Recent Synthetic Studies Leading to Structural Revisions of Marine Natural Products

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Examples of Syntheses Leading to Structural Revisions of Marine Natural Products
2.1. Macrocyclic Marine Natural Products
2.2. Etheric Marine Natural Products
2.3. Other Examples
2.4. Our Experiences with Pericosines
3. Summary
- Sample Availability: Not available
References and Notes
- Newman, DJ; Cragg, GM. Natural products from marine invertebrates and microbes as modulators of antitumor targets. Curr Drug Targets 2006, 7, 279–304. [Google Scholar]
- Newman, DJ; Cragg, GM. Advanced preclinical and clinical trials of natural products and related compounds from marine sources. Curr Med Chem 2004, 11, 1693–1713. [Google Scholar]
- Butler, MS. Natural products to drugs: natural product derived compounds in clinical trials. Nat Prod Rep 2005, 22, 162–195. [Google Scholar]
- Usami, Y; Ueda, Y. Synthetic study toward antitumour natural product pericosine A. Chem Lett 2005, 34, 1062–1063. [Google Scholar]
- Usami, Y; Ueda, Y. Stereoselective syntheses of diastereomers of antitumor natural product pericosine a from (−)-quinic acid. Synthesis 2007, 3219–3225. [Google Scholar]
- Usami, Y; Horibe, Y; Takaoka, I; Ichikawa, H; Arimoto, M. First total synthesis of (−)-pericosine A from (−)-shikimic acid: Structure Revision and determination of the absolute configuration of antitumor natural product pericosine A. Synlett 2006, 1598–1600. [Google Scholar]
- Usami, Y; Takaoka, I; Ichikawa, H; Horibe, Y; Tomiyama, S; Ohtsuka, M; Imanishi, Y; Arimoto, M. First total synthesis of (+)- and (−)-pericosine A: Determination of absolute stereo structure. J Org Chem 2007, 72, 6127–6134. [Google Scholar]
- Usami, Y; Ichikawa, H; Arimoto, M. Synthetic efforts for stereo structure determination of cytotoxic marine natural product pericosines as metabolites of Periconia sp. from sea hare. Int J Mol Sci 2008, 9, 401–421. [Google Scholar]
- Nicholas, GM; Phillips, AJ. Marine natural products: Synthetic aspects. Nat Prod Rep 2005, 22, 144–161. [Google Scholar]
- Nicholas, GM; Phillips, AJ. Marine natural products: Synthetic aspects. Nat Prod Rep 2006, 23, 79–99. [Google Scholar]
- Morris, JC; Nicholas, GM; Phillips, AJ. Marine natural products: synthetic aspects. Nat Prod Rep 2007, 24, 87–108. [Google Scholar]
- Morris, JC; Phillips, AJ. Marine natural products: synthetic aspects. Nat Prod Rep 2008, 25, 95–117. [Google Scholar]
- Morris, JC; Phillips, AJ. Marine natural products: synthetic aspects. Nat Prod Rep 2009, 26, 245–265. [Google Scholar]
- Nicolaou, KC; Snyder, SA. Chasing molecules that were never there: Misassigned natural products and the role of chemical synthesis in modern structure elucidation. Angew Chem Int Ed 2005, 44, 1012–1044. [Google Scholar]
- Kinnel, RB; Gehrken, H-P; Scheuer, PJ. Palau'amine: A cytotoxic and immunosuppressive hexacyclic bisguanidine antibiotic from the sponge Stylotella agminata. J Am Chem Soc 1993, 115, 3376–3377. [Google Scholar]
- Köeck, M; Grube, A; Seiple, IB; Baran, PS. The pursuit of Palau'amine. Angew Chem Int Ed 2007, 46, 6586–6594. [Google Scholar]
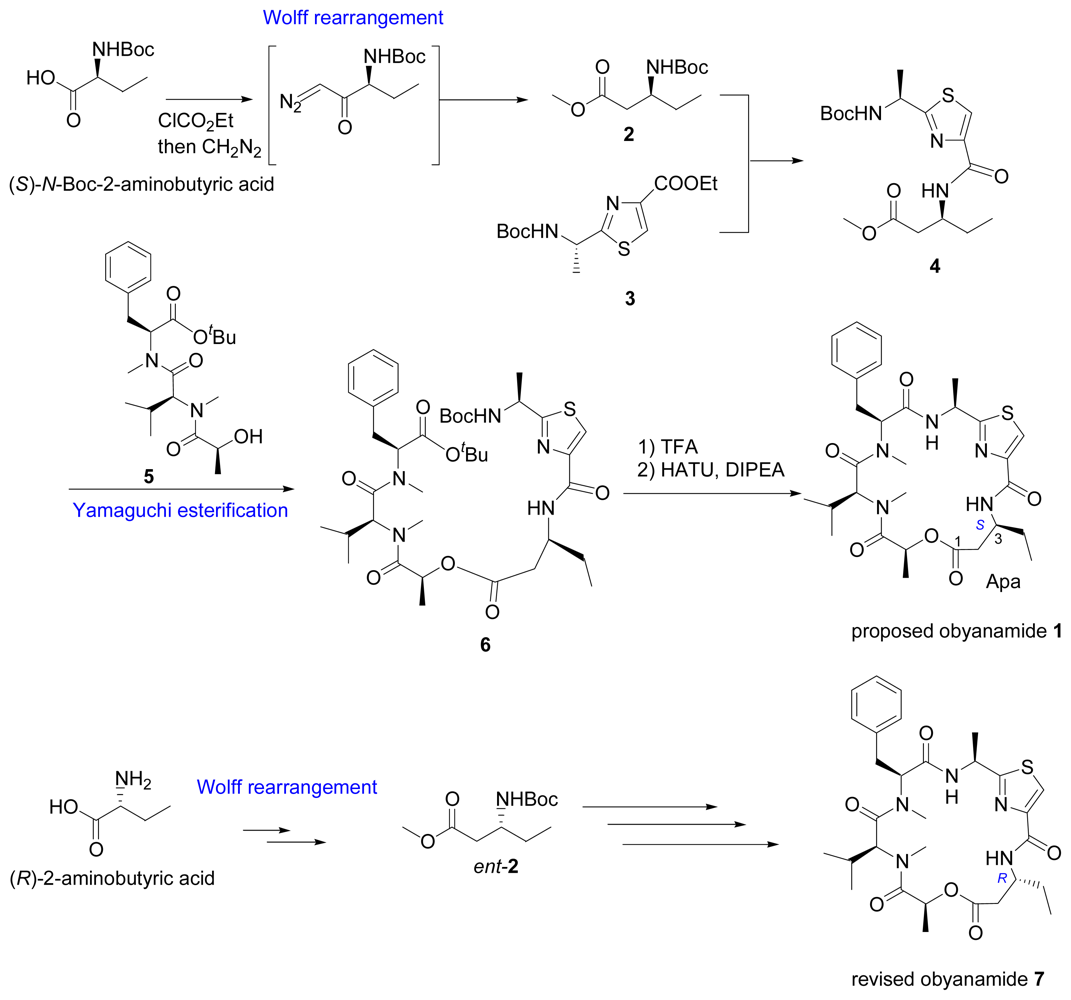
- Williams, PG; Yoshida, WY; Moore, RE; Paul, VJ. Isolation and structure determination of obyanamide, a novel cytotoxic cyclic depsipeptide from the marine cyanobacterium Lyngbya confervoides. J Nat Prod 2002, 65, 29–31. [Google Scholar]
- Zhang, W; Song, N; Li, Z-Z; Li, Y-X. Synthesis of obyanamide, a marine cytotoxic cyclic depsipeptide. Chin Chem Lett 2006, 17, 285–288. [Google Scholar]
- Zhang, W; Ma, Z-H; Mei, D; Li, C-X; Zhang, X-L; Li, Y-X. Total synthesis and reassignment of stereochemistry of obyanamide. Tetrahedron 2006, 62, 9966–9972. [Google Scholar]
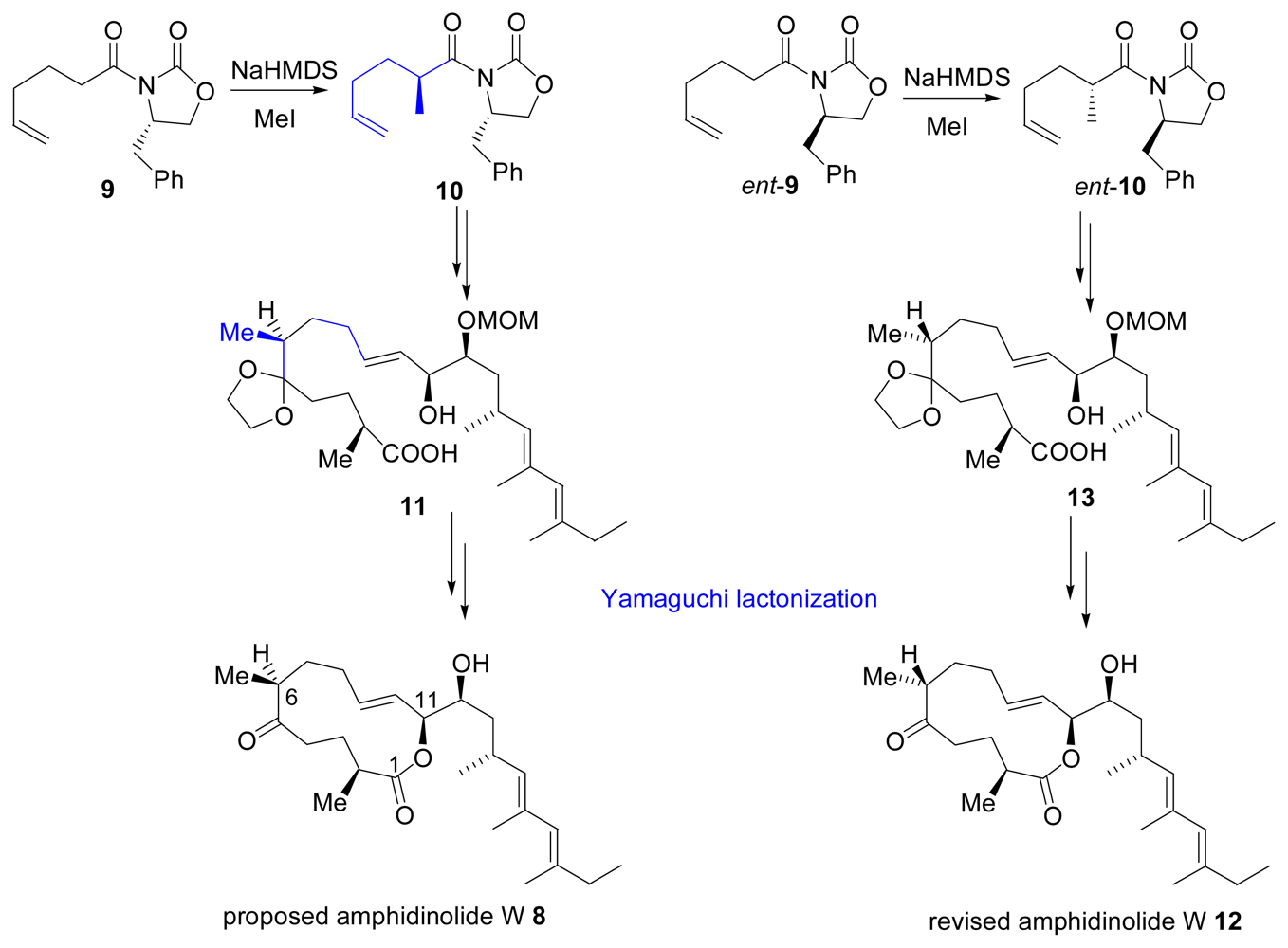
- Shimbo, K; Tsuda, M; Izui, N; Kobayashi, J. Amphidinolide W, a new 12-membered macrolide from dinoflagellate Amphidinium sp. J Org Chem 2002, 67, 1020–1023. [Google Scholar]
- Ghosh, AK; Gong, G. Total synthesis and structural revision of (+)-amphidinolide W. J Am Chem Soc 2004, 126, 3704–3705. [Google Scholar]
- Ghosh, AK; Gong, G. Total synthesis and revision of C6 stereochemistry of (+)-amphidinolide W. J Org Chem 2006, 71, 1085–1093. [Google Scholar]
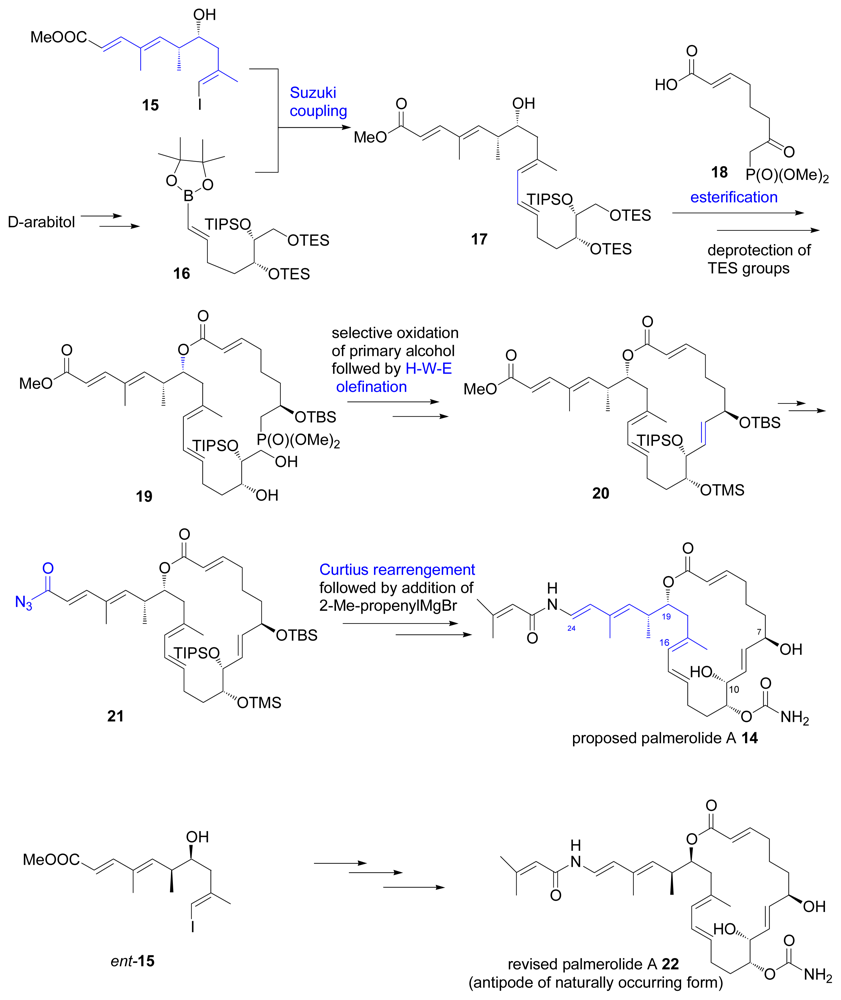
- Diyabalanage, T; Amsler, CD; McClintock, JB; Baker, J. Palmerolide A, a cytotoxic macrolide from the antarctic tunicate Synoicum adareanum. J Am Chem Soc 2006, 128, 5630–5631. [Google Scholar]
- Jiang, X; Liu, B; Lebreton, S; de Brabander, JK. Total synthesis and structure revision of the marine metabolite palmerolide A. J Am Chem Soc 2007, 129, 6386–6387. [Google Scholar]
- Nicolaou, KC; Guduru, R; Sun, Y-P; Banerji, B; Chen, DY-K. Total synthesis of the originally proposed and revised structures of palmerolide A. Angew Chem Int Ed 2007, 46, 5896–5900. [Google Scholar]
- Nicolaou, KC; Sun, Y-P; Guduru, R; Banerji, B; Chen, DY-K. Total synthesis of the originally proposed and revised structures of palmerolide A and isomers thereof. J Am Chem Soc 2008, 130, 3633–3644. [Google Scholar]
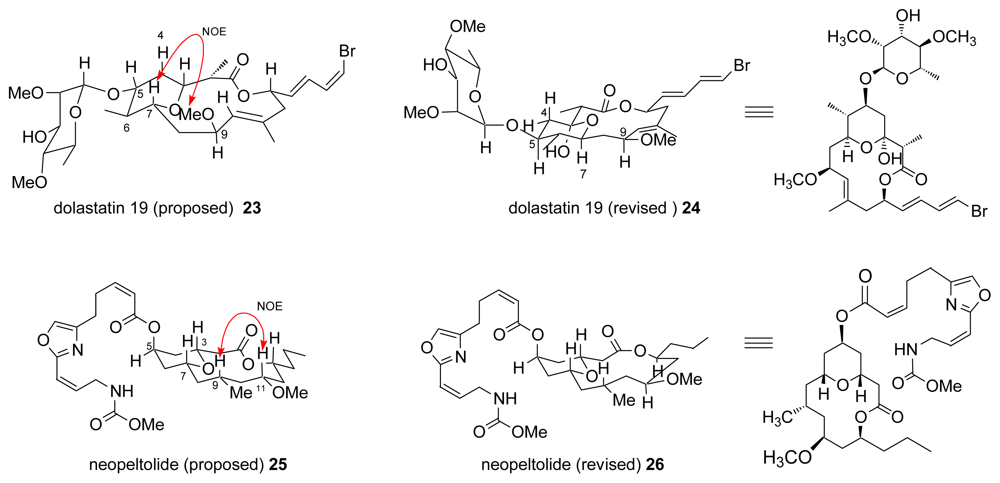
- Pettit, GR; Xu, J-P; Doubek, DL; Chapuis, J-C; Schmidt, JM. Antineoplastic agents. 510. Isolation and structure of dolastatin 19 from the Gulf of California sea hare Dolabella auricularia. J Nat Prod 2004, 67, 1252–1255. [Google Scholar]
- Paterson, I; Findlay, AD; Florence, GJ. Total synthesis and stereochemical reassignment of (+)-dolastatin 19. Org Lett 2006, 8, 2131–2134. [Google Scholar]
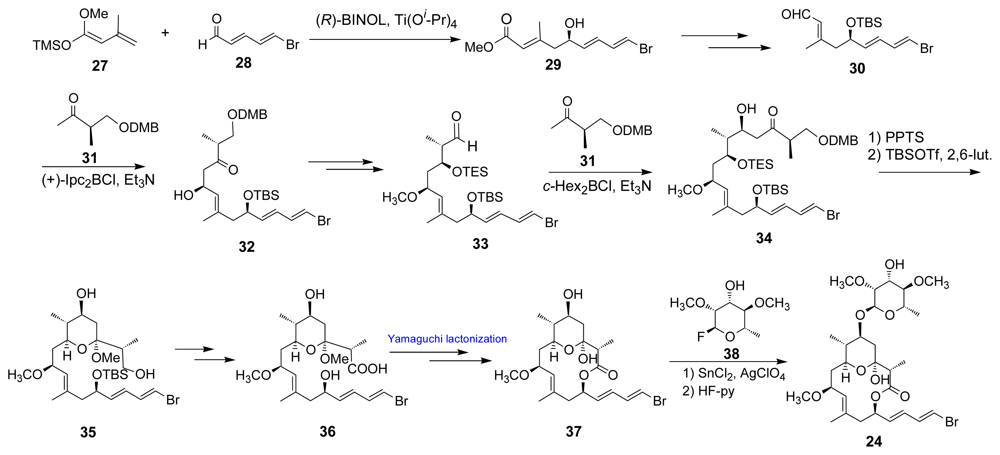
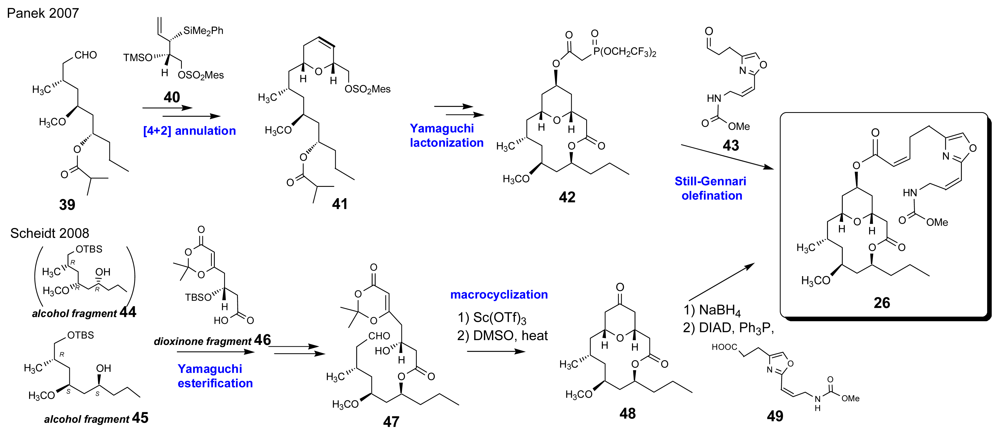
- Wright, AE; Botelho, JC; Guzman, E; Harmody, D; Linley, P; McCarthy, PJ; Pitts, TP; Pomponi, SA; Reed, JK. Neopeltolide, a macrolide from a lithistid sponge of the family Neopeltidae. J Nat Prod 2007, 70, 412–416. [Google Scholar]
- Youngsaye, W; Lowe, JT; Pohlki, F; Ralifo, P; Panek, JS. Total synthesis and stereochemical reassignment of (+)-neopeltolide. Angew Chem Int Ed 2007, 46, 9211–9214. [Google Scholar]
- Custar, DW; Zabawa, TP; Scheidt, KA. Total synthesis and structural revision of the marine macrolide neopeltolide. J Am Chem Soc 2008, 130, 804–805. [Google Scholar]
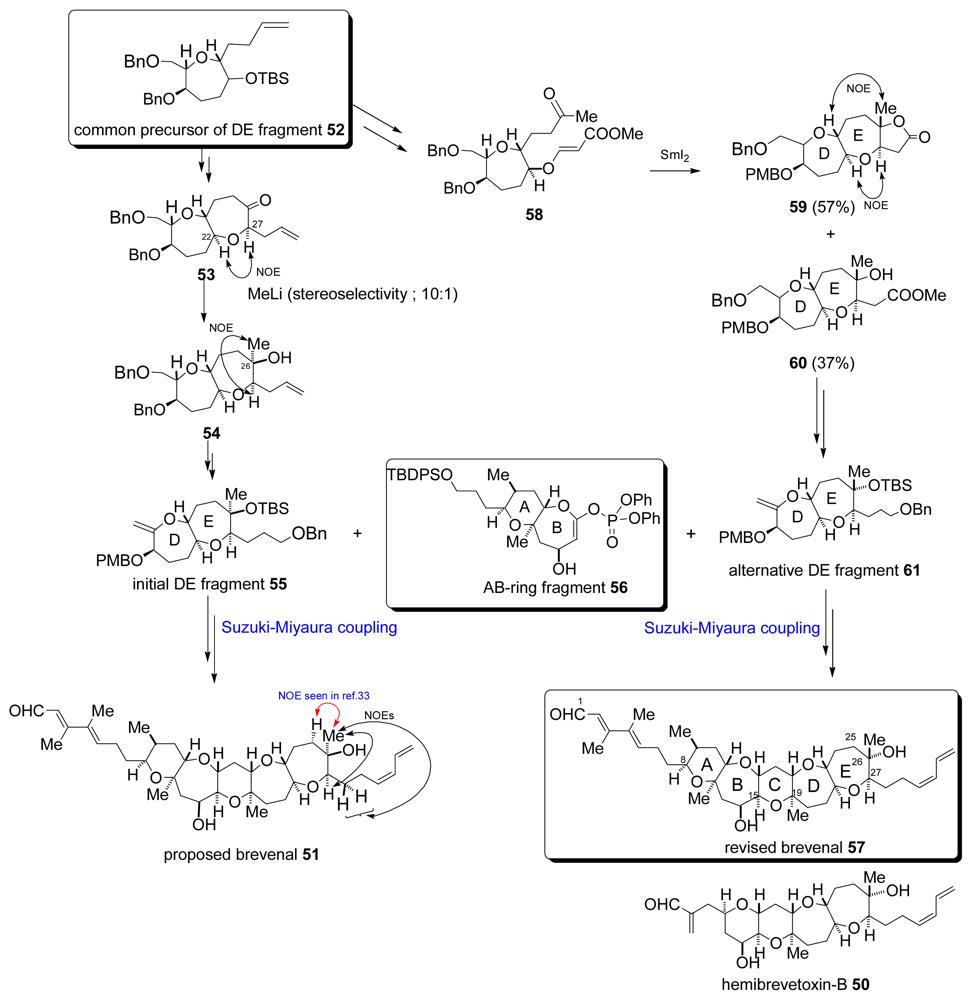
- Bourdelais, AJ; Campbell, S; Jacocks, H; Naar, J; Wright, JLC; Carsi, J; Baden, DG. Brevenal is a natural inhibitor of brevetoxin action in sodium channel receptor binding assays. Cell Mol Neurobiol 2004, 24, 553–563. [Google Scholar]
- Bourdelais, AJ; Jacocks, HM; Wright, JLC; Bigwarfe, PM, Jr; Baden, DG. A new polyether ladder compound produced by the dinoflagellate Karenia brevis. J Nat Prod 2005, 68, 2–6. [Google Scholar]
- Fuwa, H; Ebine, M; Sasaki, M. Total synthesis of the proposed structure of brevenal. J Am Chem Soc 2006, 128, 9648–9650. [Google Scholar]
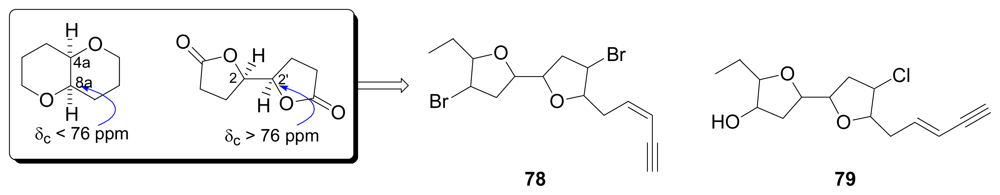
- Prasad, AVK; Shimizu, T. The structure of hemibrevetoxin-B; A new type of the Gulf of Mexico red tide organism. J Am Chem Soc 1989, 111, 6476–6477. [Google Scholar]
- Fuwa, H; Ebine, M; Bourdelais, AJ; Baden, DG; Sasaki, M. Total Synthesis, structure revision, and absolute configuration of (−)-brevenal. J Am Chem Soc 2006, 128, 16989–16999. [Google Scholar]
- Hall, JG; Reiss, JA. Elatenyne — a pyrano[3,2-b]pyranyl vinyl acetylene from the red alga Laurencia elata. Aust J Chem 1986, 39, 1401–1409. [Google Scholar]
- Wright, AD; Konig, GM; Denys, R; Sticher, O. Seven new metabolites from marine red alga Laurencia majuscule. J Nat Prod 1993, 56, 394–401. [Google Scholar]
- Sheldrake, HM; Jamieson, C; Burton, JW. The changing faces of halogenated marine natural products: total synthesis of the reported structures of elatenyne and an enyne from Laurencia majuscula. Angew Chem Int Ed 2006, 45, 7199–7202. [Google Scholar]
- Sheldrake, HM; Jamieson, C; Pascu, SI; Burton, JW. Synthesis of the originally proposed structures of elatenyne and an enyne from Laurencia majuscule. Org Biomol Chem 2009, 7, 238–252. [Google Scholar]
- Encarnacion, RD; Sandoval, E; Malmstrom, J; Christophersen, C. Calafianin, a bromotyrosine derivative from the marine sponge Aplysina gerardogreeni. J Nat Prod 2000, 63, 874–875. [Google Scholar]
- Ogamino, T; Nishiyama, S. Synthesis and structural revision of calafianin, a member of the spiroisoxazole family isolated from the marine sponge, Aplysina gerardogreeni. Tetrahedron Lett 2005, 46, 1083–1086. [Google Scholar]
- Ogamino, T; Obata, R; Tomoda, H; Nishiyama, S. Total synthesis, structural revision, and biological evaluation of calafianin, a marine spiroisoxazoline from the sponge, Aplysina gerardogreeni. Bull Chem Soc Jpn 2006, 79, 134–139. [Google Scholar]
- Bardhan, S; Schmitt, DC; Porco, JA, Jr. Total synthesis and stereochemical assignment of the spiroisoxazoline natural product (+)-calafianin. Org Lett 2006, 8, 927–930. [Google Scholar]
- Gavagnin, M; Mollo, E; Cimino, G; Ortea, J. A new γ-dihydropyrone-propionate from the caribbean sacoglossan Tridachia crispata. Tetrahedron Lett 1996, 37, 4259–4262. [Google Scholar]
- Jeffery, DW; Perkins, MV; White, JM. Synthesis of the putative structure of tridachiahydropyrone. Org Lett 2005, 7, 1581–1584. [Google Scholar]
- Sharma, P; Griffiths, N; Moses, JE. Biomimetic synthesis and structural revision of (±)-tridachiahydropyrone. Org Lett 2008, 10, 4025–4027. [Google Scholar]
- Atta-Ur-Rahman; Choudhary, MI; Hayat, S; Khan, AM; Ahmed, A. Two new aurones from marine brown alga spatoglossum variabile. Chem Pharm Bull 2001, 49, 105–107. [Google Scholar]
- Venkateswarlu, S; Panchagnula, GK; Gottumukkala, AL; Subbaraju, GV. Synthesis, structural revision, and biological activities of 4′-chloroaurone, a metabolite of marine brown alga Spatoglossum variabile. Tetrahedron 2007, 63, 6909–6914. [Google Scholar]
- Numata, A; Iritani, M; Yamada, T; Minoura, K; Matsumura, E; Yamori, T; Tsuruo, T. Novel antitumor metabolites produced by a fungal strain from a sea hare. Tetrahedron Lett 1997, 38, 8215–8218. [Google Scholar]
- Yamada, T; Iritani, M; Ohishi, H; Tanaka, K; Doi, M; Minoura, K; Numata, A. Pericosines, antitumor metabolites from the sea hare-derived fungus Periconia byssoides. Structures and biological activities. Org Biomol Chem 2007, 5, 3979–3986. [Google Scholar]
- Usami, Y; Mizuki, K; Ichikawa, H; Arimoto, M. Determination of the absolute configuration of the cytotoxic marine natural product pericosines D. Tetrahedron: Asymmetry 2008, 19, 1460–1463. [Google Scholar]
- Usami, Y; Suzuki, K; Mizuki, K; Ichikawa, H; Arimoto, M. Synthesis of (−)-pericosine B, the antipode of the cytotoxic marine natural product. Org Biomol Chem 2009, 7, 315–318. [Google Scholar]
- Usami, Y; Ohsugi, M; Mizuki, K; Ichikawa, H; Arimoto, M. Facile and efficient synthesis of naturally occuring carbasugars (+)-pericosines A and C. Org Lett 2009, 11, 2699–2701. [Google Scholar]














© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Usami, Y. Recent Synthetic Studies Leading to Structural Revisions of Marine Natural Products. Mar. Drugs 2009, 7, 314-330. https://doi.org/10.3390/md7030314
Usami Y. Recent Synthetic Studies Leading to Structural Revisions of Marine Natural Products. Marine Drugs. 2009; 7(3):314-330. https://doi.org/10.3390/md7030314
Chicago/Turabian StyleUsami, Yoshihide. 2009. "Recent Synthetic Studies Leading to Structural Revisions of Marine Natural Products" Marine Drugs 7, no. 3: 314-330. https://doi.org/10.3390/md7030314
APA StyleUsami, Y. (2009). Recent Synthetic Studies Leading to Structural Revisions of Marine Natural Products. Marine Drugs, 7(3), 314-330. https://doi.org/10.3390/md7030314