

A DFT Computational Study of Type-I Clathrates A8Sn46−x (A = Cs or NH4, x = 0 or 2)


Abstract
:
1. Introduction
2. Materials and Methods
2.1. DFT Calculations
2.2. Electronic Properties Calculation Method
2.3. Electron Localization Function Method
2.4. Phonon Calculations Method
3. Results
3.1. Structural Optimization and Calculation of Formation Enthalpies
3.2. Electronic Properties
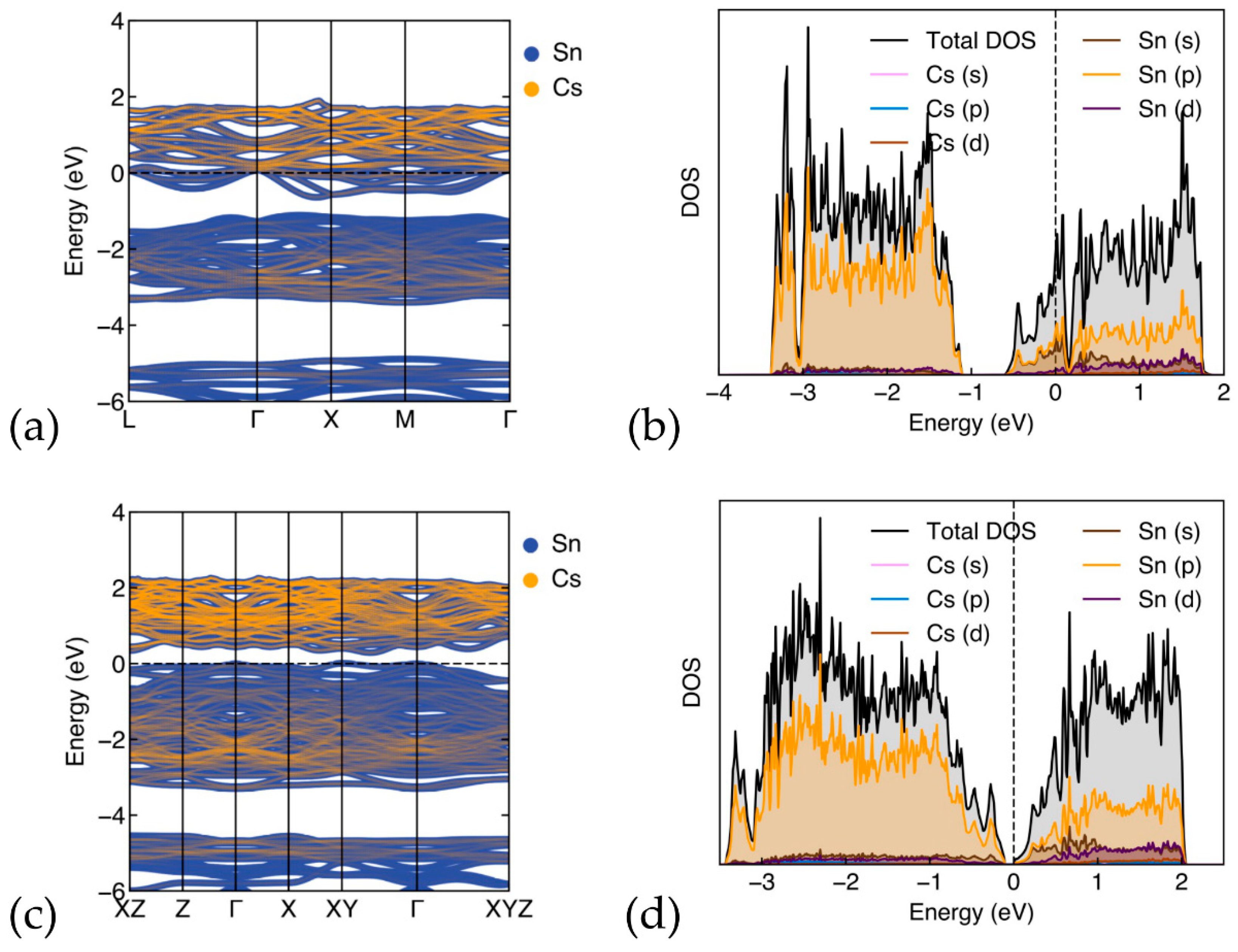
3.2.1. Band-Structure and Density of States for Cs8Sn46−x
3.2.2. Band-Structure and Density of States for (ΝH4)8Sn46−x
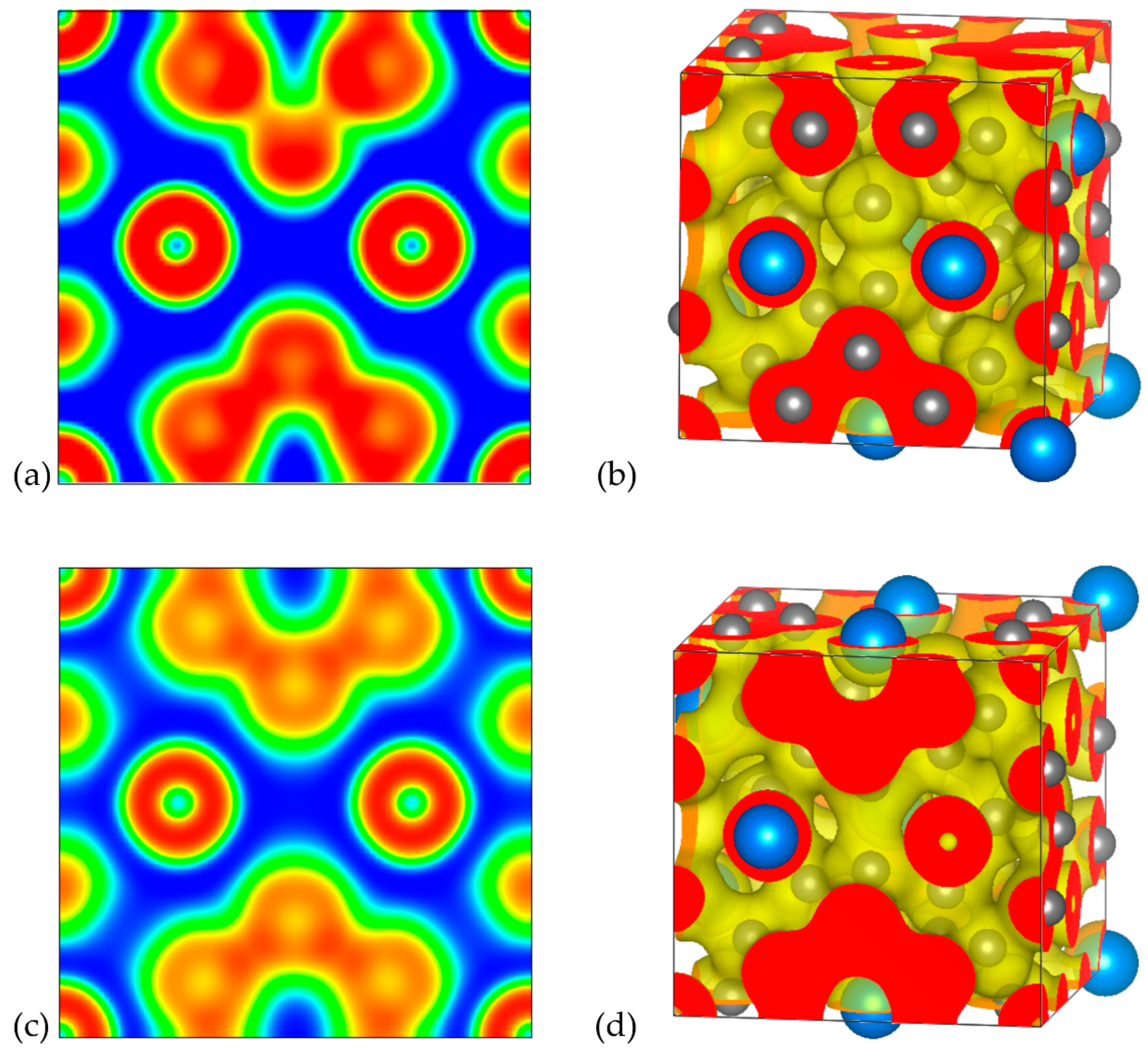
3.2.3. Electron Localization Function
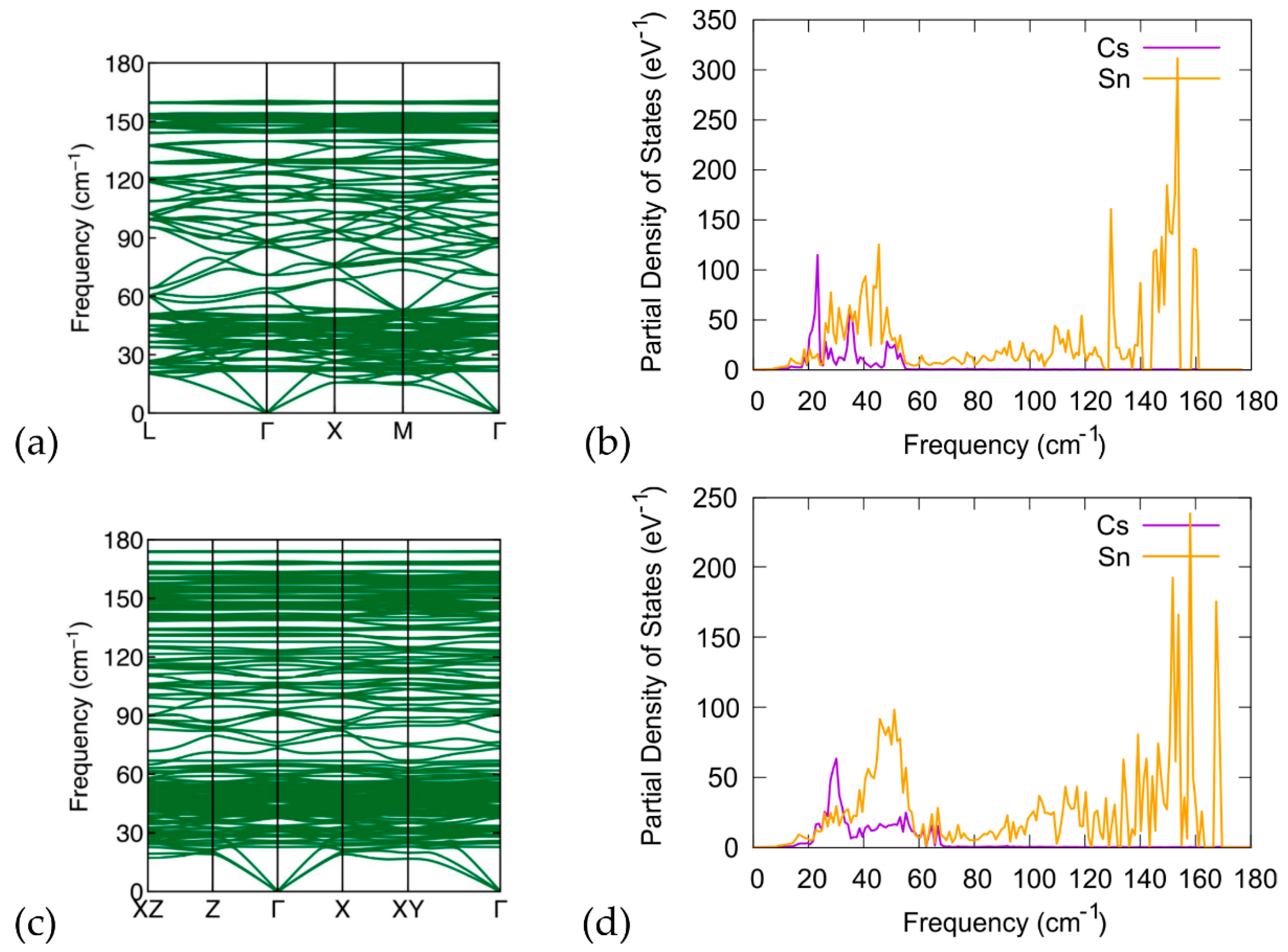
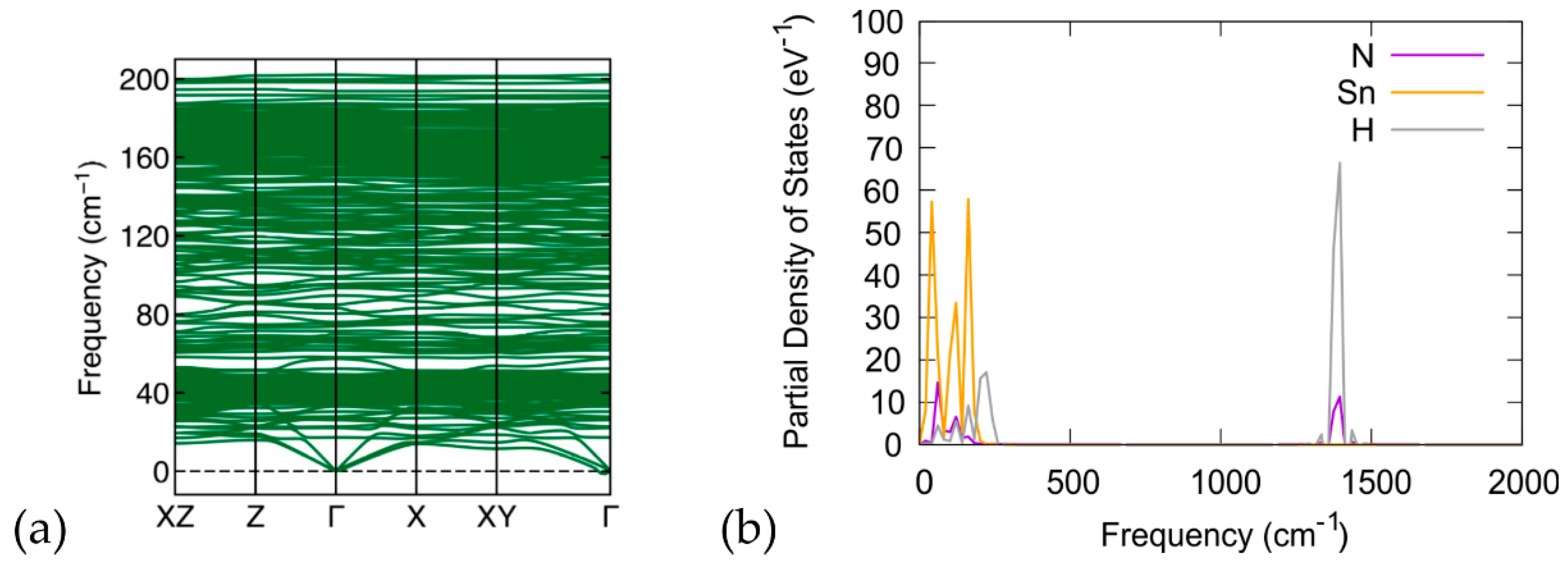
3.3. Phonon Calculations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rogl, P. Chapter 32. Formation and Crystal Chemistry of Clathrates. In Thermoelectrics Handbook Macro to Nano; CRC Taylor & Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Konvir, K.; Shevelkov, A. Semiconducting clathrates: Synthesis, structure and properties. Russ. Chem. Rev. 2004, 73, 923–938. [Google Scholar] [CrossRef]
- Guloy, A.M.; Ramlau, R.; Tang, Z.; Schnelle, W.; Baitinger, M.; Grin, Y. A guest-free germanium clathrate. Nature 2006, 443, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Freer, R.; Ekren, D.; Ghosh, T.; Biswas, K.; Qiu, P.; Wan, S.; Chen, L.; Han, S.; Fu, C.; Zhu, T.; et al. Key properties of inorganic thermoelectric materials—Tables (version 1). J. Phys. Energy 2022, 4, 02200. [Google Scholar] [CrossRef]
- Corbett, J. Polyatomic Zintl Anions of the Post-Transition Elements. Chem. Rev. 1985, 85, 383–397. [Google Scholar] [CrossRef]
- Baitinger, M.; Böhme, B.; Wagner, F.R.; Schwarz, U. Zintl Defects in Intermetallic Clathrates. Z. Anorg. Allg. Chem. 2020, 646, 1034–1041. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Carbogno, C.; Böhme, B.; Baitinger, M.; Grin, Y.; Scheffler, M. Formation of Vacancies in Si- and Ge-based Clathrates: Role of Electron Localization and Symmetry Breaking. Phys. Rev. Lett. 2017, 118, 236401. [Google Scholar] [CrossRef]
- Kauzlarich, S.M.; Brown, S.R.; Snyder, G.J. Zintl phases for thermoelectric devices. Dalton Trans. 2007, 2099–2107. [Google Scholar] [CrossRef]
- Yang, L.; Chen, Z.-G.; Dargusch, M.S.; Zou, J. High Performance Thermoelectric Materials: Progress and Their Applications. Adv. Energy Mater. 2017, 8, 1701797. [Google Scholar] [CrossRef]
- Slack, G. Chapter 34. New Materials and Performance Limits for Thermoelectric Cooling. In Handbook of Thermoelectrics; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Iversen, B.; Palmqvist, A.E.C.; Cox, D.E.; Nolas, G.S.; Stucky, G.D.; Blake, N.P.; Metiu, H. Why are Clathrates Good Candidates for Thermoelectric Materials? J. Solid State Chem. 2000, 149, 455–458. [Google Scholar] [CrossRef]
- Myles, C.; Dong, J.; Sankey, O. Rattling guest atoms in Si, Ge, and Sn-based type-II clathrate materials. Phys. Status Solidi B 2003, 239, 26–34. [Google Scholar] [CrossRef]
- Wu, J.; Xu, J.; Tanigaki, K. Universal van der Waals-type interactions in rattler containing cage materials. arXiv 2018, arXiv:1802.09228. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, W.; Gao, W.; Mori, T. A material catalogue with glass-like thermal conductivity mediated by crystallographic occupancy for thermoelectric application. Energy Environ. Sci. 2021, 14, 3579–3587. [Google Scholar] [CrossRef]
- Laurita, G.; Fabini, D.H.; Stoumpos, C.C.; Kanatzidis, M.G.; Seshadri, R. Chemical tuning of dynamic cation off-centering in the cubic phases of hybrid tin and lead halide perovskites. Chem. Sci. 2017, 8, 5628–5635. [Google Scholar] [CrossRef] [PubMed]
- von Schnering, H.G.; Kröner, R.; Baitinger, M.; Peters, K.; Nesper, R.; Grin, Y. Crystal structure of the defect clathrate Cs8Sn44. Z. Kristallogr. NCS 2000, 215, 205–206. [Google Scholar]
- Miller, G.J. Chapter 1. Structure and Bonding at the Zintl Border. In Chemistry, Structure and Bonding of Zintl Phases and Ions; VCH Publishers, Inc.: Hoboken, NJ, USA, 1996. [Google Scholar]
- Myles, C.; Dong, J.; Sankey, O. Structural and electronic properties of tin clathrate materials. Phys. Rev. B 2001, 64, 165202. [Google Scholar] [CrossRef]
- Dubois, F.; Fässler, T. Ordering of Vacancies in Type-I Tin Clathrate: Superstructure of Rb8Sn44□2. J. Am. Chem. Soc. 2005, 127, 3264. [Google Scholar] [CrossRef]
- Kaltzoglou, A.; Hoffmann, S.; Fässler, T. Order-disorder phase transition in type-I clathrate Cs8Sn44. Eur. J. Inorg. Chem. 2007, 2007, 4162–4167. [Google Scholar] [CrossRef]
- Kaltzoglou, A.; Fässler, T.; Christensen, M.; Johnsen, S.; Iversen, B.; Presniakov, I.; Sobolev, A.; Shevelkov, A. Effects of the order-disorder phase transition on the physical properties of A8Sn44 (A = Rb, Cs). J. Mater. Chem. 2008, 18, 5630–5637. [Google Scholar] [CrossRef]
- Christensen, S.; Bjerg, L.; Kaltzoglou, A.; Juranyi, F.; Fässler, T.; Unruh, T.; Christensen, M. Guest host interaction and low energy host structure dynamics in tin clathrates. J. Appl. Phys. 2013, 113, 084902. [Google Scholar] [CrossRef]
- Candolfi, C.; Koza, M.M.; Aydemir, U.; Carrillo-Cabrera, W.; Grin, Y.; Steglich, F.; Baitinger, M. Vibrational dynamics of the type-I clathrates A8Sn44 (A = Cs, Rb, K) from lattice-dynamics calculations, inelastic neutron scattering, and specific heat measurements. J. Appl. Phys. 2020, 127, 145104. [Google Scholar] [CrossRef]
- Kieslich, G.; Sun, S.; Cheetham, A.K. Solid-state principles applied to organic–inorganic perovskites: New tricks for an old dog. Chem. Sci. 2014, 5, 4712–4715. [Google Scholar] [CrossRef]
- Sidey, V. On the effective ionic radii for ammonium. Acta Crystallogr. 2016, B72, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Vainshtein, B.K. Refinement of the Structure of the Group NH4 in the Structure of Ammonium Chloride. Tr. Instituta Krist. Akad. Nauk. SSSR 1956, 12, 18–24. [Google Scholar]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Kresse, I.G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Ganose, A.M.; Jackson, A.J.; Scanlon, D.O. sumo: Command-line tools for plotting and analysis of periodic ab initio calculations. J. Open Source Softw. 2018, 3, 717. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Fuentealba, P.; Chamorro, E.; Santos, J.C. Chapter 5. Understanding and using the electron localization function. Theor. Asp. Chem. React. 2007, 19, 57–85. [Google Scholar] [CrossRef]
- Burdett, J.K.; McCormick, T.A. Electron Localization in Molecules and Solids: The Meaning of ELF. J. Phys. Chem. A 1998, 102, 6366–6372. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The Electron Localization Function. Angew. Chem. Int. Ed. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Togo, A.; Chaput, L.; Tadano, T.; Tanaka, I. Implementation strategies in phonopy and phono3py. J. Phys. Condens. Matter 2023, 35, 353001. [Google Scholar] [CrossRef]
- Togo, A. First-principles Phonon Calculations with Phonopy and Phono3py. J. Phys. Soc. Jpn. 2023, 92, 012001. [Google Scholar] [CrossRef]
- Shimizu, H.; Imai, T.; Kume, T.; Sasaki, S.; Kaltzoglou, A.; Fässler, T. Raman spectroscopy study of type-I clathrates A8Sn44 (A = Rb, Cs) and Rb8Hg4Sn42. Chem. Phys. Lett. 2008, 464, 54–57. [Google Scholar] [CrossRef]
- Dopilka, A.; Peng, X.; Chan, C.K. Ab Initio Investigation of Li and Na Migration in Guest-Free, Type I Clathrates. J. Phys. Chem. C 2019, 123, 22812–22822. [Google Scholar] [CrossRef]
- Alvarez, S. Polyhedra in (inorganic) chemistry. Dalton Trans. 2005, 2209–2233. [Google Scholar] [CrossRef]
- Egbele, P.O.; Shoko, E.; Joubert, D.P. Structural and thermoelectric properties of the type-I Sn clathrates Cs8Sn46-n(n=0,2) from Density Functional Theory (DFT). MRS Adv. 2018, 3, 1031–1038. [Google Scholar] [CrossRef]
- Nolas, G.S.; Chakoumakos, B.C.; Mahieu, B.; Long, G.J.; Weakley, T.J.R. Structural Characterization and Thermal Conductivity of Type-I Tin Clathrates. Chem. Mater. 2000, 12, 1947–1953. [Google Scholar] [CrossRef]
- Tse, J.S.; Li, Z.; Uehara, K. Phonon band structures and resonant scattering in Na8Si46 and Cs8Sn44 clathrates. Europhys. Lett. 2001, 56, 261. [Google Scholar] [CrossRef]
- Lu, M.; Tian, F.; Zhou, Q.; Cui, T. First principle studies of ammonium chloride under high pressure. RSC Adv. 2021, 11, 5149–5155. [Google Scholar] [CrossRef] [PubMed]
- Neiner, D.; Okamoto, N.L.; Condron, C.L.; Ramasse, Q.M.; Yu, P.; Browning, N.D.; Kauzlarich, S.M. Hydrogen Encapsulation in a Silicon Clathrate Type I Structure: Na5.5(H2)2.15Si46: Synthesis and Characterization. J. Am. Chem. Soc. 2007, 129, 13857–13862. [Google Scholar] [CrossRef]
- Karttunen, A.J.; Fassler, T.F. Semiconducting Clathrates Meet Gas Hydrates: Xe24[Sn136]. Chem. Eur. J. 2014, 20, 6693–6698. [Google Scholar] [CrossRef]
- Shin, K.; Kumar, R.; Udachin, K.A.; Alavi, S.; Ripmeester, J.A. Ammonia clathrate hydrates as new solid phases for Titan, Enceladus, and other planetary systems. Proc. Natl. Acad. Sci. USA 2012, 109, 14785–14790. [Google Scholar] [CrossRef]
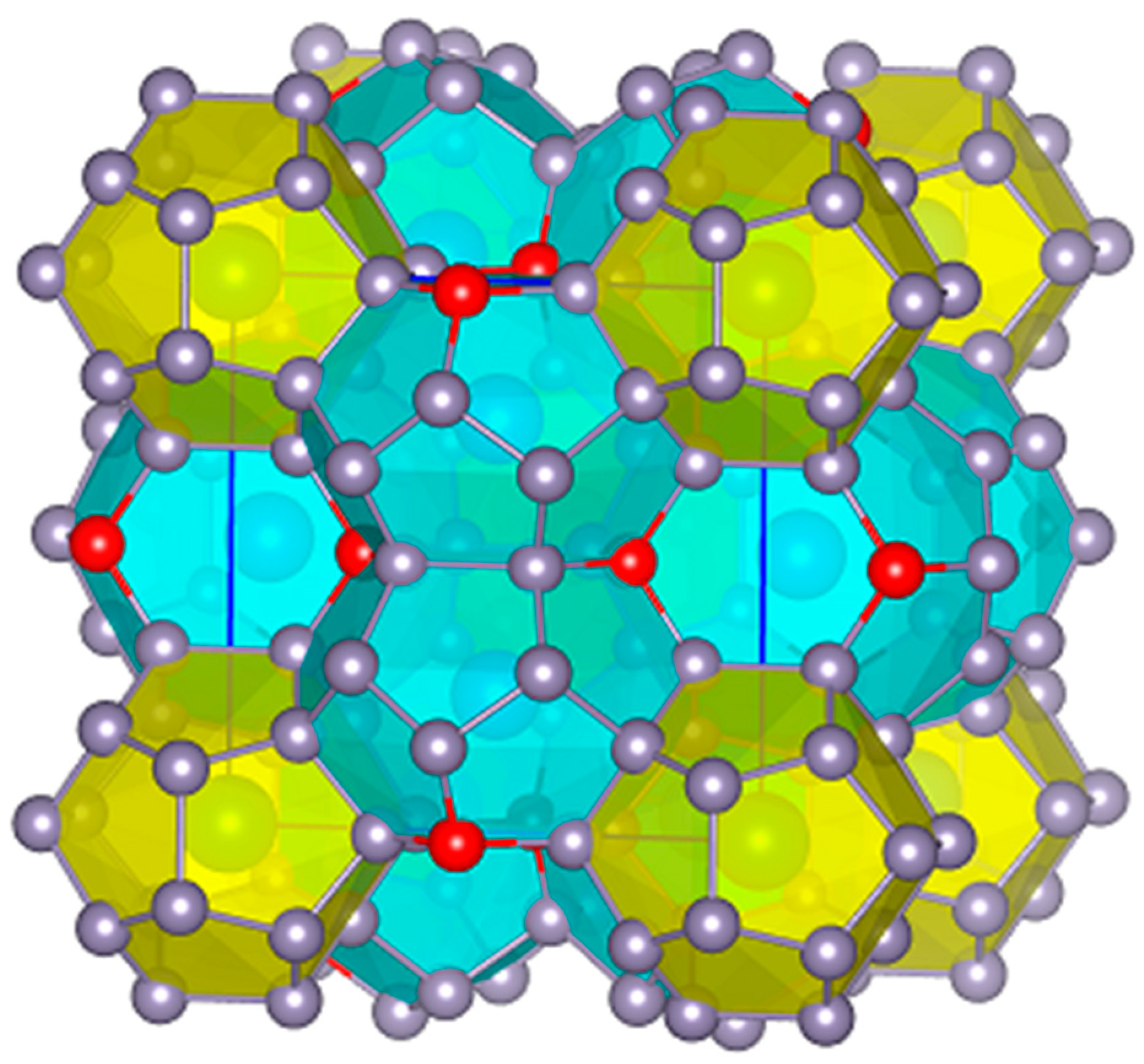

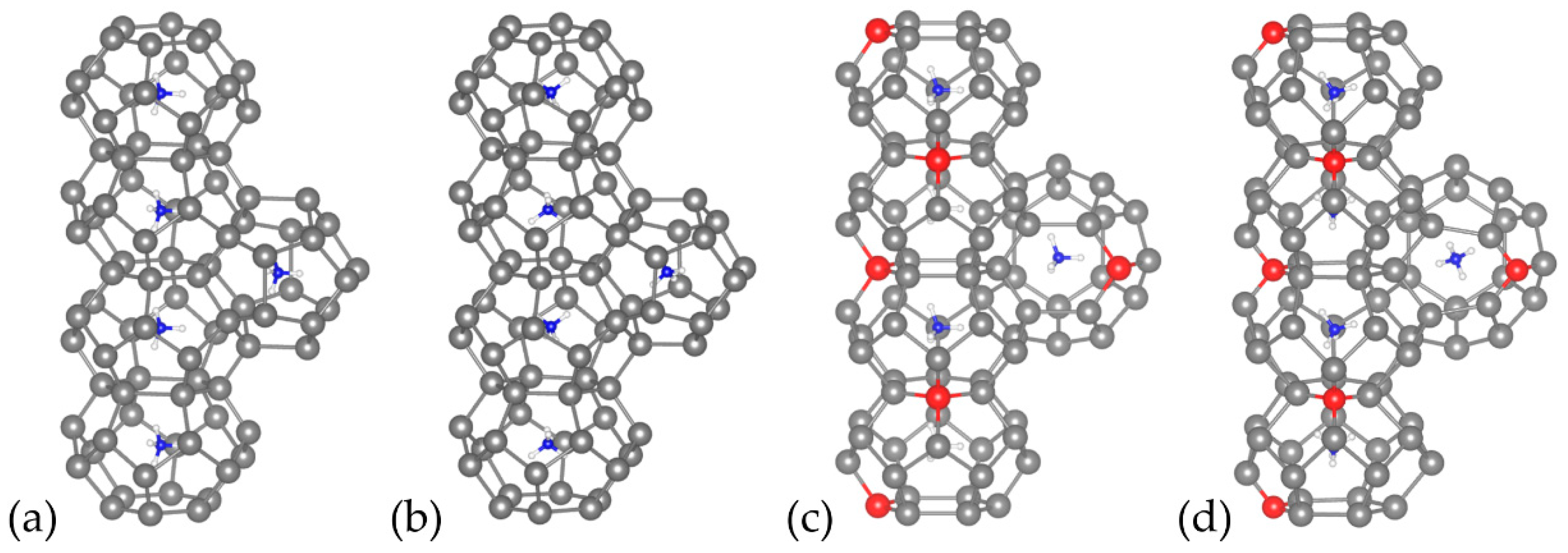



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Closest Distance between Vacancies (Å) | a (Å) | b (Å) | c (Å) | Volume (Å3) | Vacancy Eformation (eV) | Compound Eformation (eV) |
|---|---|---|---|---|---|---|---|
| Cs8Sn46 | − | 12.497 | 12.497 | 12.497 | 1951.75 | − | −8.948 |
| Cs8Sn44 (model A) | 8.154 | 12.100 | 12.135 | 12.137 | 1782.13 | −0.411 | −9.359 |
| Cs8Sn44 (model B) | 6.165 | 12.176 | 12.101 | 12.122 | 1785.98 | 0.125 | −8.832 |
| Cs8Sn44 (model C) | 7.407 | 12.157 | 12.069 | 12.157 | 1783.71 | −0.555 | −9.503 |
| (NH4)8Sn46 | − | 12.384 | 12.406 | 12.406 | 1906.062 | − | −6.689 |
| (NH4)8Sn44 (model A) | 8.154 | 12.016 | 12.044 | 12.051 | 1743.993 | −0.5063 | −7.195 |
| (NH4)8Sn44 (model B) | 6.165 | 12.001 | 11.982 | 12.255 | 1760.361 | 0.3164 | −6.372 |
| (NH4)8Sn44 (model C) | 7.407 | 12.083 | 11.957 | 12.075 | 1744.555 | −0.9322 | −7.621 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kelaidis, N.; Klontzas, E.; Kaltzoglou, A. A DFT Computational Study of Type-I Clathrates A8Sn46−x (A = Cs or NH4, x = 0 or 2). Materials 2024, 17, 4595. https://doi.org/10.3390/ma17184595
Kelaidis N, Klontzas E, Kaltzoglou A. A DFT Computational Study of Type-I Clathrates A8Sn46−x (A = Cs or NH4, x = 0 or 2). Materials. 2024; 17(18):4595. https://doi.org/10.3390/ma17184595
Chicago/Turabian StyleKelaidis, Nikolaos, Emmanuel Klontzas, and Andreas Kaltzoglou. 2024. "A DFT Computational Study of Type-I Clathrates A8Sn46−x (A = Cs or NH4, x = 0 or 2)" Materials 17, no. 18: 4595. https://doi.org/10.3390/ma17184595