (δ
H 1.16), setting the ring fusion
trans, as observed in all other briarane diterpenes [
11], with H
3-20 occupying the same face of the ring fusion as H-10. The remaining stereogenic carbon, C-7, lacked a proton, but correlation between the 7-OH/11-OH overlapping hydroxyl signal in the
1H NMR spectrum, δ
H 3.52, and H-10 must result from 7-OH rather than 11-OH, which has already been shown to be on the opposite face of the tricycle from H-10. These stereocenters, as well as the configuration of the C-7 hemiacetal, were confirmed by X-ray crystallography analysis (
Figure 2,
Table S1), which facilitated the assignment of the absolute configuration of bathyptilone A as depicted in
1.
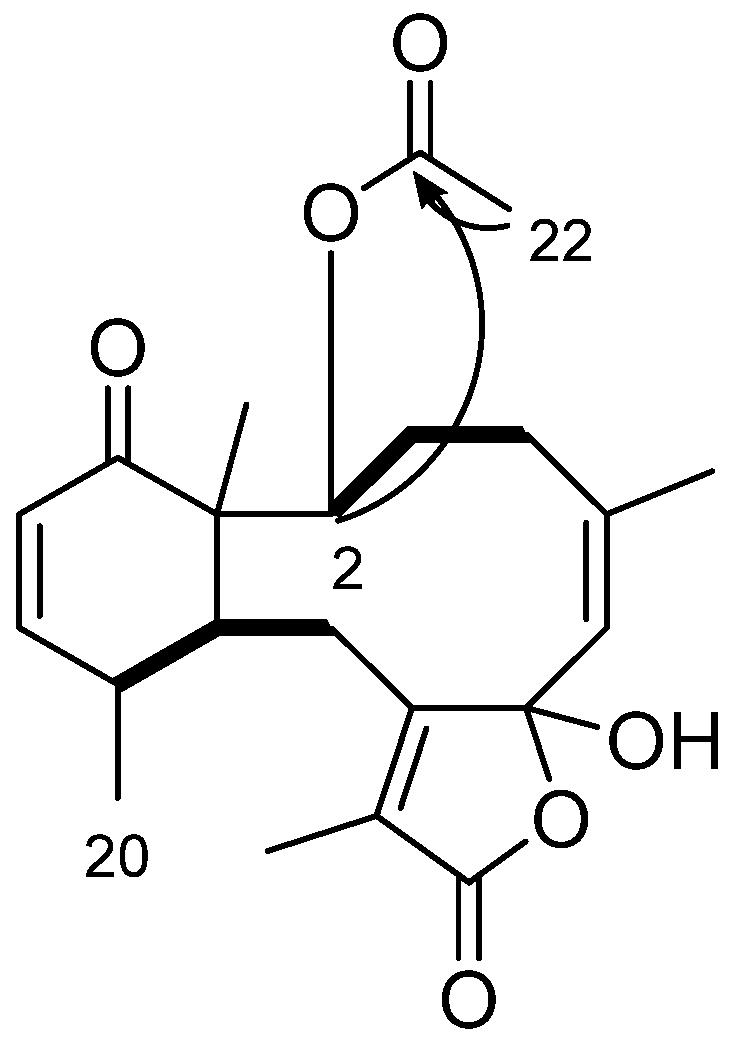
The molecular formula of bathyptilone B (
2) was established as C
22H
28O
6 by HRESIMS and supported by
1H and
13C NMR data (
Table 2), which appears to differ by the acetylation of bathyptilone A (
1). This was confirmed by observation of
13C NMR signals for the two additional carbons as an ester-type carbonyl at δ
C 169.5 (C-21) and an aliphatic methyl at δ
C 21.2 (C-22), the latter bearing protons that were found in the
1H NMR spectrum as a 3H singlet at δ
H 2.03 (H
3-22). This substituent could be placed at C-2 (δ
C 77.9) based on a deshielded proton at δ
H 5.89 (H-2), which correlated in the HMBC spectrum to carbons that matched their bathyptilone A analog, including δ
C 52.5 (C-1) and δ
C 41.4 (C-10), as well as the new carbon at C-21 (
Figure 3). With new oxidation at C-2, relative to bathyptilone A, C-11 (δ
C 38.0) was compared between
1 and
2 and found to be proton bearing rather than oxygen bearing.
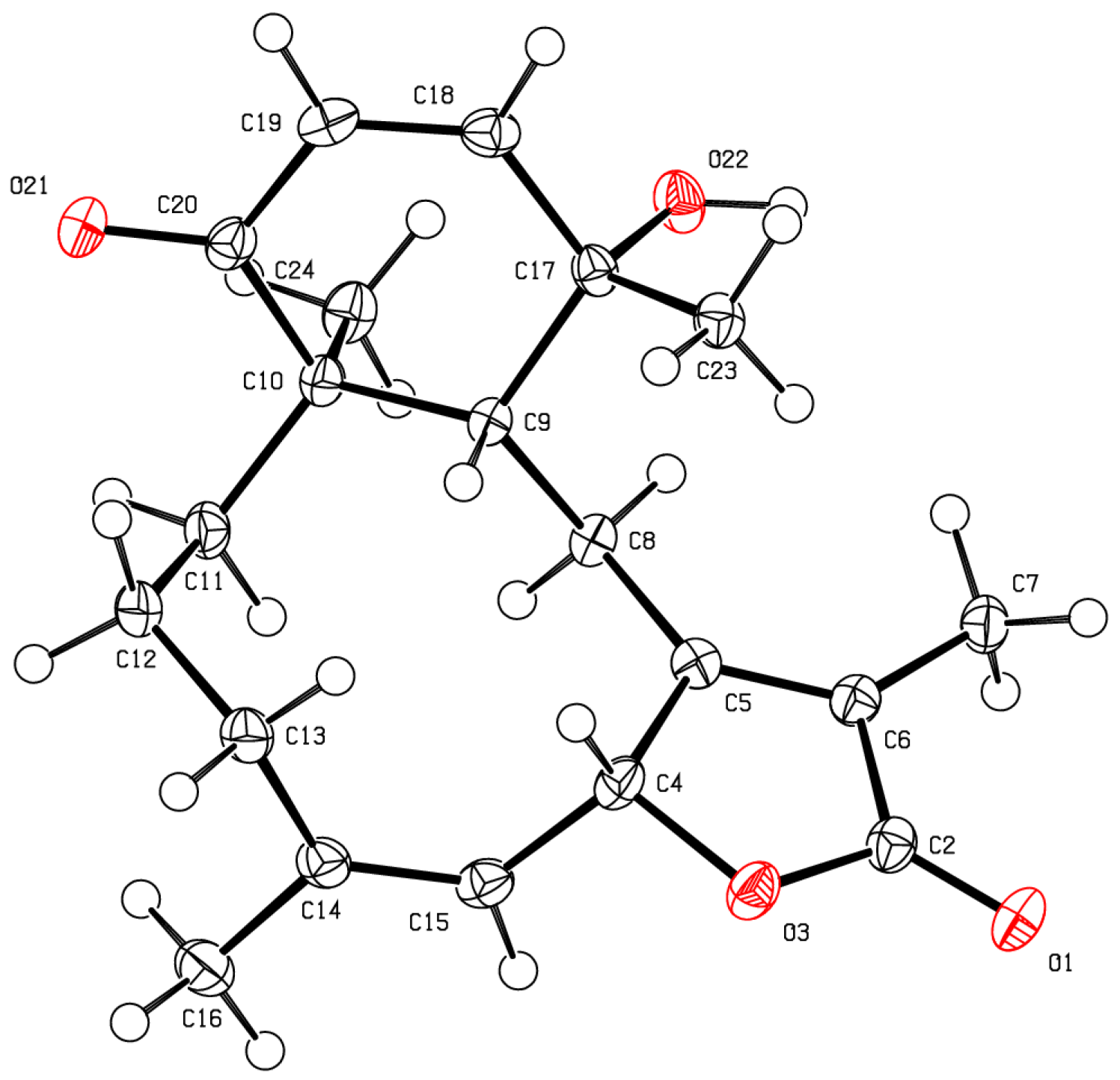
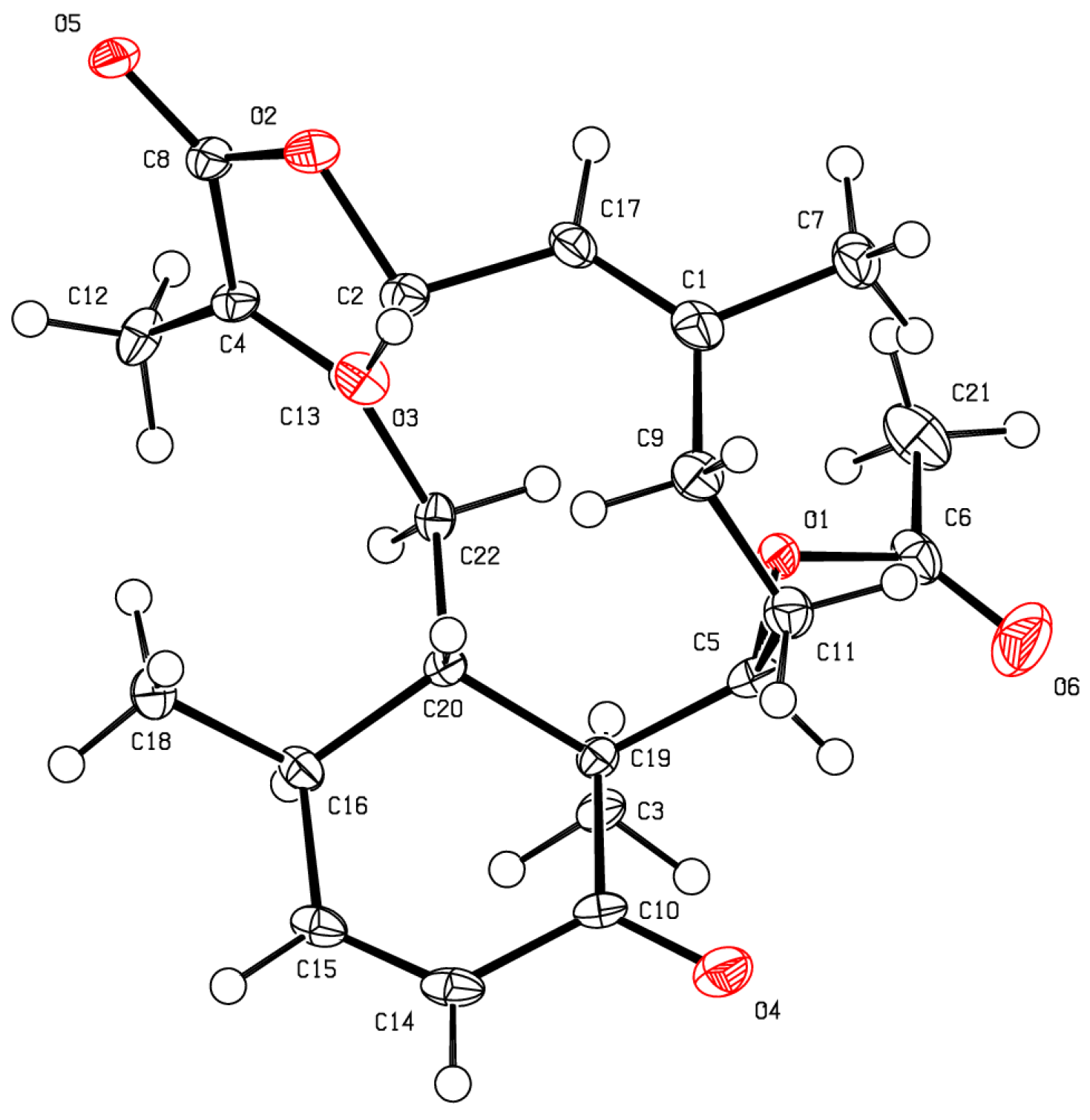
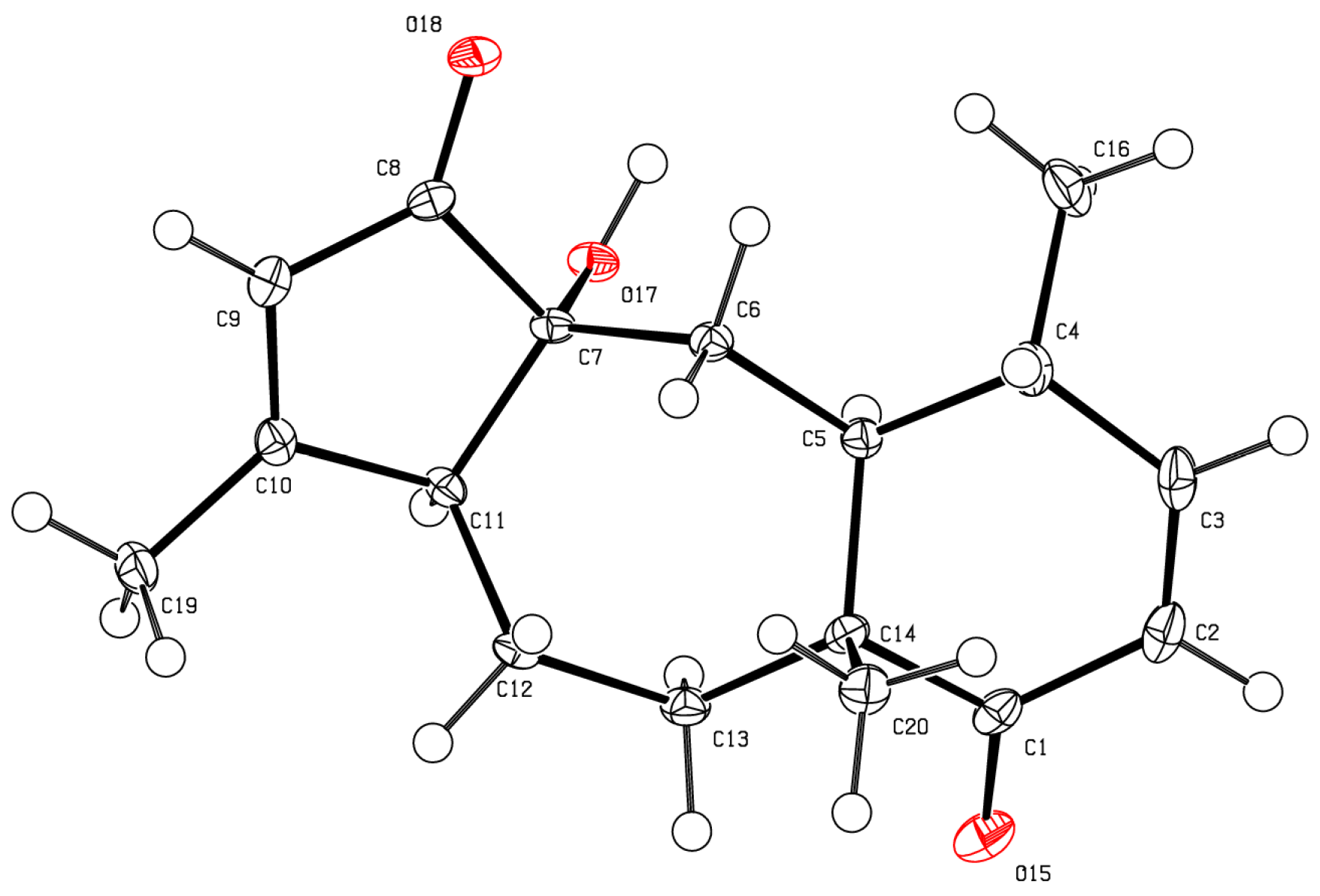
As for bathyptilone A (
1), bathyptilone B (
2) was evaluated by 2D-NOE to determine the configuration of the asymmetric centers. H
3-15 (δ
H 1.11) correlates to both H-2 (δ
H 5.89) and H-11 (δ
H 2.45), placing them on the same face of the bicyclic ring system, while H-10 (δ
H 3.43) only correlated to H-3a (δ
H 3.93). This evaluation supported the usual observation of the
trans configured C-1/10 bridge in briarane-type scaffolds. X-ray crystallography (
Figure 4) corroborated these assignments.
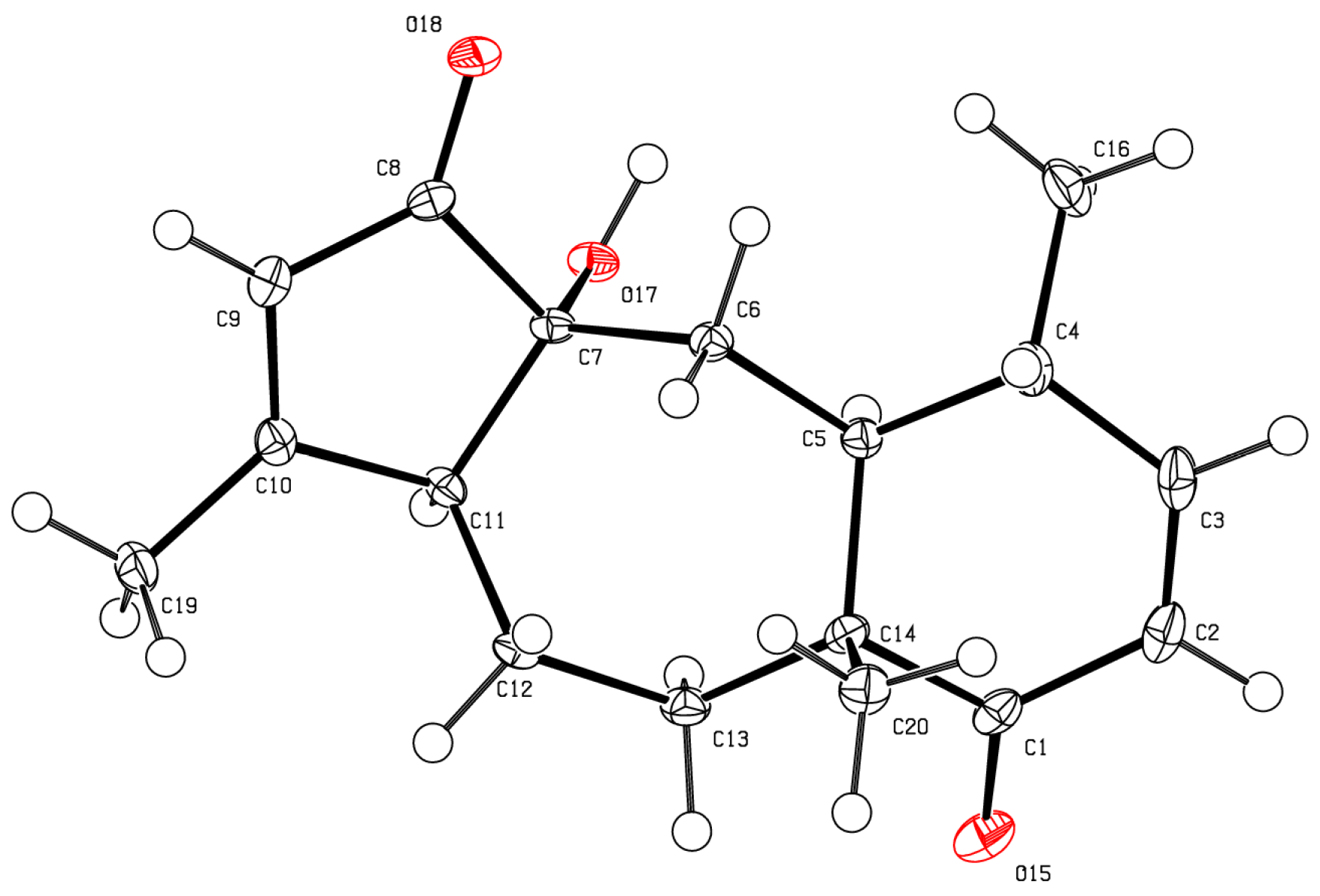
Analysis of the crystalline bathyptilone C (
3) with HRESIMS and 1D
1H and
13C NMR spectra yielded a molecular formula of C
20H
26O
4. The
1H NMR spectrum of
3 showed shifts similar to compound
1 with the addition of a methine at δ
H 5.66 (H-7). The
13C NMR spectrum was closely matched to that of bathyptilone A, with the major difference being the upfield shift of C-7 (δ
C 79.2), which indicated the replacement of the bathyptilone A hemiacetal functionality by an alcohol function. Stereochemical analysis using NOE correlations, as well as comparison of shifts to bathyptilone A (
1), established bathyptilone C as the C-7 reduced analog of
1, an assignment which was confirmed by X-ray analysis (
Figure 5).
A fourth compound (
4) isolated from
Anthoptilum grandiflorum was determined to have the molecular formula C
17H
22O
3 based on HRESIMS. 1D
1H and
13C NMR spectra (
Table 3) suggested that compound
4 was a terpenoid, despite the odd carbon count. There were seven degrees of unsaturation that were accounted for by the presence of two ketone carbonyls, two olefins and a tricyclic ring system. Initially, COSY was used to identify two isolated spin systems. Starting with the most deshielded protons, δ
H 6.57 (H-12) and δ
H 6.07 (H-13) were coupled and H-12 further coupled to aliphatic methine δ
H 2.08 (H-11). The spin system bifurcated at H-11, with the system terminating in one direction with a methyl group at δ
H 1.09 (H
3-17) but extending the system in the other direction to a methylene at δ
H 1.35 (H-10). The second spin system established by COSY was found as a methine at δ
H 2.20 (H-4) coupled to δ
H 1.20 (H-3a). Proton H-4 was extended by HMBC to an α,β-unsaturated ketone based on correlation to δ
C 177.5 (C-5), δ
C 133.2 (C-6), δ
C 205.4 (C-7), and δ
C 99.5 (C-8). Proton H-4 further correlated in the HMBC spectrum to a vinyl methyl at δ
H 2.09 (H
3-16). The remaining methyl group, δ
C 19.3 (C-15), was assigned based on HMBC correlations to δ
C 52.8 (C-1), δ
C 31.9 (C-2) and δ
C 202.9 (C-14). The complete two-dimensional structure of
4 was completed by connecting the first COSY spin system to C-1, C-8 and C-14, as depicted in
Figure 6.
The stereochemistry of enbepeanone A (
4) was assigned from NOESY. Proton H-10 showed a strong correlation to the methyl doublet, which indicated their proximity; however, the methyl singlet of H-15 did not have a NOESY correlation to H-10 as noted for the briarane scaffolds in the bathyptilones. The cyclopentenone stereocenters, and their relationship to the cyclohexenone, were established by X-ray crystallographic analysis (
Figure 7). The crystallographic metadata (
Table S4) facilitated the assignment of the absolute configuration of enbepeanone A as shown.
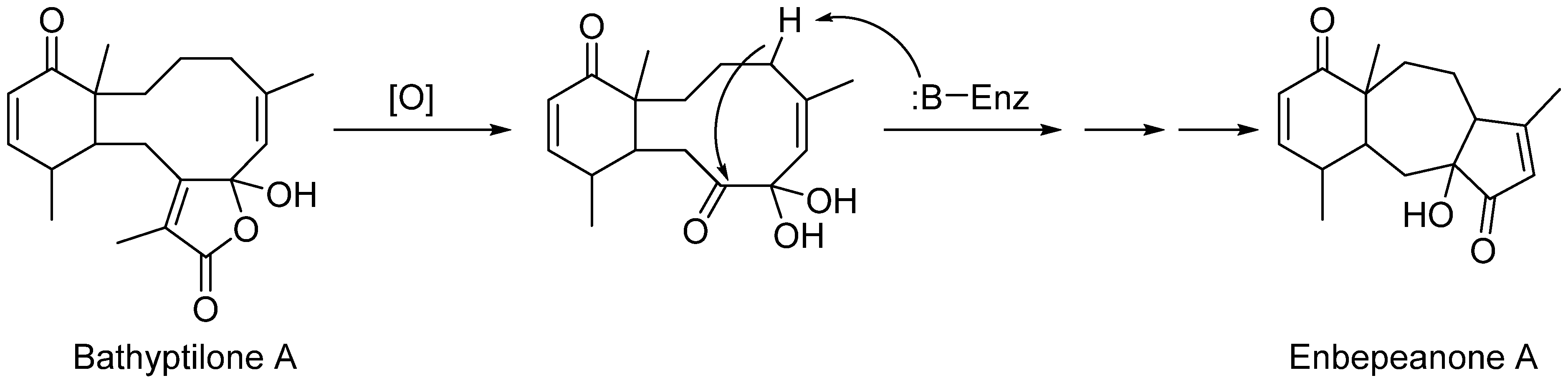
Enbepeanone A (
4) is a trinorditerpene seemingly derived from the oxidation of a briarane skeleton such as bathyptilone A (
1). Oxidative cleavage of the C-8 pendant isopropyl group from
1 provides an intermediate suited to enzyme-catalyzed cyclopentanulation that would produce the enbepeanone A skeleton (
Scheme 2). This is the first example of such a trinorditerpene bearing this carbon skeleton.
Several mammalian cell types were analyzed for sensitivity to these diterpenes (
Figure 8). Of the compounds tested, bathyptilone A (
1) was highly toxic to a pluripotent embryonal carcinoma cell line, NTera-2 (NT2). This cell line was isolated from a lung metastasis of testicular cancer; however, these cells exhibit many properties of precursor neuronal cells like the formation of neurons and the presence of homeobox clusters in the genome [
18]. Due to their similarity with developing neuronal cells, this cell line is accepted as a valid system for the assessment of neurotoxicity [
19]. Compound
1 effectively killed the NT2 cells at with an IC
50 of 29 nM. A recent publication describes the promising antiproliferative effect of gedunin with an IC
50 as low as 13.5 µM against NT2 cells [
20]. Compounds
2–
4 were also tested against the NT2 cell line, but their activities were not significant. Based on this finding, we speculate that bathyptilone A may prove to be an effective therapeutic strategy for specifically targeting neuronal cancers.
3. Experimental Section
3.1. General Experimental Procedure
Optical Rotations were measured by a Rudolph Instruments (Hackettstown, NJ, USA) AutoPol IV polarimeter at 589 nm. UV absorptions were measured by an Agilent (Santa Clara, CA, USA) Cary 60 UV-Vis spectrophotometer; wavelengths are recorded in nm. IR spectra were recorded with a PerkinElmer (Waltham, MA, USA) Spectrum Two with a UATR (single-reflection diamond) sample introduction system; peaks are reported in cm−1. NMR spectra were recorded at 298 K on an Agilent (Santa Clara, CA, USA) Varian Direct Drive 500 or Varian Direct Drive 800 MHz NMR spectrometers. Chemical shifts are reported with the use of the residual CDCl3 signals (δH 7.27 ppm; δC 77.0 ppm) as internal standards for 1H and 13C NMR spectra. The 1H and 13C NMR assignments were supported by COSY, HSQC, HMBC, and NOESY experiments. The high-resolution mass spectra were recorded on an Agilent (Santa Clara, CA, USA) 6230 TOF LC/MS. Semi-preparative and analytical HPLC was performed on a Shimadzu (Columbia, MD, USA) LC-20 AT system equipped with an evaporative light-scattering detector (ELSD) and a ultraviolet detector using a Luna column (5 μm, 250 × 10 mm) and a YMC (Kyoto, Japan) packed column (10 µm, 150 × 4 mm). MPLC was performed on a Teledyne Isco (Lincoln NE) CombiFlash Rf 200i equipped with an ELSD using a RediSep Rf silica 120 g flash column with silica gel 230–400 mesh ASTM used to load the sample.
3.2. Animal Material
Specimens of the pennatulacean octocoral
Anthoptilum grandiflorum were collected in April 2013, north of Burdwood Bank (54°14′56.9″ S, 59°00′00.6″ W), by trawling on the R/V Nathaniel B. Palmer from depths of between 662 and 944 meters. A specimen voucher is retained at the Scripps Institution of Oceanography Benthic Invertebrate collection (SIO-BIC Co2892, field number S22350). A piece of tissue from this voucher was extracted with a DNeasy kit (Qiagen) and was amplified for the mitochondrial msh1 (muts homolog) to confirm identification. We used the primers ND42599F/mut3458R [
21,
22] to produce amplicons and outsourced these for sequencing (AGRF Perth node). The resulting bi-directional sequence was compared to other available Pennatulacea sequences in GenBank using a Maximum-Likelihood analysis at the W-IQ-tree server [
23]. W-IQ-Tree incorporates the ModelTest [
24], which selected the K3P + G4 as the best-fit model, and which is automatically applied. The tree topology was assessed using 1000 ultrafast bootstrap replicates and shows that our specimen has an identical msh1 sequence to a specimen of
Anthoptilum grandiflorum from Greenland (
Supplementary information) [
25]. The species has been recorded as having a worldwide distribution [
26], although this study represents the southernmost record of this species.
3.3. Isolation of Bathyptilones and Enbepeanone
The Anthoptilum grandiflorum was immediately frozen after collection and later freeze-dried at our lab. The lyophilized organism (84.7 g) was then broken into smaller pieces and exhaustively extracted via 3 cycles of DCM (3.5 L) in a Soxhlet extractor. The crude extract (11.7 g) was partitioned through liquid–liquid extraction using DCM and water. The organic layer (10.4 g) was then concentrated, resulting in a brownish residue, and then re-suspended in DCM to be dried onto silica gel for flash chromatography using MPLC. A concentration gradient of hexane and ethyl acetate was extended over a period of time to elute 8 fractions (A–H). Fraction E (191.0 mg) eluted in 9:11 hexane/ethyl acetate and was subjected to further purification in normal phase HPLC on a silica gel column to afford 11 fractions. Fractions 4, 6, and 9 afforded the tricyclic diterpene compounds, bathyptilone A (1, 3.6 mg), B (2, 2.1 mg), C (3, 1.8 mg) and enbepeanone A (4, 0.7 mg) with further purification on a C18 reverse-phase HPLC column. The crystallization of these compounds occurred through slow evaporation using the mobile-phase solvents.
Bathyptilone A (
1): Crystalline solid; [α]
−18.3 (
c 0.11, CHCl
3); UV
λmax (EtOH) (log ε) 271 nm; IR 3650, 3356, 2980, 2646, 2322, 2286, 2166, 1738, 1656, 1446, 1378, 1212, 1114, 934, 892, 834, 754 cm
−1;
1H NMR (500 MHz, CDCl
3) and
13C NMR (125 MHz, CDCl
3), see
Table 1; HRESIMS
m/z 347.1853 [M + H]
+ (347.1859 calculated for C
20H
27O
5).
Bathyptilone B (
2): Crystalline solid; [α]
−40.2 (
c 0.11, CHCl
3); IR 3346, 2980, 2886, 1736, 1662, 1456, 1378, 1230, 938, 752 cm
−1;
1H NMR (800 MHz, CDCl
3), see
Table 2 and
13C NMR (200 MHz, CDCl
3), see
Table 2; HRESIMS
m/z 411.1775 [M + Na]
+ (411.1784 calculated for C
22H
28O
6Na).
Bathyptilone C (
3): Crystalline solid; [α]
−16.2 (
c 0.11, CHCl
3);
1H NMR (800 MHz, CDCl
3), see
Table 2 and
13C NMR (200 MHz, CDCl
3), see
Table 2; HRESIMS
m/z 330.1842 [M + H]
+ (330.1831 calculated for C
20H
27O
4).
Enbepeanone A (
4): Crystalline solid; [α]
−11.0 (
c 0.11, CHCl
3); IR 3424, 3190, 2646, 2322, 1650, 1020, 742, 512, 456, 414 cm
−1;
1H NMR (500 MHz, CDCl
3) and
13C NMR (125 MHz, CDCl
3), see
Table 3; HRESIMS
m/z 275.1640 [M + H]
+ (275.1647 calculated for C
17H
23O
3).
3.4. X-Ray Crystallography
The X-ray diffraction data were measured on a Bruker D8 Venture PHOTON 100 CMOS system (Madison, WI, USA) equipped with a Cu K
α INCOATEC ImuS micro-focus source (
λ = 1.54178 Å). Indexing was performed using Apex3 [
27]. Data integration and reduction were performed using SaintPlus 6.01 [
28]. Absorption correction was performed by a multi-scan method implemented in SADABS [
29]. Space groups were determined using XPREP implemented in APEX3 [
27]. Structures were solved using SHELXT [
30] and refined using SHELXL-2017 [
31,
32,
33] (full-matrix least-squares on F
2) through the OLEX2 interface program [
34]. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms of –OH groups were freely refined. The remaining hydrogen atoms were placed in geometrically calculated positions and were included in the refinement process using a riding model with isotropic thermal parameters. Crystal data and refinement conditions are shown in
Supplementary Tables S1–S5. The absolute configuration for compounds
1 and
3 was established based on the Flack parameter value and verified additionally with Bijvoet–Pair Analysis and Bayesian Statistics Methods [
35,
36] (
Table S5). P2 ≈ 1 for all cases and this represents the probability that the current model is correct assuming the possibility that one out of two enantiomers is present. The results of the absolute configuration assignments for compound
2 are not conclusive
. Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 1,944,295 for 1, CCDC 1,944,292 for 2, CCDC 1,944,293 for 3 and CCDC 1,944,294 for 4. Copies of the data can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK.
Crystallographic data for bathyptilone A (1). C20H26O5, M = 346.41, crystal size 0.093 × 0.038 × 0.034 mm3, orthorhombic, a = 8.7926(2) Å, b = 12.2611(2) Å, c = 16.4222(3) Å, α = β = γ = 90°, V = 1770.43 (6) Å3, T = 100.01 K, space group P212121, Z = 4, Dcalcd = 1.300 g/cm3, µ = 0.753 mm−1, F (000) = 744.0, 3123 independent reflections (Rint = 0.0453, Rsigma = 0.0213). The final R1 = 0.0288 (I ≥ 2σ (I)), wR2 = 0.0674 (I ≥ 2σ (I)), R1 = 0.0312 (all data), wR2 = 0.0688 (all data), S = 1.088. The Flack parameter was −0.04 (6).
Crystallographic data for bathyptilone B (2). C22H28O6, M = 388.44, crystal size 0.04 × 0.02 × 0.005 mm3, monoclinic, a = 9.5139(4) Å, b = 10.3255(5) Å, c = 10.4615(4) Å, α = γ = 90°, β = 90.924° (3), V = 1027.56 (8) Å3, T = 100.02 K, space group P21, Z = 2, Dcalcd = 1.255 g/cm3, µ = 0.744 mm−1, F (000) = 416.0, 3614 independent reflections (Rint = 0.1118, Rsigma = 0.1052). The final R1 = 0.0576 (I≥2σ (I)), wR2 = 0.1076 (I ≥ 2σ (I)), R1 = 0.0859 (all data), wR2 = 0.1184 (all data), S = 1.041. The Flack parameter was 0.2 (3).
Crystallographic data for bathyptilone C (3). C20H26O4, M = 330.41, crystal size 0.467 × 0.058 × 0.055 mm3, orthorhombic, a = 8.2529(2) Å, b = 10.8806(3) Å, c = 19.0878(5) Å, α = β = γ = 90°, V = 1714.02 (8) Å3, T = 99.99 K, space group P212121, Z = 4, Dcalcd = 1.280 g/cm3, µ = 0.707 mm−1, F (000) = 712.0, 3597 independent reflections (Rint = 0.0725, Rsigma = 0.0359). The final R1 = 0.0356 (I ≥ 2σ (I)), wR2 = 0.0852 (I ≥ 2σ (I)), R1 = 0.0398 (all data), wR2 = 0.0885 (all data), S = 1.071. The Flack parameter was 0.03 (9).
Crystallographic data for enbepeanone A (4). C17H22O3, M = 274.34, crystal size 0.2 × 0.06 × 0.02 mm3, orthorhombic, a = 7.1483(2) Å, b = 7.7391(2) Å, c = 25.4330(7) Å, α = β = γ = 90°, V = 1406.99 (7) Å3, T = 100.0 K, space group P212121, Z = 4, Dcalcd = 1.295 g/cm3, µ = 0.698 mm−1, F (000) = 592.0, 2884 independent reflections (Rint = 0.0551, Rsigma = 0.0432). The final R1 = 0.0374 (I ≥ 2σ (I)), wR2 = 0.0789 (I ≥ 2σ (I)), R1 = 0.0455 (all data), wR2 = 0.0825 (all data), S = 1.105. The Flack parameter was −0.03 (13).
3.5. Biological Assays
Ntera-2, MiaPaca-2, HeLa and U2OS cell lines. Several cell lines were tested for sensitivity to the tricyclic diterpenes, the cell lines tested were HeLa (ATCC CCL-2™), MiaPaCa-2 (ATCC CRL-1420™, U2OS (ATCC HTB-96™), and NT2 (ATCC CRL-1973™). All cells used were seeded into 96-well plates (~25 cells per well) and allowed to attach for 48 h under normal growth conditions (described below). Compounds were administered at the indicated concentrations and the cells were allowed to grow for 10 days. All cell lines used were grown in Dulbecco’s Modification of Eagle’s Medium (DMEM), with 4.5 g/L glucose and l-glutamine (Corning, 10-107-CV) supplemented with 10% Fetal Bovine Serum (Seradigm, 3100-500) and 1% penicillin–streptomycin–glutamine (HyClone, SV30082.01). All cell lines were grown at 37 °C with 5% CO2 in a FORMA Series II Water Jacket CO2 Incubator (Thermo). After the growth period, the cells were fixed with a 10% methanol + 10% acetic acid solution for 15 minutes at room temperature. The fixed cells were stained with crystal violet for 5 minutes and distained by washing with water. The cells were dried overnight. Crystal violet was removed from the cells with Sorensen buffer (0.1M sodium citrate, 50% ethanol), the colorimetric intensity of each solution was quantified using Gen5 software on a Synergy 2 (BioTek, Winooksi, VT, USA) plate reader (OD at 595 nm). Error bars are representative of 3 independent experiments.

 and
and 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
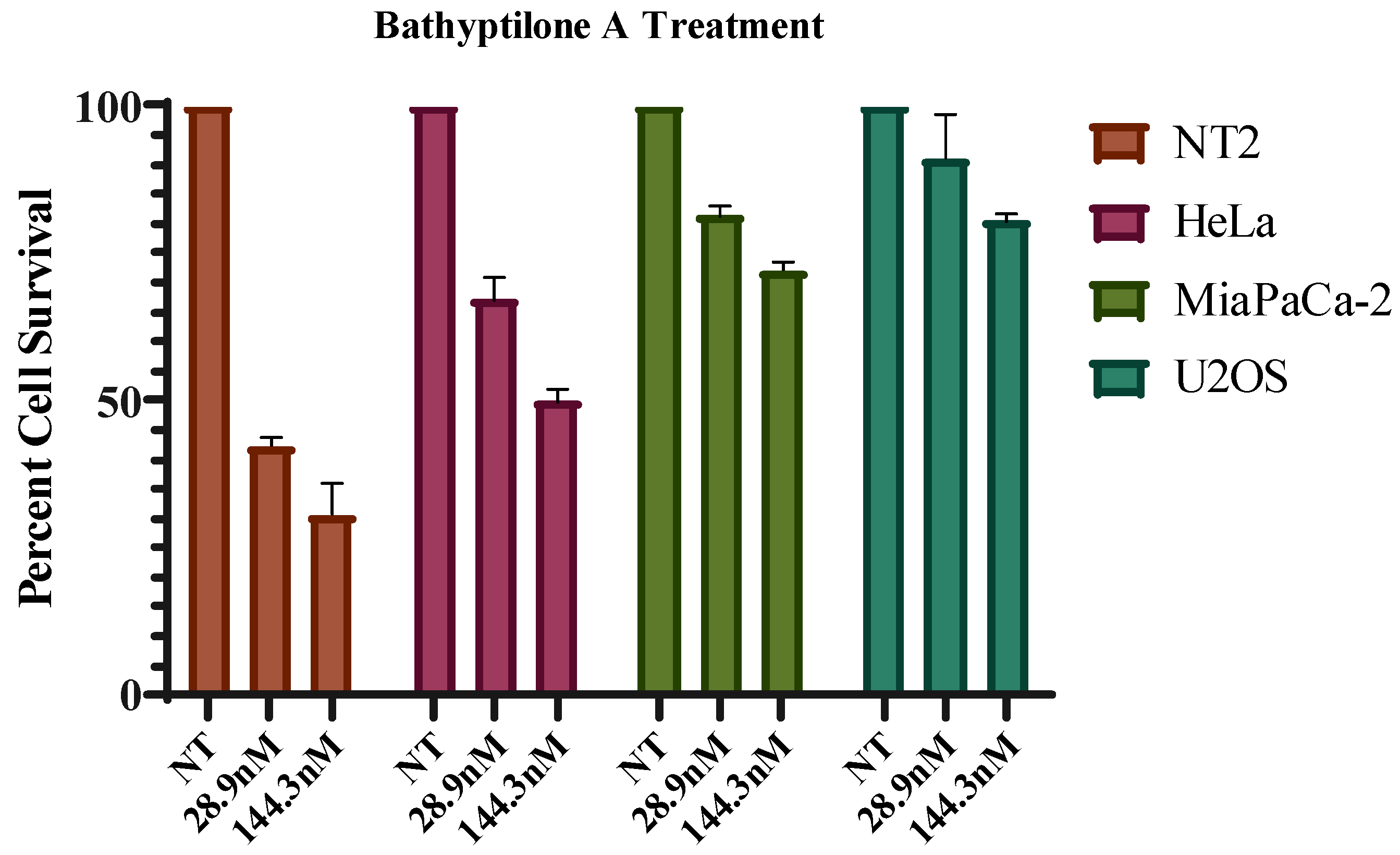{kind=link}
{kind=link}
{kind=link}