1. Introduction
The mineral mayenite, also known as C12A7 and chemically Ca
12Al
14O
33, has gained attention in the last two decades due to the high degree of functionality afforded by the crystal structure. C12A7 crystallizes in the highly symmetric
space group with
a = ≈11.98 Å and two formula units per unit cell resulting in 118 atoms within the unit cell. The functionality of the material results from the clathrate structure where the unit cell is made up of a positively charged framework of twelve interconnected cages and occluded stabilizing anions. These anions within the cage are weakly bound to the framework leading to a [framework]: occluded-anion notation,
. Manipulating cationic doping of the framework and mobile occluded anions in C12A7 leads to potential applications in inorganic and organic synthesis; surface treatments; catalytic reactions; as a high purity and high density anionic source; transparent conductive oxide; direct writable transparent wires and media; luminescent for displays, lighting, or photoelectric devices; gas and biomass reforming; and even an antibacterial agent among numerous other applications; Liao et al. present a comprehensive review on the potential applications of the stoichiometric
as well as its derivatives [
1].
Hosono et al. have conducted extensive research on C12A7 most notably on the formation of the
derivative [
2,
3]. The stabilizing
anion is replaced by electrons that are localized in the “potential well” created by the cationic cage leading to the classification of an electride. C12A7 was the first room-temperature stable inorganic electride bringing the realization and application of electrides to ambient conditions [
4]. The formation of the electride seems to be robust with many different formation routes, all focusing on processing C12A7 in the presence of a preferentially oxidized sacrificial target, known in this context as an “oxygen getter” [
5]. The most practical methods involve utilizing carbonaceous environments due to experimental ease and lack of post processing of previously synthesized and consolidated samples. Kim and Hosono have published a review on the characteristics of the electride C12A7 [
3].
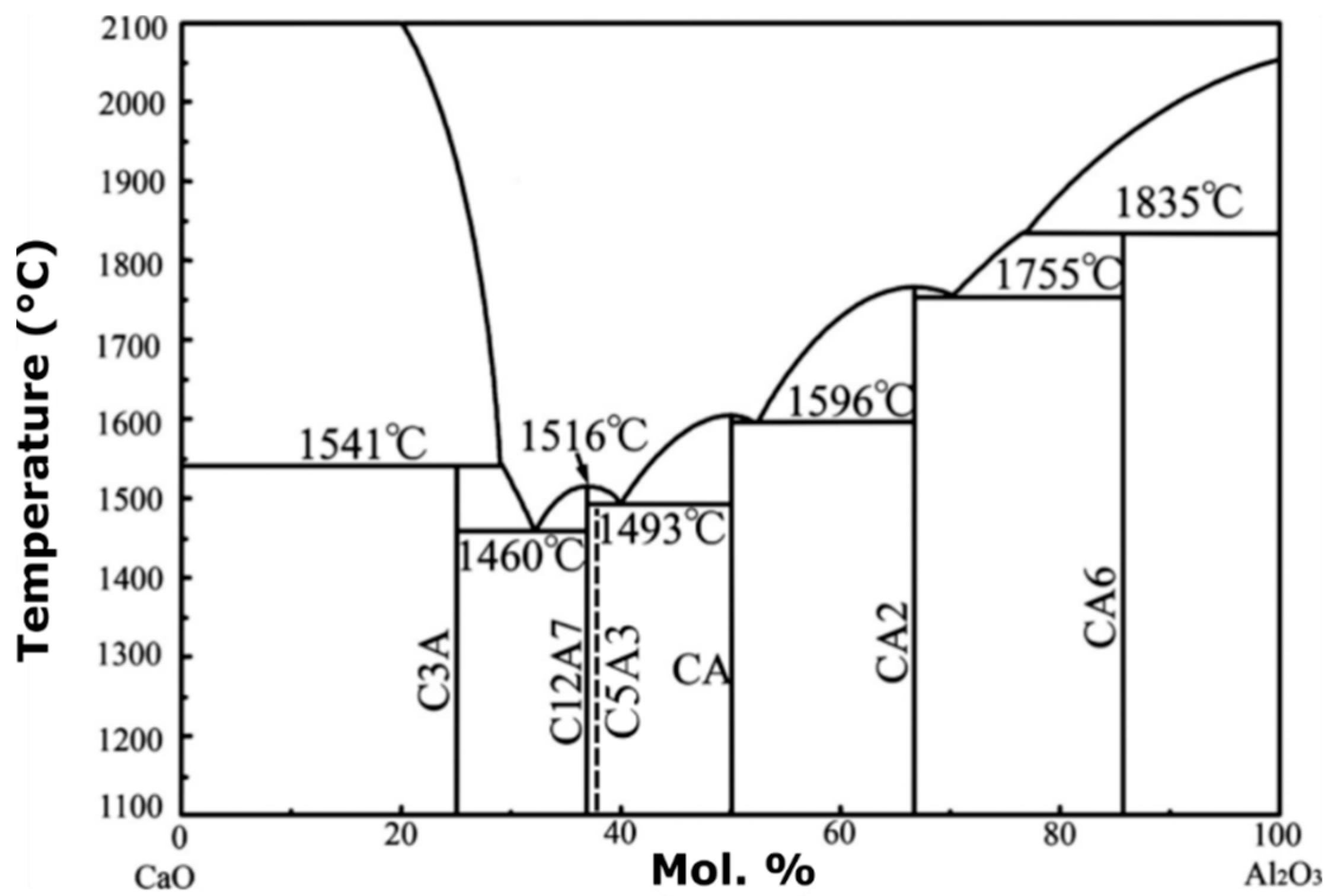
The stoichiometric compound,
is an invariant binary compound in the CaO–Al
2O
3 system at approximately 37 mol%
(
Figure 1). In the shorthand notation in
Figure 1, C represents CaO, while A refers to the sesquioxide Al
2O
3. Gfellar and Salasin and Rawn have published reviews on the structure of C12A7 and electride C12A7 [
5,
6]. On either side of the invariant C12A7 is a two-phase region containing C12A7 and two other invariant binary compounds
(C3A) and
(CA). The compound
(C5A3) is meta-stable and is frequently discussed in the literature when dealing with the C12A7 system. It is interesting to note that the shorthand notation provides a convenient means to write mass balance reactions for the formations of calcium aluminate compounds found in the binary CaO–Al
2O
3 phase diagram.
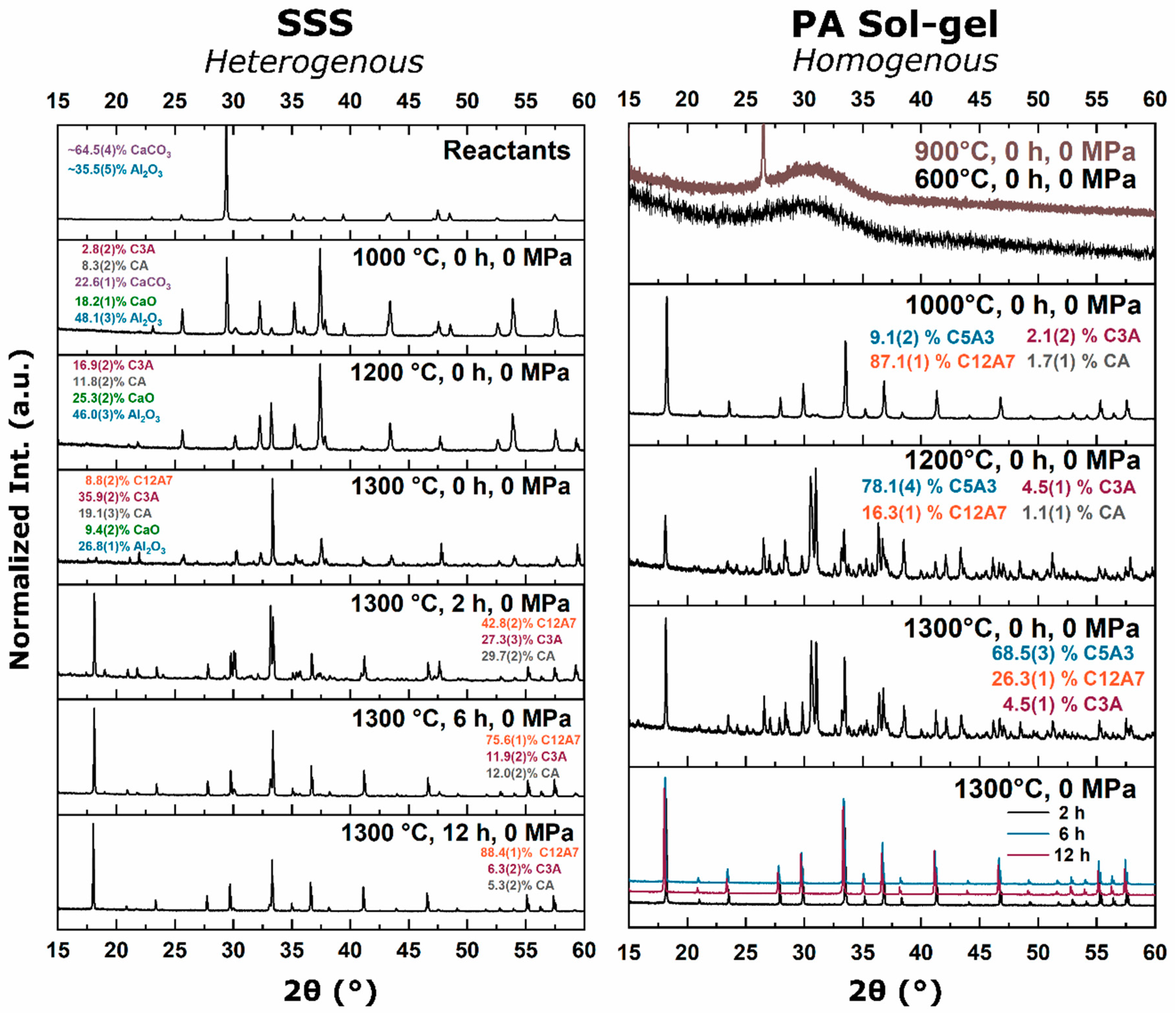
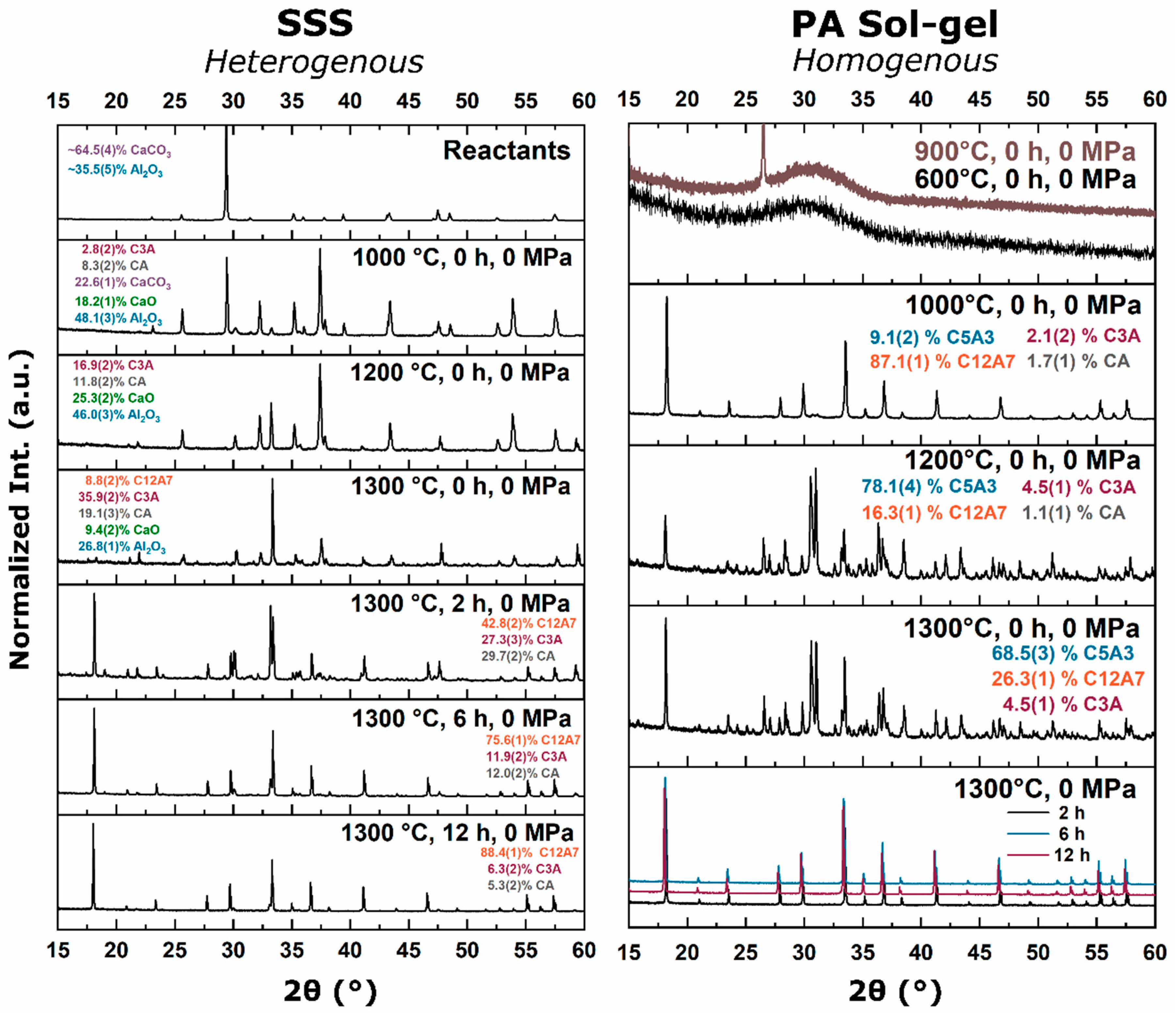
Thermodynamic solid-state synthesis (SSS) investigations into the formation of C12A7 from
and
found that the first phases to crystalize are C5A3 and C3A [
7,
8]. The C5A3 concentration increases until approximately 950 °C, after which C5A3 is consumed in favor of the formation of C12A7. C5A3 and C12A7 are close in stoichiometry,
, and crystal structure. Both structures contain octahedrally coordinated Ca and tetrahedrally coordinated Al cations, but C5A3 is an ordered layered structure, while C12A7 is a disordered clathrate structure (
Figure 2). As the temperature is increased, the formation of CA occurs until approximately 1200 °C when C3A and CA combine to form C12A7 [
7]. These observations suggest there are two formation pathways from solid-state reactants: (1) C5A3 + C3A at low temperatures (<900 °C) when cationic species are well-mixed [
9,
10], and (2) C3A + CA at high temperatures (>1100 °C), observed when long diffusion lengths of reactants are present [
7,
8]. Recent in situ kinetic studies suggest that different pathways might occur with C12A7 being a kinetically favorable phase to form directly at lower temperature and C5A3 only observed as a decomposition product of C12A7 [
11].
The relationship between C3A, CA, and C5A3 is further elucidated when investigating the decomposition of C12A7. The presence of occluded anions is heavily correlated to the stability of the clathrate structure, and in the absence of any template anions, i.e., under dry and/or reducing conditions, C12A7 is not thermodynamically favorable and decomposes. This was first observed in single crystal growth experiments where moisture or oxygen was needed to nucleate the C12A7 framework [
3]. The decomposition products were reported to vary based on process temperature and were either C5A3 + C3A or C3A + CA; a historical summary of formation products in various oxidizing, reducing, inert, dry, and hydrated atmospheres has been compiled by Kim et al. [
3,
12]. As the breadth of electride formation research grows, the decomposition of C12A7 continues to limit electride formation at elevated processing temperatures for long process durations. Palacios et al. observed the electride formation and subsequent decomposition of electride-C12A7 powder to C5A3 and C3A through in situ neutron diffraction in a V sample holder heated to 1100 °C under vacuum [
13]. Ali et al. observed C12A7 decomposition during electride formation of float zone (FZ) single crystals sealed in an evacuated quartz ampule with Ti powder and fired at 1200 °C for 48 h [
14]. Eufinger et al. conducted a study to identify the diffusion of various anions in the C12A7 structure and discovered a correlation between humidity and decomposition of the C12A7 framework [
15]. A decomposition analysis found that under dry conditions, C12A7 decomposes at temperatures above 1050 °C, which is correlated with the critical mobility limit for Ca ions [
15]. This decomposition was observed in both inert and oxidative atmospheres, where decomposition times were greater than 24 h. The decomposition products of C3A + C5A3 or CA + C3A were observed and correlated with the temperature and atmosphere conditions during the initial temperature ramp [
15]. Decomposition was found to nucleate at the macroscopic crystal defects, which agrees well with the calculated Avrami exponent by Palacios et al., suggesting that the limiting rate step of decomposition is the growth of the decomposed products around the C12A7 phase and not the random nucleation of C5A3 products [
13,
15]. These decomposition times and temperatures agree with those observed in the reduction experiments, but the vacuum and reducing environments may lead to an increase in the rate of the decomposition reaction. Just as in the formation of C12A7, there are two decomposition pathways: (1) C5A3 + C3A, and (2) C3A + CA.
Conversion to
occurs primarily via the reaction of the occluded oxygen with an oxygen getter [
5]. As the electride formation occurs, sample color transitions from white, through green, to black, and the electrical conductivity transitions through an insulating, semi-conducting, and metallic conducting regime, respectively [
3]. One oxygen getter reduction method involves processing the synthesized C12A7 powder, single crystal, thin film, consolidated sample, etc., in a C crucible under flowing inert gas. This generates a strong reducing atmosphere in the crucible leading to the proposed reduction of the sample via the following reaction
[
16]. This is favorable leading, to a conversion with no post processing, but the degree of reduction attainable is not well-reported or discussed and it appears that only limited conversion of
to
occurs. The other proposed C based reduction method involves the replacement of the occluded
with
which has a similar valence and ionic radii with 1.4 and 1.2 Å, respectively [
17,
18]. The carbide anion is then hypothesized to be instantaneously unstable or unstable during cooling and decomposes to either solid C or
gas creating anionic vacancies and forming the electride. The pathway to anionic exchange is not clear and appears to not be an exchange but a full recrystallization of the decomposition products to C12A7, indicating that decomposition of
needs to occur before
formation and electride conversion can occur [
17]. The realization of a full CA recrystallization process yields promise for direct formation of electride-C12A7 from reactants. The direct formation of electride-C12A7 from C3A + CA reactant mixtures has been presented as well as from a carbon-rich Pechini sol-gel precursor [
17,
19,
20]. These studies show a clear plateau in electrical conductivity but little theory for this plateau in electronic properties or characterization of the underlying atomic structure has been presented.
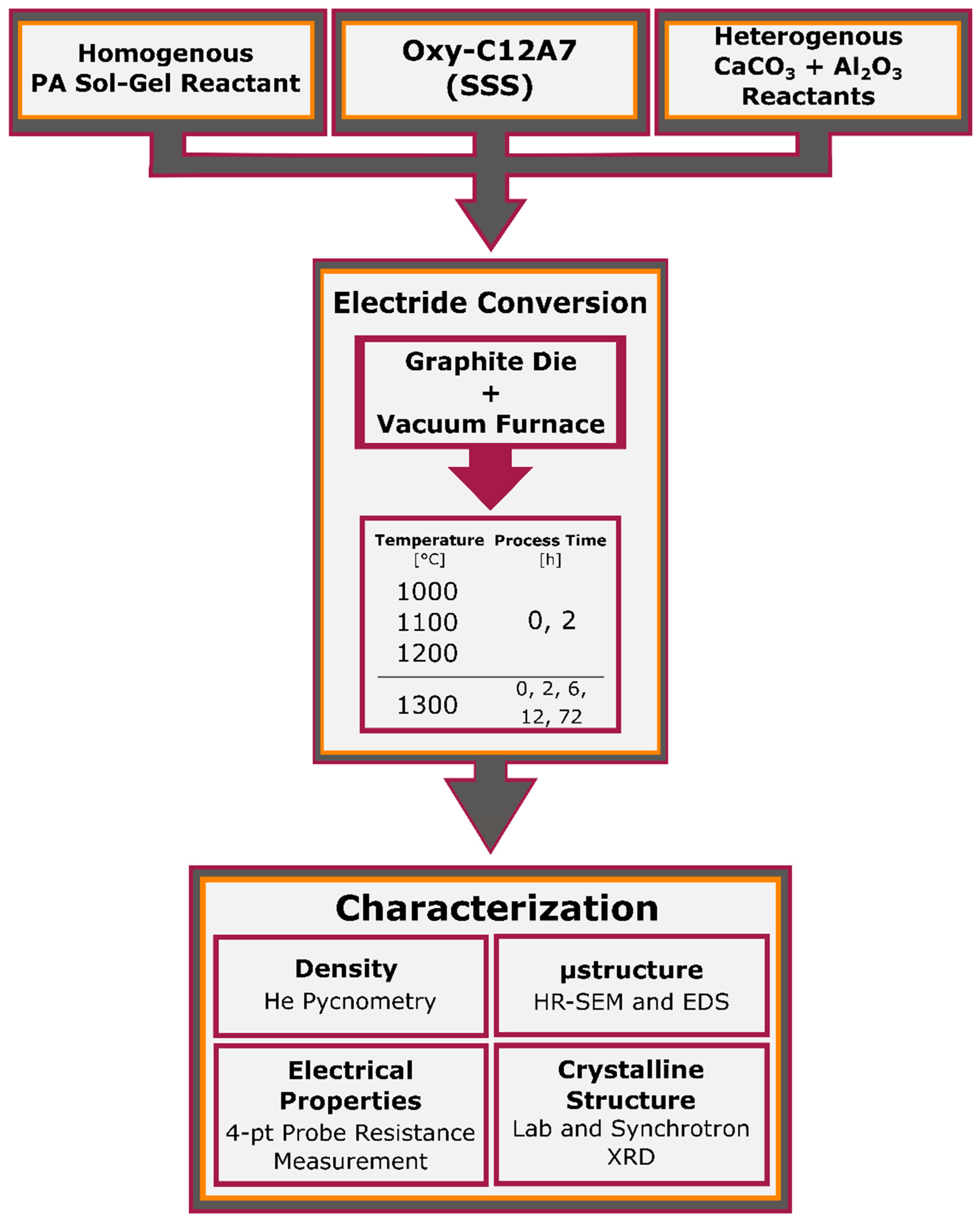
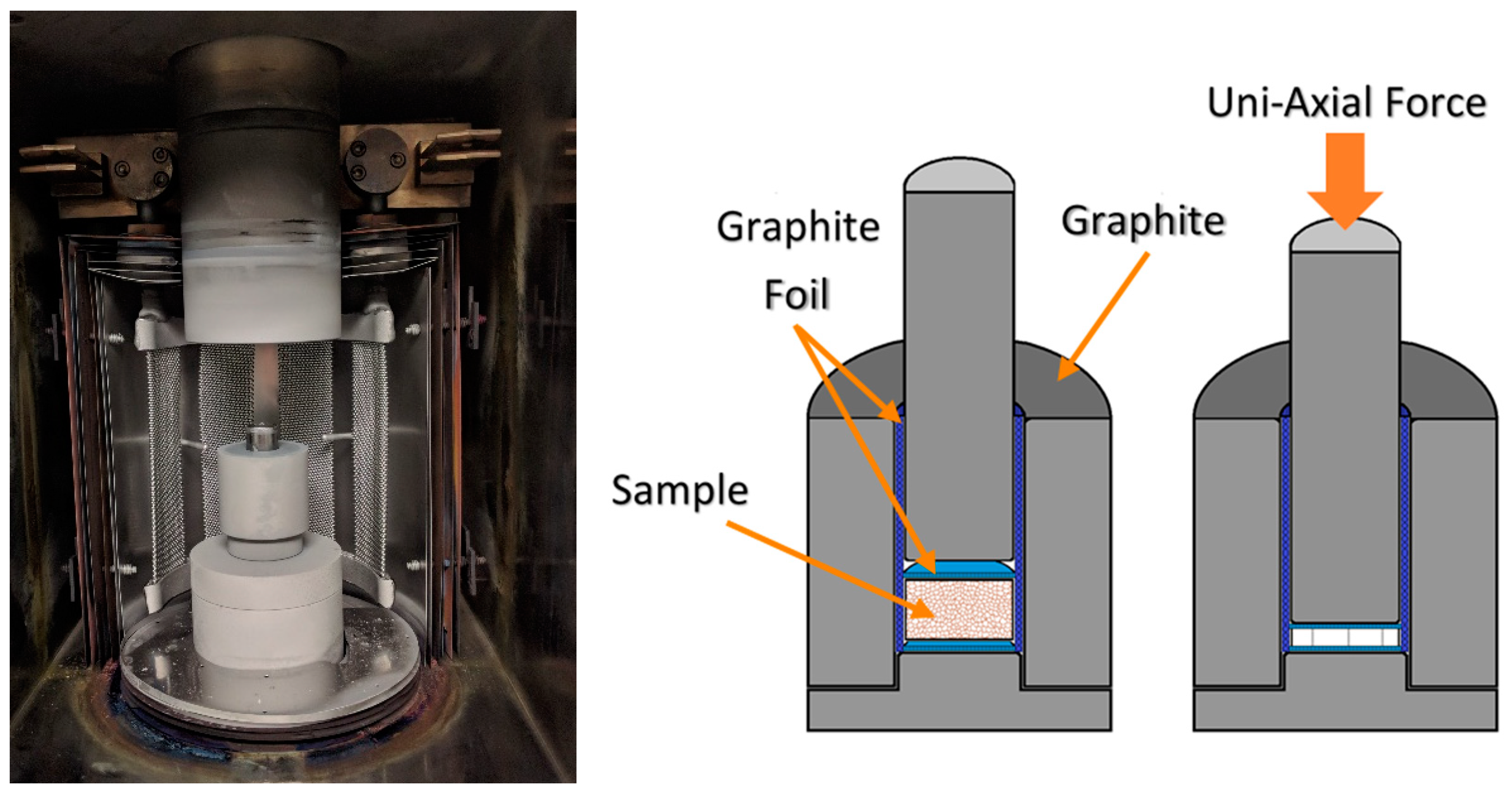
This report seeks to study the processing–structure–property relationships of the conversion of oxy-C12A7 to electride-C12A7, as well as the direct formation of electride-C12A7 from both conventional solid-state (CaCO
3 + Al
2O
3) and non-carbonaceous polymer-assisted sol-gel reactants. The polymer-assisted sol-gel reactant mixture benefits from short diffusion pathways achieved in the Pechini synthesis, previously implemented by Khan et al. to create a carbon-C12A7 electride composite, but instead utilizes the environmental carbon instead of reactant carbon to create a consolidated C12A7 sample with low carbon content [
20]. The experiments reported here make use of a carbonaceous environment within a high vacuum furnace that results in a carbonaceous dry (low
) low-pressure (low
) environment. Temperature and time are controlled to investigate the formation, stability, and degree of reduction obtainable by the carbonaceous reduction processes. The changes in electronic and atomic structure for the conversion from oxy-C12A7 to electride-C12A7 and direct formation of electride-C12A7 from heterogenous and homogenous reactants can be analyzed post-processing: X-ray diffraction (XRD) is used to characterize the change in the atomic structure, scanning electron microscopy (SEM) is used to evaluate microstructural changes, and electrical resistivity measurements associate the atomic and microstructural features to electronic properties determining the degree of reduction. The results of this characterization are then used to develop a reduction model involving the formation of a mixed C- and O-occupied C12A7 caged structure; C is sourced from the outward solid-state diffusion of C from the sample holder and O is sourced from the extra oxygen present in the solid reactants. The O is explicitly not sourced from any gas phase reactant.
4. Discussion
The electride-C12A7 phase was achieved through conversion of oxy-C12A7 and through direct formation from heterogenous SSS and homogeneous sol-gel reactants. The degree of electron concentration agrees well with other reports and the structural evolution as a function of process time and temperature elucidated three key points for discussion: what is responsible for the change in phase equilibria at 1300 °C leading to C12A7 phase stability, what is the mechanism for electride formation, and what leads to the plateau in the degree of reduction?
4.1. High Temperature C12A7 Stability
The thermodynamic phase equilibria changed as a function of process temperature and time. The as-synthesized oxy-C12A7 starting phase was stable up to 1000 °C followed by instability and decomposition as the process temperature was raised to 1200 °C. This decomposition is well documented in the C12A7 literature and coupled to the instability of the “free” occluded O
2− anion and activation of Ca diffusion above 1050 °C, leading to a change in thermodynamic equilibria [
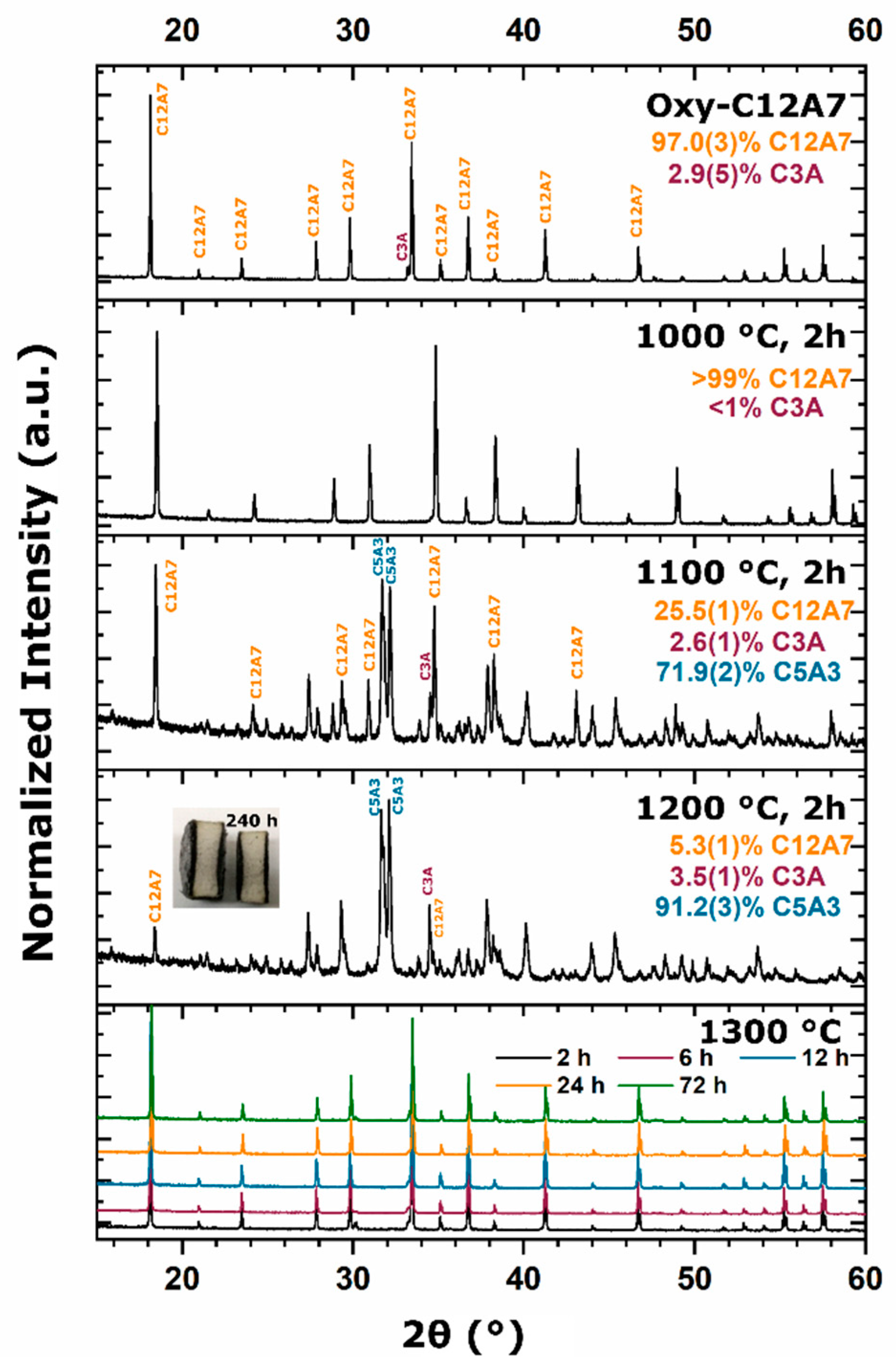
15]. This thermodynamic equilibrium was also observed from reactant mixtures. For heterogenous reactants no C12A7 formation was observed as the temperature was raised to 1200 °C with C3A and CA being favorable. Homogenous reactants led to C12A7 formation at low temperatures (≤1000 °C) due to kinetic favorability, but at higher temperatures, C12A7 decomposition was observed and C5A3 became the thermodynamic favorable phase. A shift in phase equilibria came when the process temperature was increased to 1300 °C. C12A7 phase purity was observed as processing time was increased regardless of the starting point.
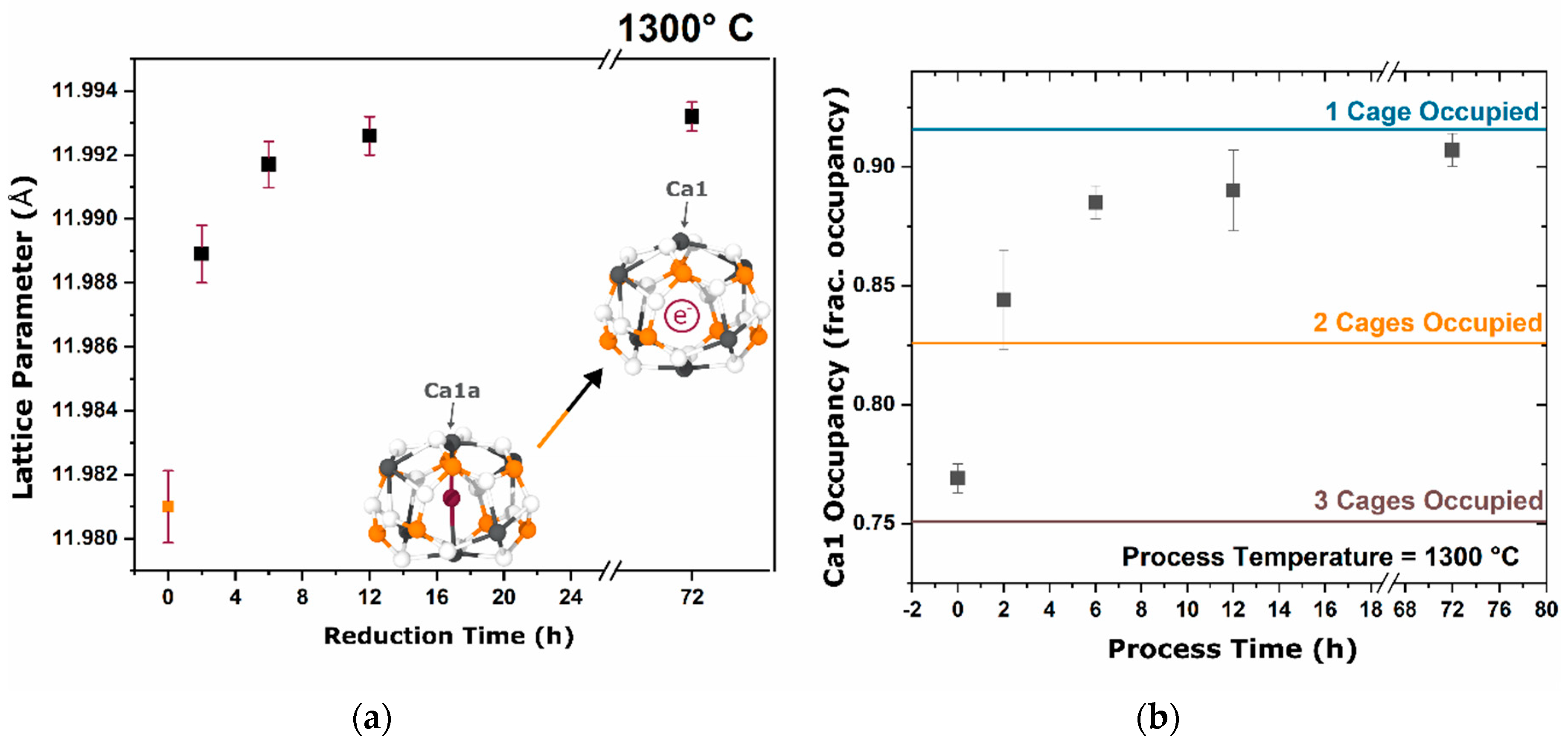
During the furnace ramp, the decomposition of previously synthesized oxy-C12A7 to C5A3 and C3A did not occur instantaneously, and as the temperature was rapidly increased to 1300 °C, at a ramp rate of 480 °C/h, full decomposition would not occur before 1300 °C. Once at 1300 °C the oxy-C12A7 structure was mostly retained but actively decomposing. The secondary phases of C3A and CA were observed, indicating a change in the decomposition kinetics brought on by higher cationic diffusion, but after 6 h, C12A7 phase purity was achieved correlated with approaching the asymptotic values of conductivity, lattice parameter, and occupied cages. This suggests that a decomposition of oxy-C12A7 and reformation back to C12A7 was fundamental to achieving the electride phase.
This reformation process was corroborated for the oxy-C12A7 conversion process through a multi-step processing experiment. The sample was initially fired isothermally at 1200 °C for 2 h before increasing the process temperature to 1300 °C for 2 h. Full decomposition to C5A3 occurred at the lower temperature and the presence of the C12A7 structure at the higher temperature corroborated the recrystallization process. The atomic structure lattice parameter and sample conductivity were greater than that when directly processing at 1300 °C for 2 h, but slightly less than that of a sample processed at 1300 °C for 6 h. This suggested that the rate of approaching the asymptotic atomic and electron structure was not dependent on the time spent at 1300 °C, but on the total time spent at a temperature where decomposition could occur and the degree of off-stoichiometry from C12A7 of the decomposed products; the more off-stoichiometric the intermediary calcium aluminate phases, the more diffusion was necessary to form C12A7.
The exact transformation dynamics were obscured by the presence of oxy-C12A7 and the approach to the asymptotic values could be related to an average of the oxy-C12A7 and the newly formed electride-C12A7 phase. The direct formation of electride-C12A7 from reactant mixtures was not obscured by the presence of previously synthesized oxy-C12A7. The fact that there was no approach to the asymptotic values suggested that the electride forms from a direction crystallization process. Structural characterization showed that the asymptotic structural values observed in the conversion of oxy-C12A7 to electride-C12A7 were the intrinsic starting values of the electride-C12A7 formed through the direct formation process. These structural observations led to the question of “what thermodynamic change or activated kinetic process leads to this change in equilibria?” Oxy-C12A7, full electride-C12A7, or a mixed oxy/electride-C12A7 structure would all be unstable under these experimental conditions (>1050 °C in a dry reducing atmosphere) indicating that a high temperature stable anionic species must be aiding in phase stability [
13,
14,
15].
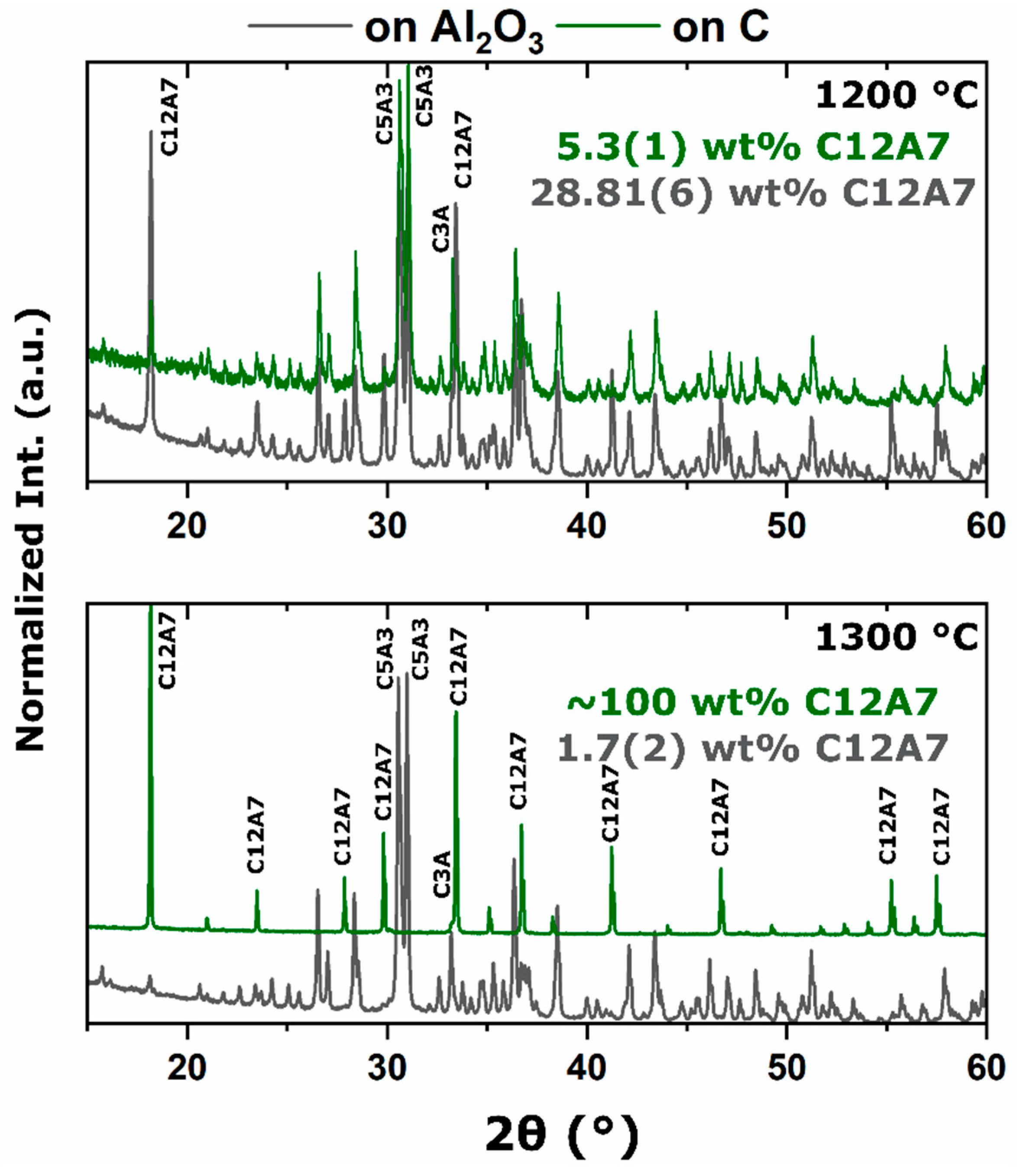
As the processing temperature was raised from 1200 to 1300 °C, the formation and stability of C12A7 was observed from the three starting points. The rise in temperature was correlated with a visible color change of samples from the as-synthesized white to a black; however, when the consolidated samples were ground, the powder color was dark green; the ultimate conductivity achieved was too low for the change to black to be correlated to an increase in free Drude carriers, indicating that the change to black resulted from another reason [
31]. This observation coupled with the observance of C in the microstructure indicated the increase in system energy activated the diffusion of C whose availability as an anionic species led to high temperature C12A7 stability; the diffusion of C into hot-pressed materials (similar experimental conditions, but with an addition of pressure) is well documented with the carbonaceous source of the die [
32,
33].
To evaluate this theory, the carbon source was removed and replaced with alumina and no observed stabilization occurred; phase equilibria consisted of C5A3 and C3A at 1300 °C. At 1200 °C, the kinetics of decomposition to C5A3 and C3A were sluggish compared to when the sample was directly in contact with C, indicating that the presence of sample–die interfacial carbon played a role in the instability of the oxygen anion. This correlates well with the observation that decomposition increased with a shift toward more reducing environments from a dry oxidizing, to vacuum, and ultimately to a carbonaceous vacuum environment [
15,
34]. The activation of carbon diffusion and phase equilibria achieved when removing the carbon source clearly indicated that a solid-state interaction of C led to the C12A7 phase.
The structural observations from this solid-state experimentation match well with the equilibria theory proposed in characterizing electride-C12A7 synthesized from high temperature melts [
17]. No inference can be made on the species of carbon, but in the solid-state process, the diffusion of carbide species in C12A7 is unlikely. The cages may act like nano-reactors forming the carbide anion, but diffusional processes likely require the disassociation and interstitial diffusion of individual carbon species. This is a similar process to that proposed for hydroxide diffusion [
21].
4.2. Electride Formation Mechanism
The structural investigation provided strong experimental evidence for the necessity of C for high temperature C12A7 crystallization, but the mechanism behind electride conversion and the asymptotic behavior in electride conversion were still difficult to elucidate with a high degree of confidence. The theory for electride conversion put forth from melt formed electride-C12A7 is that the
anion is responsible for C12A7 crystallization and anion instability is observed during cooling or instantaneously after nucleating the cages [
17]. In this study anion instability required a diffusionless phase transformation as the temperature was quickly quenched to below 600 °C; this was further limited due to the slow dissociative diffusion process proposed for carbon diffusion. Instantaneous instability after nucleating the cages did not explain the retained phase stability of electride-C12A7 for extended time durations in a thermodynamic regime where decomposition would occur. The electron is not capable of sustaining the C12A7 framework and decomposition was observed in non-carbonaceous environments [
13,
14]. The second consideration is the electride conversion process, which does not continue to full electron concentration, resulting in semi-conducting behavior reported in other carbonaceous reduction studies [
3,
16,
35]. The aforementioned model does not accurately characterize the structural behavior reported here and another model is needed to characterize the electride formation observed in this report.
An alternative model proposed by Jiang et al. assumes that carbide anion diffusion into the C12A7 particles will not occur and that the reduction process occurs through the interfacial reaction of occluded O and microstructural carbon. Two issues that remain unresolved with this model are: (1) the formation of oxy-C12A7 at higher temperature, where it is thermodynamically unstable, and (2) the long duration of phase stability under conditions where neither oxy-C12A7 nor electride-C12A7 are thermodynamically favored.
We propose a new theory that builds upon the two current theories where C12A7 formation occurs with mixed C and O occluded anions and the electride conversion occurs through interaction of these occluded species and interfacial carbon species. Previously, the theory that carbide was responsible for the crystallization of C12A7 from a high temperature melt assumed no other templating anions besides C were available. This is a reasonable assumption in the sense that even if there was excess oxygen in the melt, it is not thermodynamically favorable to form oxy-C12A7, but in the solid-state system, extra-framework oxygen is accounted for in the chemical equilibria. The main formation pathways observed for C12A7 formation occur through the interaction of off-stoichiometry calcium aluminate phases:
If stoichiometrically balanced utilizing the framework stoichiometry (Ca12Al14O32), an excess of oxygen is needed. Support for this theory is observed during the direct formation of the homogeneous sol-gel reactants where C12A7 formation occurs between 900°C and 1000 °C where carbon diffusion has yet to be activated and the energy for oxy-C12A7 thermodynamic instability has yet to be reached; the only available anion for C12A7 formation is oxygen contained within the reactants. The presence of oxygen in the second theory by Jiang is plausible with a realized oxygen source and interfacial reduction can occur; however, the stability of the oxy/electride-C12A7 structure is not explained if we assume a single anion model.
It was previously assumed that carbide diffusion into the C12A7 particle is unlikely, but the diffusion of mono-atomic carbon species is likely. Structural analysis shows that the cage “windows” are 3.7 Å in diameter and carbon diffusion is readily observed interstitially in Fe, where nearest neighbor bond distances are <3 Å [
36,
37]. The idea of mixed anion stability is observed in the C12A7 phase space [
15,
28,
36]. Under humid conditions, a mixture of oxygen- and hydroxide-occupied cages leads to phase stability above 1050 °C where oxy-C12A7 instability is observed under dry conditions [
15]. With the presence of oxygen and carbon species, phase stability is understood and there is a clear electride formation process. The negative enthalpy of formation for the reaction of oxygen and C drives the formation of anion vacancies and electron injections; this may occur through the interaction of occluded species, as well as interfacial species, as proposed by Jiang et al. [
19].
4.3. Plateau in Electride Formation
The final point for consideration is the driving factor behind the ultimate level of reduction reaching a semi-conducting state rather than the metallic type conductivity state achieved with other techniques. The driving factor behind electride conversion is thermodynamic equilibria and the presence of a negative enthalpy chemical reaction. In order to reach this thermodynamic equilibrium, the diffusion of occluded anions from the C12A7 phase to the oxygen getter needs to occur, resulting in anionic vacancies. The plateau in conductivity is either limited by the intrinsic reduction process, the kinetic diffusion of anionic species, or the thermodynamic driving factor. An example of the intrinsic reduction process limiting electron concentration is observed in the hydrogen gas reduction process [
38]. However, the same equilibrium observed in this study was observed when treating oxy-C12A7 in a
-reducing atmosphere; in this case there were no carbon species occupying the cage and only
, suggesting that another limiting factor should be considered [
16].
The Ca metal reduction method only achieves moderate conductivity (≈100 S·cm
−1) due to the blockage of the kinetic pathway responsible for anionic vacancy injection [
39]. In the mixed oxygen and carbon occluded anion C12A7, high oxygen mobility toward the occluded carbon species would be observed. The formation of CO would take place inside of the cage, similar to the hydration of C12A7 and hydroxide diffusion, and once the new polyatomic anion has formed, its diffusion and mobility will be dramatically hindered relative to monoatomic species; a diffusion method similar to
diffusion may occur with the dissociation of CO, interstitial diffusion of C, and then recombination in the adjacent cages [
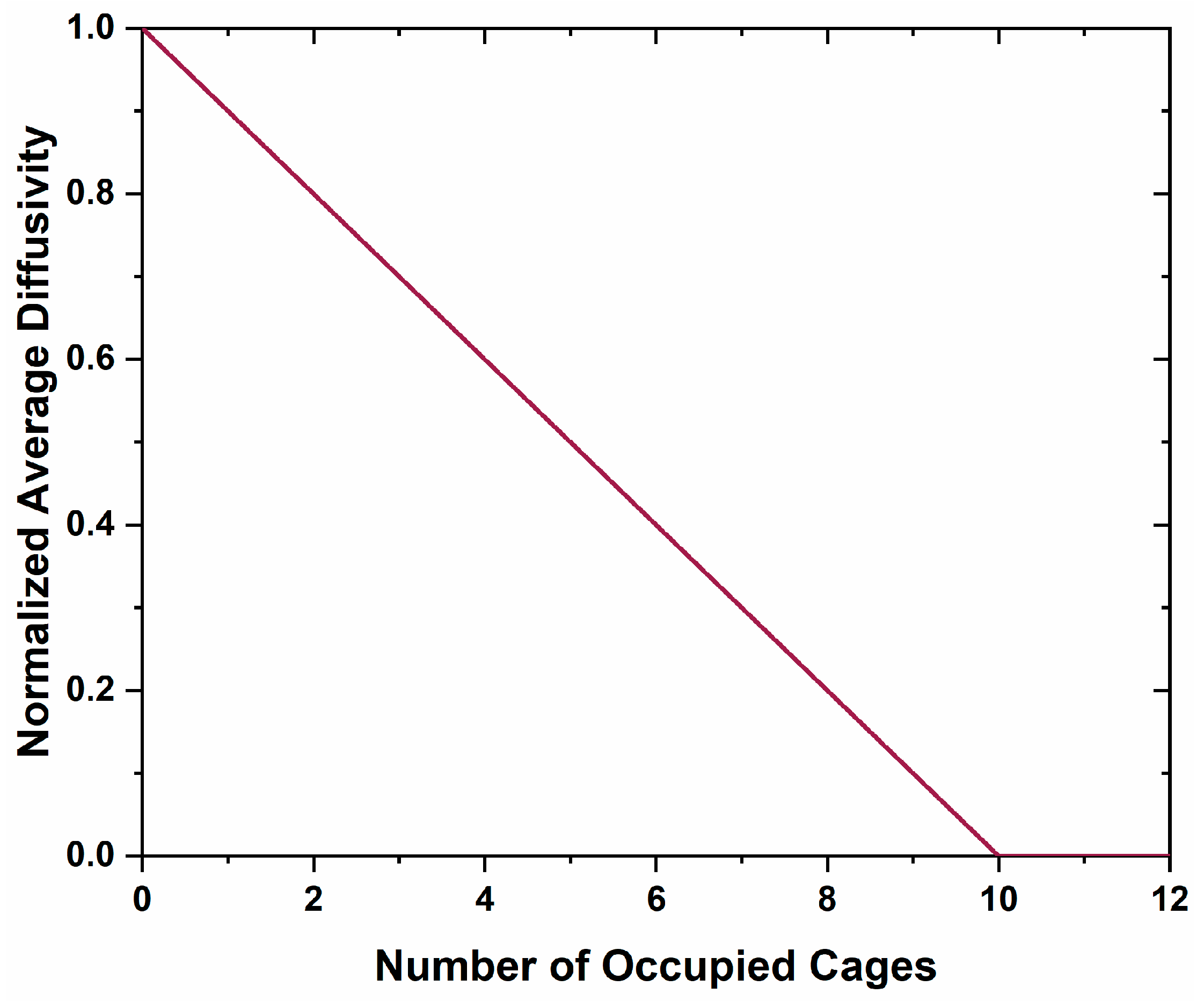
21]. The large diatomic anion will act as a blocking agent for future oxygen, carbon, and electron diffusion, limiting further formation of anion vacancies and limiting mobility of localized carriers. There is a significant difference between the blocked kinetic pathways in the Ca metal reduction method versus this method. In the former the kinetic pathway blockage is due to a layer of CaO encasing the material, where this discussion suggests a blocking of diffusion pathways by molecules within the cages. This is unlikely due to the high interconnectivity of the cages, where each cage has 12 nearest neighbor cages. Application of a percolation theory, Equations (1) and (2), of a system with 12 neighbor connectivity demonstrates that while diffusion would decrease with an increase in the number of occupied cages, it would not cease completely until 10 cages are occupied (
Figure 12) [
40].
where
is the normalized average diffusivity;
is the fraction of blocked cages;
f is the diffusivity through blocked pathways, which was set to zero; and
z is the average coordination number to nearest neighbor diffusion sites, 12 in this case [
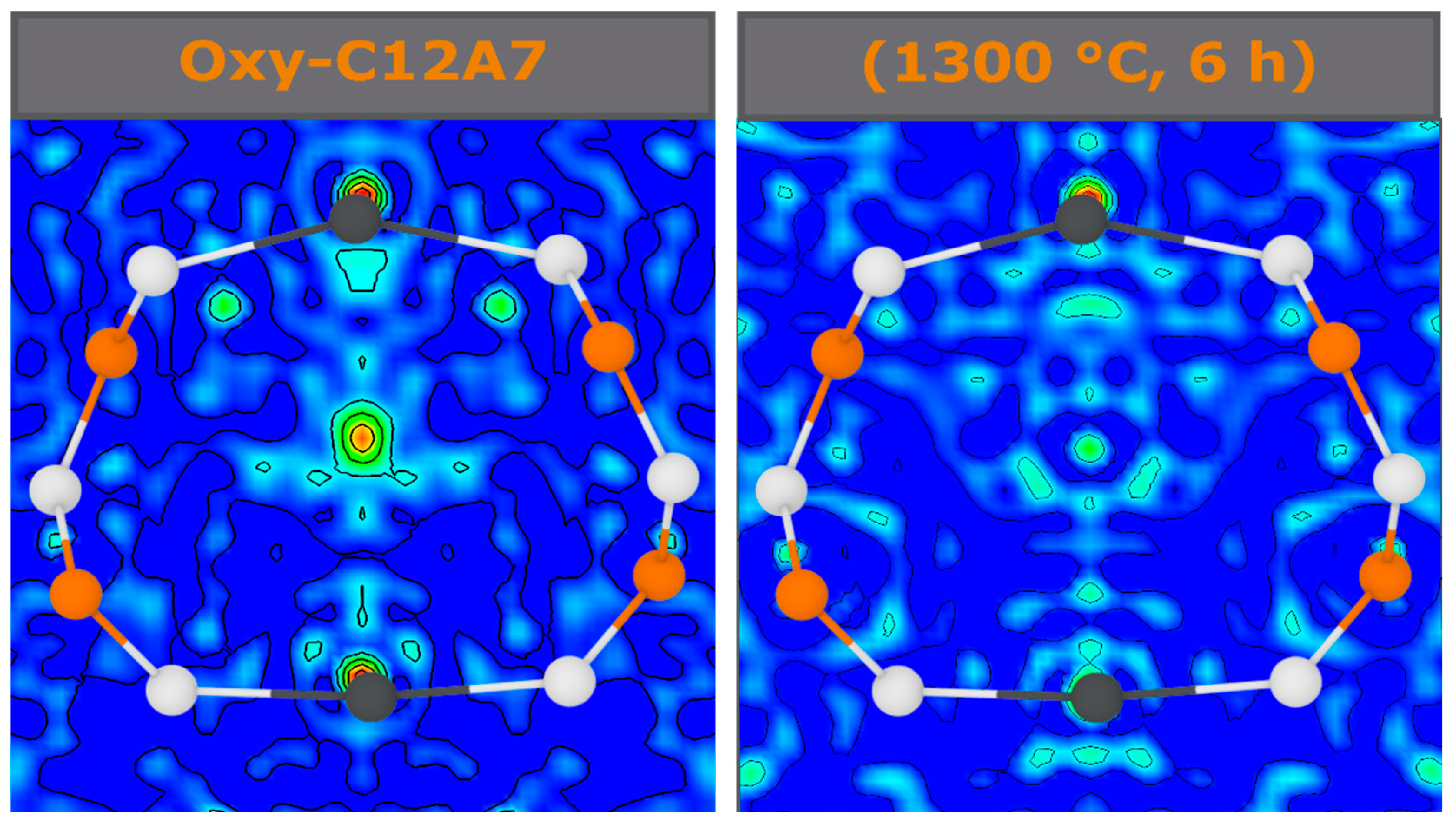
40]. The Fourier difference maps demonstrate a decrease in occluded position scattering density corresponding to cage occupancy, making the equilibria observed unlikely due to a blocking of diffusion kinetic pathways in the cage structure. Therefore, another process should be considered.
The reduction of C12A7, in all techniques, is driven by the negative enthalpy of oxidation of an external chemical species. Oxidation of Ca and Ti have large negative enthalpies of formation, −635 and −940 kJ·mol
−1, respectively [
41,
42]. The enthalpy of formation for CO and CO
2 is relatively lower at −110.53 and −393.52 kJ·mol
−1, respectively [
41]. While C12A7 is an oxidizing agent with oxygen in the cage, the full electride has a positive enthalpy of formation, indicating that the full oxygen occupation is thermodynamically preferred and oxidation of the fully converted electride has an enthalpy of formation between −425 and −600 kJ/mol [
16,
43]. This makes the electride a reducing agent (the ability for C12A7 to be both an oxidizing and a reducing agent as a function of processing is part of what leads to the observed high functionality).
Figure 13 compares the enthalpy of oxidation as a function of electride conversion with the enthalpy of oxidation for C, Ca, and Ti superimposed; the enthalpy of oxidation for electride-C12A7 decreases as the degree of electron concentration decreases and the slope of this line is −580 kJ·mol
−1 [
43]. This representation assumes that these values are comparable at elevated temperatures, that the enthalpy of formation from elemental species is a good approximation for that observed in the complex C12A7 system, and that the linear decrease in enthalpy as a function of electron concentration is valid under the carbonaceous vacuum conditions. TiO
2 and CaO have enthalpies of formation with magnitudes higher than that of the full electride, indicating that oxidation of these cations will occur in favor of oxidation of the electride-C12A7 with the highest possible electron occupation. However, for the oxidation of C, both the oxidation to CO
2 and CO have lower enthalpies of formation with magnitudes less than that of the full electride. This makes the formed electride-C12A7 a competing reduction agent and equilibrium will occur where the enthalpy of oxidation of electride-C12A7 is equal to the enthalpy of oxidation for C. At this point, the electride conversion process will cease and an equilibrium will be observed irrespective of further processing. Based on our structural observations, this coincides with one cage occupied and an electrical conductivity of ≈15 S·cm
−1; the exact ratio of O:C species and valence of these remaining species is unable to be determined in this study. The presence of the remaining C species leads to the high temperature stability at the electride-conversion equilibrium.
This model is supported by the observed structural characterization. However, future work is needed to fully identify the type and presence of occluded C species. Such a task is challenging due to the occluded position disorder, low occupation, and low Z of the anions of interest. The kinetic processes during reduction involve changes in local short-range order while retaining the long-range order of the framework. In situ diffraction techniques as a function of temperature, atmosphere, and time, probing both local and long-range order, could provide the experimental verification of the proposed reduction methods with higher resolution than the limited process data points able to be analyzed in ex situ techniques. Atomistic modelling can be employed by utilizing reactive force field potentials to determine thermodynamic properties and confirm the nano-reactor theory responsible for electride C12A7 formation.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}