1. Introduction
The computational models and methods used to obtain the thermodynamic and physical properties of systems with a certain number of particles have evolved so fast that it is feasible to analyze structures with hundreds of atoms in unitary cells, thus resulting in a more approachable task [
1,
2,
3]. However, the solution to these problems leads to new questions, challenges, and materials to study, as well as new applications of these methods and models to different areas such as medicine, chemistry, and materials science [
4,
5,
6,
7].
A typical example of applying these models and methods lies in the characterization of new compounds related to boronate esters, where the relation between the atoms and the peaks in magnetic resonance spectroscopy occasionally turns out to be difficult [
8,
9,
10]. In certain chemical reactions, the boronic acid group in some organic compounds sometimes causes the cleavage of the B–C bond, so it is necessary to protect the boronic acid group with a diol [
11]. The compounds with boronate esters represent valuable means in organic synthesis, particularly in the Suzuki–Miyaura coupling reaction [
12]. They are useful for carbohydrate detection [
13] due to their capacity to produce cyclic esters with suitable diols [
14]. They show biological activities, such as antidepressant, antiallergic, anesthetic, and anti-Alzheimer agents, as well as proteasome and lipogenic inhibitors [
15]. For this reason, when a new molecule appears, it is necessary to know its properties and possible uses.
Crystallographic and spectroscopic studies related to new compounds of boronate remain unexplored to a great extent, and only a few crystalline structures have been reported so far. The main goal of this work is to study the isotropic shielding,
H,
C,
B, NMR chemical shifts of the 1-(5-(4,5-dimethyl-1,3,2-dioxoborolan-2-yl)thiophen-2-yl)ethanone [
16] by applying density functional theory. The paper is arranged as follows: In
Section 2, the theoretical background is given. In
Section 3, the methodology is presented, whereas, in
Section 4, the results are presented and analyzed. Finally, the conclusion is given in
Section 5.
4. Results and Discussion
The structural parameters of the composite boronate were obtained by their geometrical optimization, as shown in
Table 2,
Table 3,
Table 4 and
Table 5, in a representative manner. In all Tables, we show the results using the three different used DFT techniques.
The carbon–hydrogen bond lengths in the
compound exhibit a slight variation after optimization (
Table 2). This is due to the higher freedom of motion of the hydrogen atoms, which can be compared with the carbon–carbon bond lengths (
Table 3), where the variation is smaller since the carbon atoms have stronger bonds.
Table 4 depicts the bond lengths and optimized lengths in the
compound. The variation, although not significant, is acceptable for the calculations of the reference isotropic shielding;
ppm for
C and
ppm for hydrogen (see
Table 6). The calculated values are in agreement with those reported in the literature.
Table 5 shows the bond lengths and optimized lengths in the
compound and
Table 6 shows the isotropic shielding;
ppm (reference value isotropic shielding) for
B.
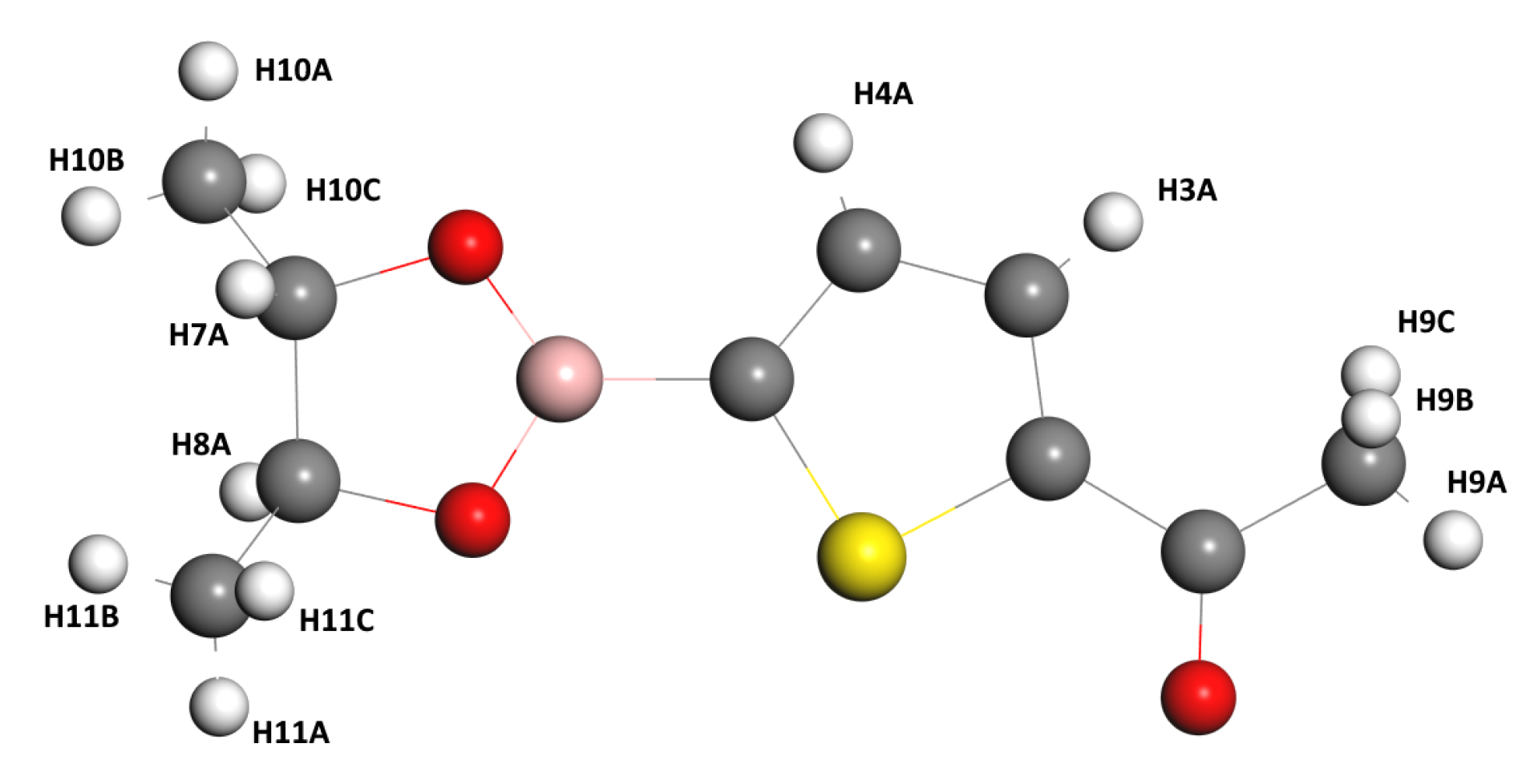
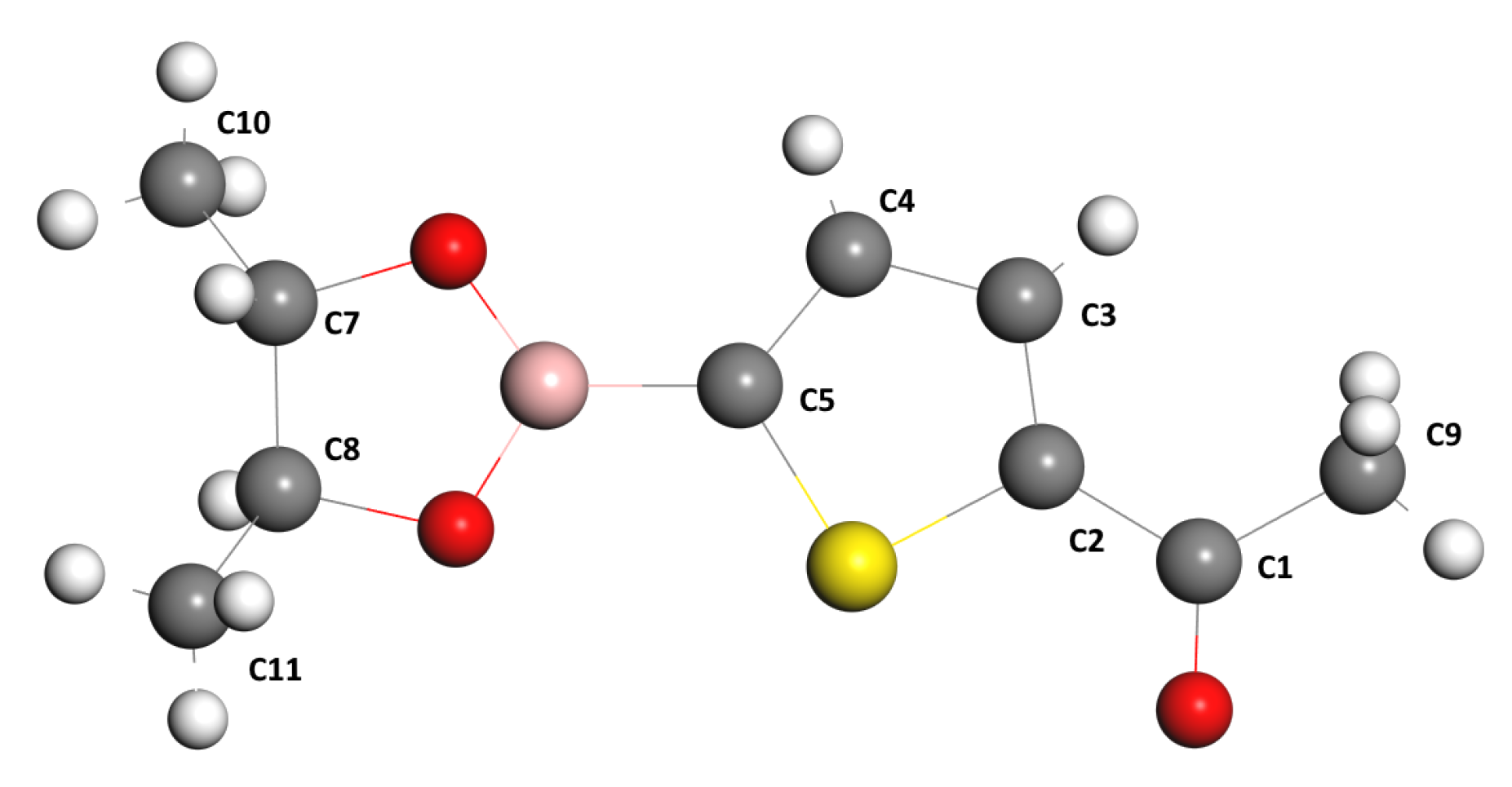
Before analyzing the results of the theoretical calculations on NMR, let us comment that all the labels of the Hydrogen atoms are referred to in
Figure 3.
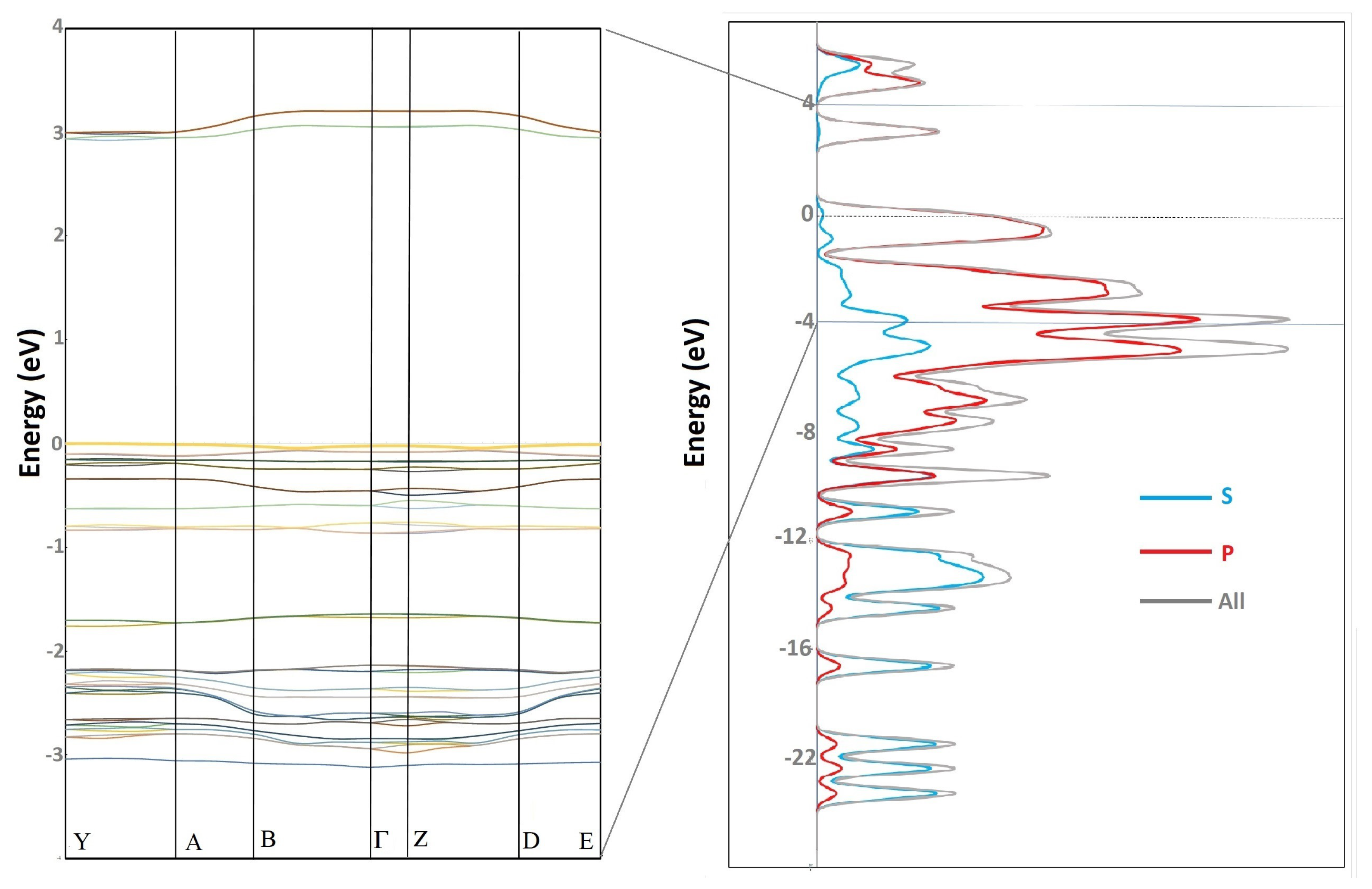
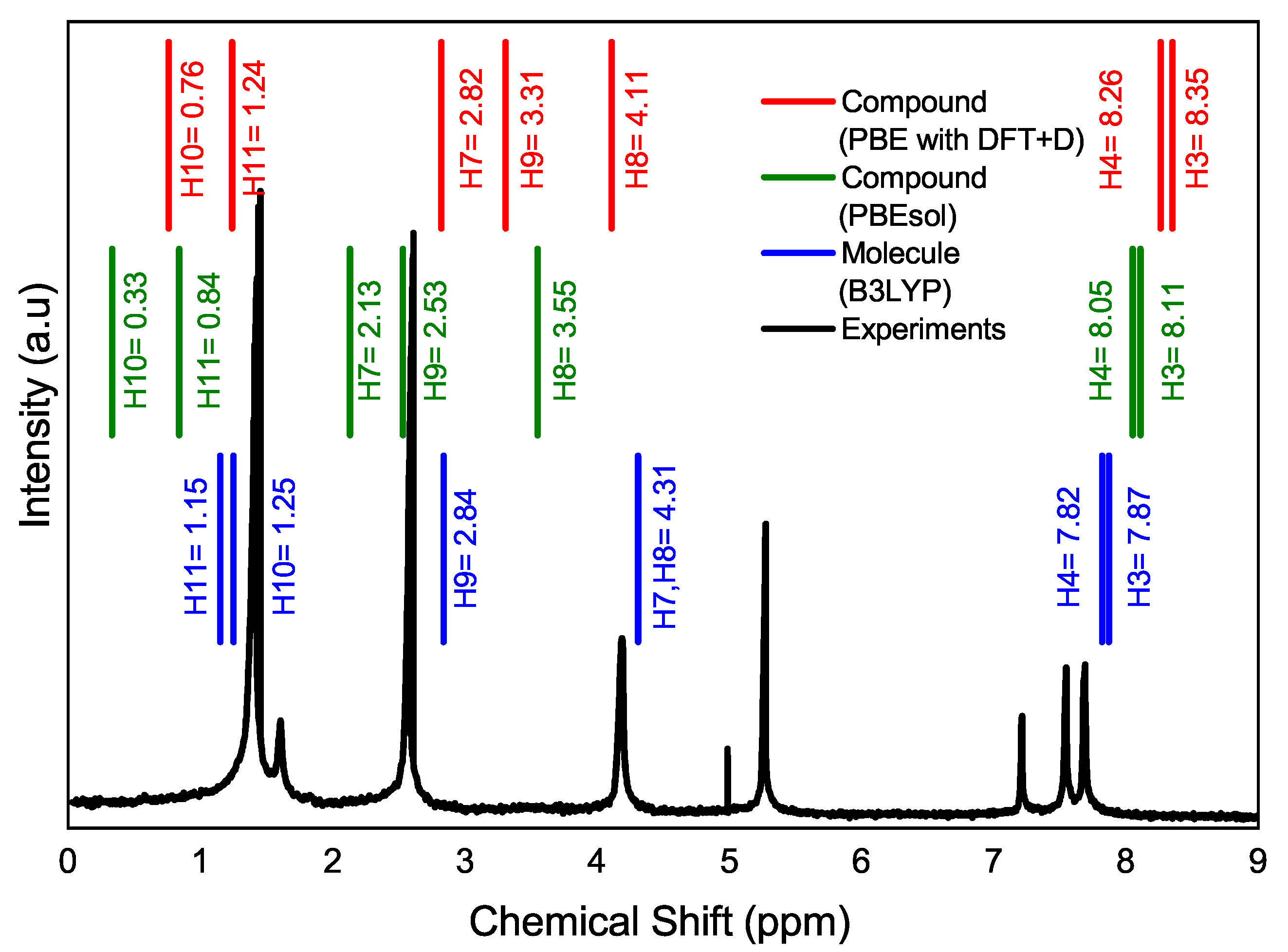
Figure 4 illustrates the magnetic resonance spectrum of the
compound with experimental data and theoretical results. Although the model in CASTEP is applied to a solid (red and green lines), the experimental resonance spectrum is from a solution of the compound since the compound is dissolved in chloroform. In fact, the difference can be noticed in the Gaussian calculations (blue line), where the atoms of H7A and H8A have the same value, and the calculations made with CASTEP (on solids, periodic lattice
Figure 2b) where the values H7A and H8A differ slightly from the experimental spectrum. It is also shown how the H3A and H4A hydrogen atoms of the thiophene ring exhibit less shielding, which means that the electrons are shifted, and their nuclei are less protected due to the inductive effect of electron-withdrawing groups (acetyl and boronic acid moieties) and the electron delocalization of the
electrons [
41]. It implies that they appear in the magnetic resonance spectrum at a higher field, followed by the dioxaborolane H7A and H8A hydrogen atoms, and at a lower field than the H3A and H4A of the thiophene ring.
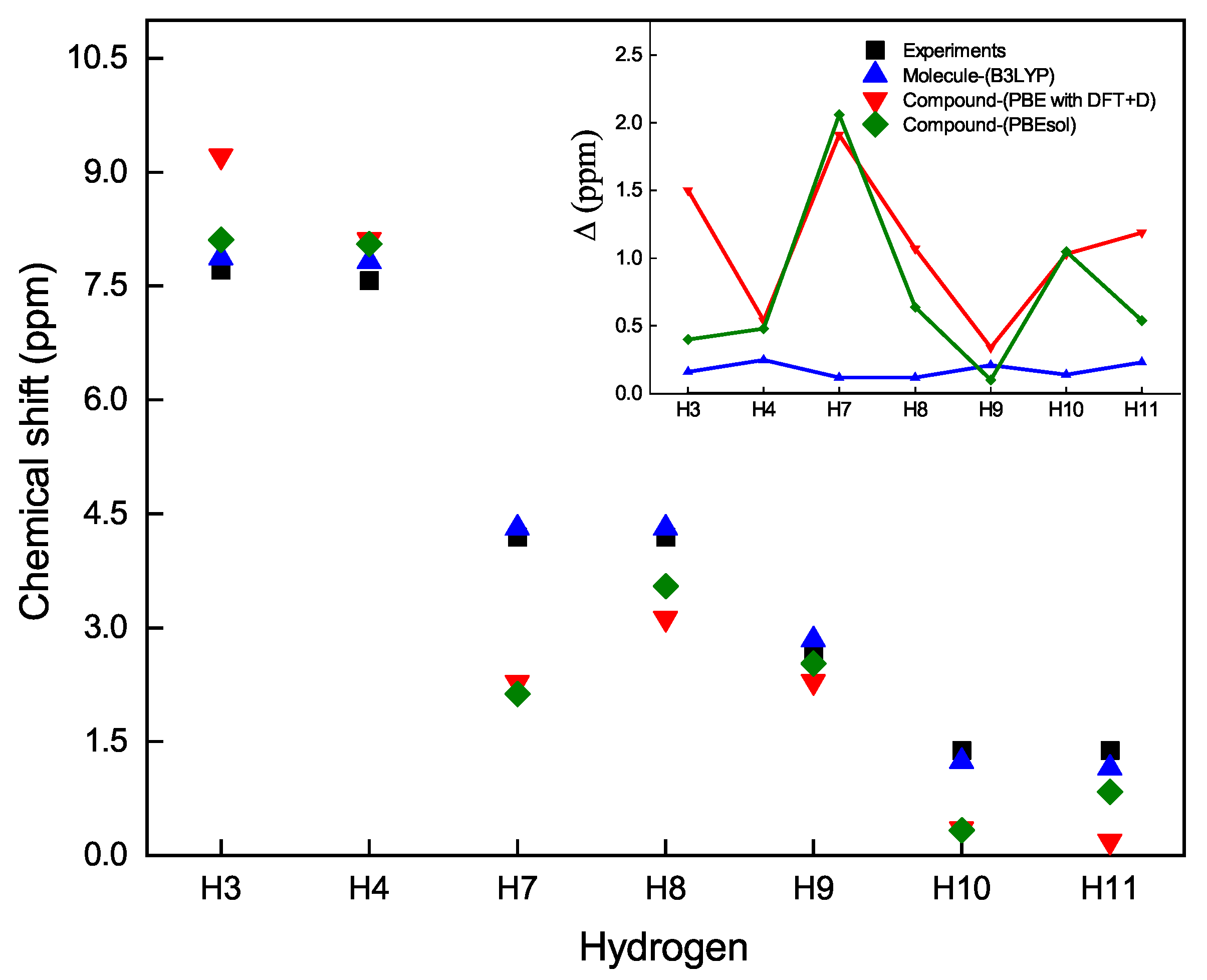
An appropriate approach to show the chemical shifts of the hydrogen atoms in the
compound is provided in
Figure 5. In this figure, one can observe how far or close the theoretical results from the experimental data are. The inset of
Figure 5 shows the deviation of the theoretical values from the experimental of the chemical shifts,
. In general, we observe that the computational model that best approximates the experimental calculations is that of the Gaussian software on the boronate ester molecule in solution.
On the other hand, we remark that the number of wave vectors k plays a crucial role in the calculation of nuclear magnetic resonance. For even greater accuracy, we should increase the number of wave vectors k, which would facilitate a better reading of the theoretical data with respect to the experimental data.
Now, let us analyze the chemical shift as a function of the carbon atoms from the NMR spectrum. For this reason, let us label the carbon atoms in
Figure 6.
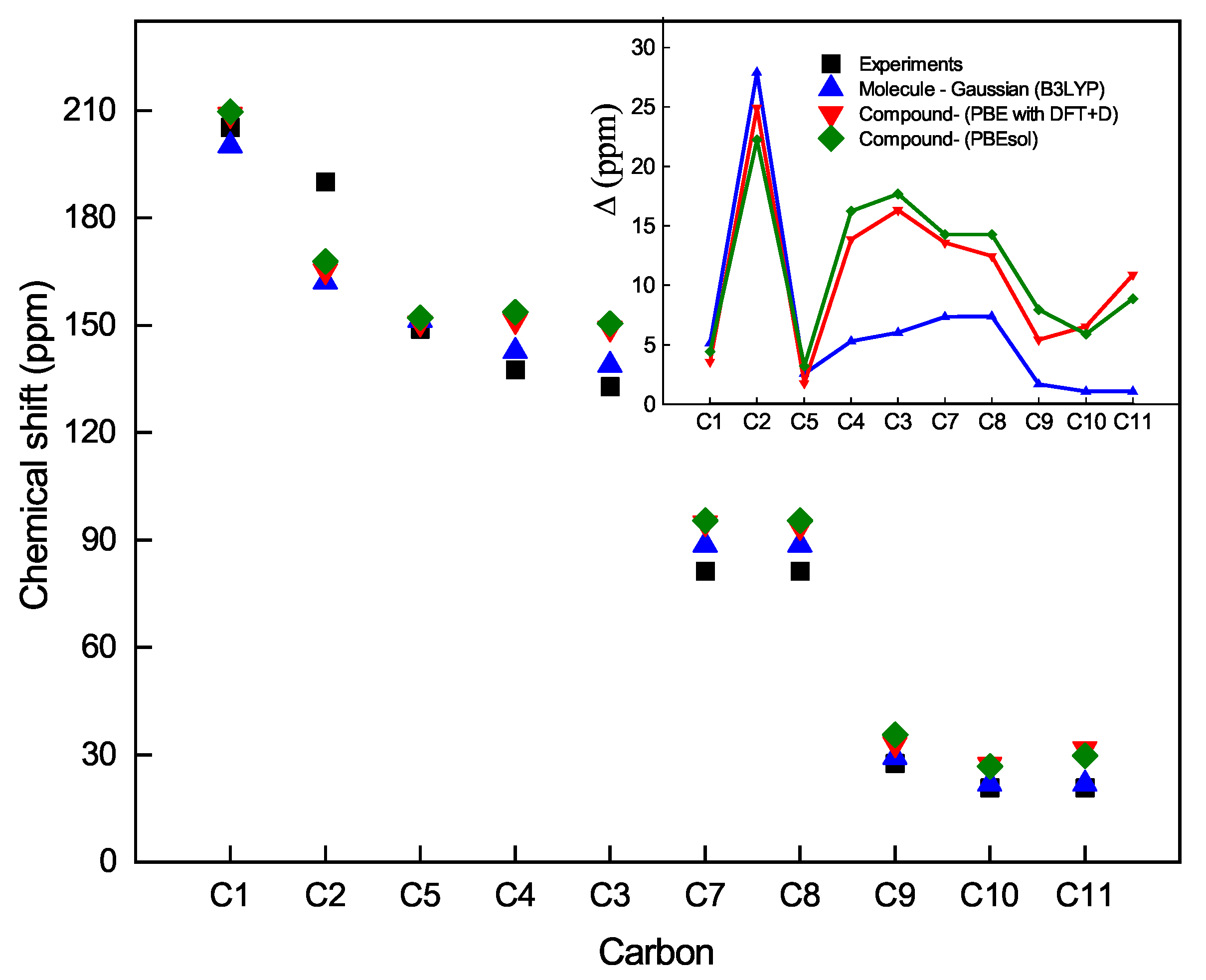
Figure 7 shows the chemical shift as a function of the carbon atoms for both theories with the three methods mentioned above and experimental data. We observe that the carbon atom C1 is less shielded. Its electrons are shifted due to the inductive effect of the oxygen O1 atom, and accordingly, the nucleus of C1 is less protected, followed by the C2-5 carbon atoms of the thiophen ring at a higher field. The C7-8 atoms and the C9 atom of the acetyl moiety are more protected than the C1 atom or less exposed to the inductive effect of oxygen atom O1. If we compare the bond lengths C7-O2 (1.465 Å, optimized length) and C8-O3 (1.461 Å, optimized length) with the double bond length C1-O1 (1.246 Å, optimized length), it can be appreciated that the C1 atom of the acetyl moiety is less shielded and, therefore, its chemical shift appears at a lower field than the C9 atom of the methyl moiety. Finally, the C10-11 atoms of the methyl groups are more shielded, and consequently, their chemical shifts appear at a higher field. Moreover, the inset of
Figure 7 shows the comparison between the theoretical and experimental chemical shifts that can be observed for the carbon atoms of the
compound. Similarly, it can be seen that the same trend of the spectrum of the hydrogen atom is repeated, where the model that best approximates the experimental calculations is the Gaussian 09 on the boronate ester molecule in solution, which the CASTEP on the solid structure of the boronate ester.
5. Conclusions
Computational studies of the compound, such as NMR, electronic band structure, and density of state, have yet to be commonly done. In this work, we propose the characterization by applying the GIPAW method in CASTEP, which works on a periodic lattice structure, and the B3LYP method with the Gaussian, which works on molecules. A brief band structure study was carried out in order to visualize its electronic distribution, its density of state, and finally, the NMR study.
The calculations of the reference shielding are compared with the data reported in the literature, obtaining a good approximation. This data can be used as a reference for the chemical shifts in the boronate esters. The 1-(5-(4,5-dimetil-1,3,2-dioxoborolan-2iltiofen-2-il) etanona was obtained from the reaction of the 5-acetyl-2-thienylboronic acid with 2,3-butanediol, where the results of their experimental resonances are also in agreement with the theoretical approach. Consequently, the chemical shifts of the H hydrogen atoms, the C carbon atoms and the B boron atoms, as well as, the the infrared spectroscopy.
Our study is expected to help guide future research, identifying conditions and methods that more accurately account for the properties of different molecules of interest to materials science.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}