
Adsorption and Catalytic Reduction of Nitrogen Oxides (NO, N2O) on Disulfide Cluster Complexes of Cobalt and Iron—A Density Functional Study



Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Binding of NO to the Carbonyl Complexes Fe2S2(CO)6 and Co2S2(CO)6
3.2. Reduction of NO in the Form of Its Dimer, (NO)2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Leclercq, B.; Kluza, J.; Antherieu, S.; Sotty, J.; Alleman, L.Y.; Perdrix, E.; Loyens, A.; Coddeville, P.; Lo Guidice, J.-M.; Marchetti, P.; et al. Air pollution-derived PM2.5 impairs mitochondrial function in healthy and chronic obstructive pulmonary diseased human bronchial epithelial cells. Environ. Pollut. 2018, 243, 1434–1449. Available online: http://refhub.elsevier.com/S0169-4332(21)01770-0/h0010 (accessed on 24 August 2024). [CrossRef]
- Anenberg, S.C.; Miller, J.; Minjares, R.; Du, L.I.; Henze, D.K.; Lacey, F.; Malley, C.S.; Emberson, L.; Franco, V.; Klimont, Z.; et al. Impacts and mitigation of excess diesel-related NOx emissions in 11 major vehicle markets. Nature 2017, 545, 467–471. Available online: http://refhub.elsevier.com/S0169-4332(21)01770-0/h0005 (accessed on 24 August 2024). [CrossRef]
- Bourassa, J.; DeGraff, W.; Kudo, S.; Wink, D.A.; Mitchell, J.B.; Ford, P.C. Photochemistry of Roussin’s Red Salt, Na2[Fe2S2(NO)4], and of Roussin’s Black Salt, NH4[Fe4S3(NO)7]. In Situ Nitric Oxide Generation to Sensitize γ-Radiation Induced Cell Death. J. Am. Chem. Soc. 1997, 119, 2853–2860. [Google Scholar] [CrossRef]
- Harrop, T.C.; Tonzetich, Z.J.; Reisner, E.; Lippard, S.J. Reactions of Synthetic [2Fe-2S] and [4Fe-4S] Clusters with Nitric Oxide and Nitrosothiols. J. Am. Chem. Soc. 2008, 130, 15602–15610. [Google Scholar] [CrossRef] [PubMed]
- Bitterwolf, T.E.; Pal, P. Synthesis of the cobalt analogues of Roussin’s red salt esters Inorg. Chim. Acta 2006, 359, 1501–1503. [Google Scholar] [CrossRef]
- Hayton, T.W.; Legzdins, P.; Sharp, W.B. Coordination and Organometallic Chemistry of Metal-NO Complexes. Chem. Rev. 2002, 102, 935–991. [Google Scholar] [CrossRef]
- Tard, C.; Pickett, C.J. Structural and Functional Analogues of the Active Sites of the [Fe]-, [NiFe]-, and [FeFe]-Hydrogenases. Chem. Rev. 2009, 109, 2245. [Google Scholar] [CrossRef] [PubMed]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A.; et al. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621. [Google Scholar] [CrossRef]
- Nann, T.; Ibrahim, S.K.; Woi, P.-M.; Xu, S.; Ziegler, J.; Pickett, C.J. Water Splitting by Visible Light: A Nanophotocathode for Hydrogen Production. Angew. Chem. Int. Ed. 2010, 49, 1574. [Google Scholar] [CrossRef]
- Harcourt, R.D. The origin of the long N-N bond in N2O2: An ab initio valence bond study. J. Mol. Str.: THEOCHEM 1990, 206, 253–264. [Google Scholar] [CrossRef]
- Dkhissi, A.; Soulard, P.; Perrin, A.; Lacome, N. “The NO Dimer”. J. Mol. Spectrosc. 1997, 183, 12–17. [Google Scholar] [CrossRef]
- East, A.L.L. The 16 valence electronic states of nitric oxide dimer. J. Chem. Phys. 1998, 109, 2185–2193. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Janebi, H.; Mousavian, P. Single Al atom anchored on defective MoS2: An efficient catalytic site for reduction of greenhouse N2O gas by CO or C2H4 molecules. Appl. Surf. Sci. 2021, 569, 151001. [Google Scholar] [CrossRef]
- Matsushima, T.; Kokalj, A. N2 emission in steady-state N2O + CO and NO + CO reactions on Ir(110) by means of angle-resolved desorption. Appl. Surf. Sci. 2017, 414, 153–162. [Google Scholar] [CrossRef]
- Farhan, S.M.; Pan, W.; Zhijian, C.; JianJun, Y. Innovative catalysts for the selective catalytic reduction of NOx with H2: A systematic review. Fuel 2024, 355, 129364. [Google Scholar] [CrossRef]
- Kocía, K.; Relia, M.; Troppováa, I.; Sihor, M.; Kupkovác, J.; Kustrowski, P.; Prausa, P. Photocatalytic decomposition of N2O over TiO2/g-C3N4 photocatalysts heterojunction. Appl. Surf. Sci. 2017, 396, 1685–1695. [Google Scholar] [CrossRef]
- Ryder, J.A.; Chakraborty, A.K.; Bell, A.T. Density Functional Theory Study of Nitrous Oxide Decomposition over Fe- and Co-ZSM-5. J. Phys. Chem. B 2002, 106, 7059–7064. [Google Scholar] [CrossRef]
- Kuo, S.C.; Zhang, Z.Y.; Ross, S.K.; Klemm, R.B.; Johnson, R.D.; Monks, P.S.; Thorn, R.P.; Stief, L.J. Discharge flow-photoionization mass spectrometric study of HNO: Phtoionization efficiency spectrum and ionization energy and proton affinity of NO. J. Phys. Chem. A 1997, 101, 4035. [Google Scholar] [CrossRef]
- Hunter, E.P.; Lias, S.G. Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update. J. Phys. Chem. Ref. Data 1998, 27, 413–656. [Google Scholar] [CrossRef]
- Uzunova, E.L.; Mikosch, H. Electronic, magnetic structure and water splitting reactivity of the iron-sulfur dimers and their hexacarbonyl complexes: A density functional study. J. Chem. Phys. 2014, 141, 044307. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J. Chem. Phys. 1996, 104, 1040. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200. [Google Scholar] [CrossRef]
- Wachters, A.J.H. Gaussian basis set for molecular wavefunctions containing third-row atoms. J. Chem. Phys. 1970, 52, 1033. [Google Scholar] [CrossRef]
- Hay, P.J. Gaussian basis sets for molecular calculations. The representation of 3d orbitals in transition-metal atoms. J. Chem. Phys. 1977, 66, 4377. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W. Highly correlated systems. Excitation energies of first row transition metals Sc–Cu. J. Chem. Phys. 1989, 91, 1062. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270. [Google Scholar] [CrossRef]
- Halgren, T.A.; Lipscomb, W.N. The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem. Phys. Lett. 1977, 49, 225. [Google Scholar] [CrossRef]
- Peng, C.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comp. Chem. 1996, 17, 49. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 1981, 14, 363. [Google Scholar] [CrossRef]
- Hratchian, H.P.; Schlegel, H.B. Accurate reaction paths using a Hessian based predictor–corrector integrator. J. Chem. Phys. 2004, 120, 9918. [Google Scholar] [CrossRef] [PubMed]
- Bauernschmitt, R.; Ahlrichs, R. Stability analysis for solutions of the closed shell Kohn–Sham equation. J. Chem. Phys. 1996, 104, 9047. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454. [Google Scholar] [CrossRef]
- Furche, F.; Ahlrichs, R. Adiabatic time-dependent density functional methods for excited state properties. J. Chem. Phys. 2002, 117, 7433. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.; Handy, N. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899. [Google Scholar] [CrossRef]
- Weinhold, F.; Carpenter, J.E. The Structure of Small Molecules and Ions; Plenum: New York, NY, USA, 1988. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]

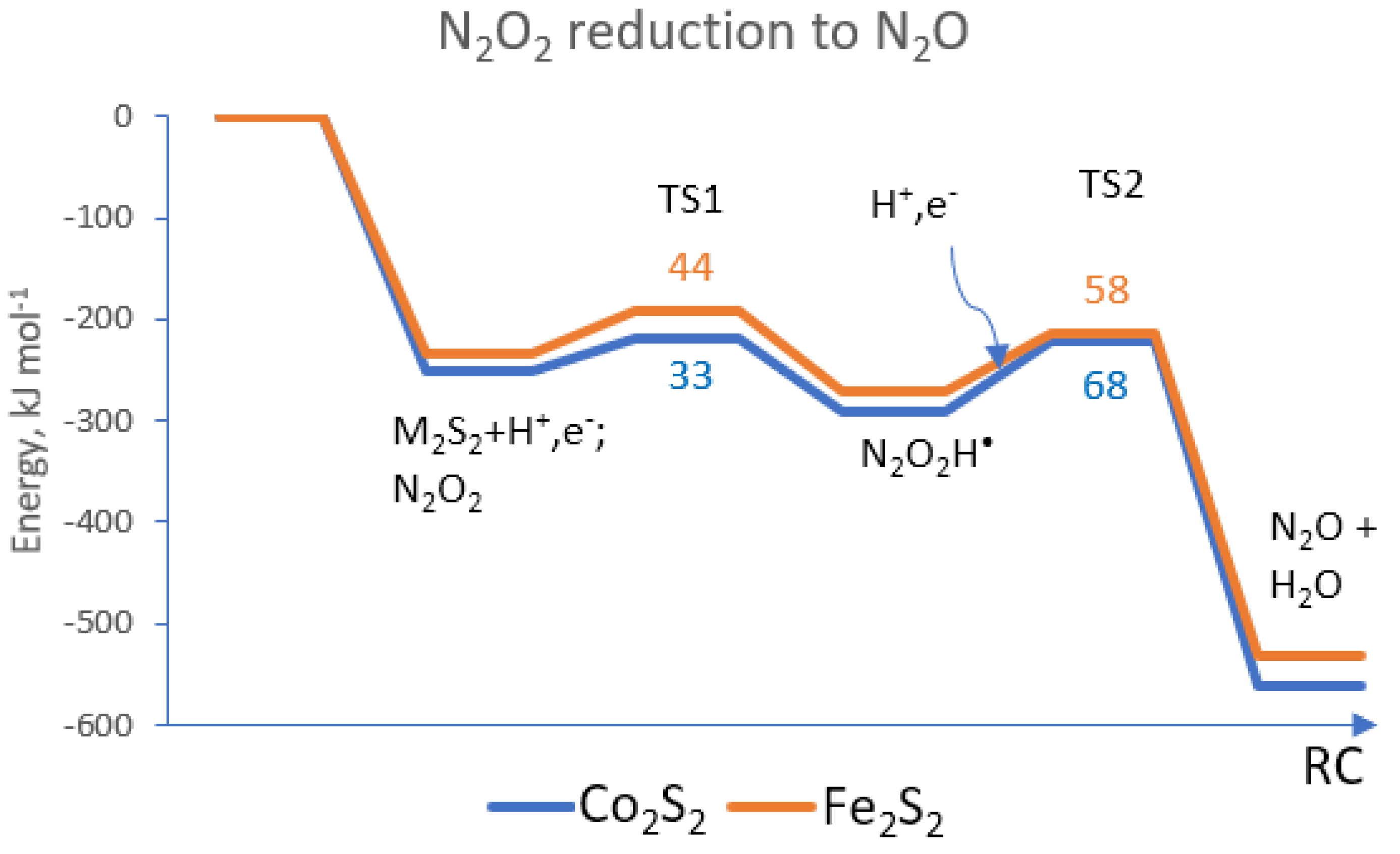
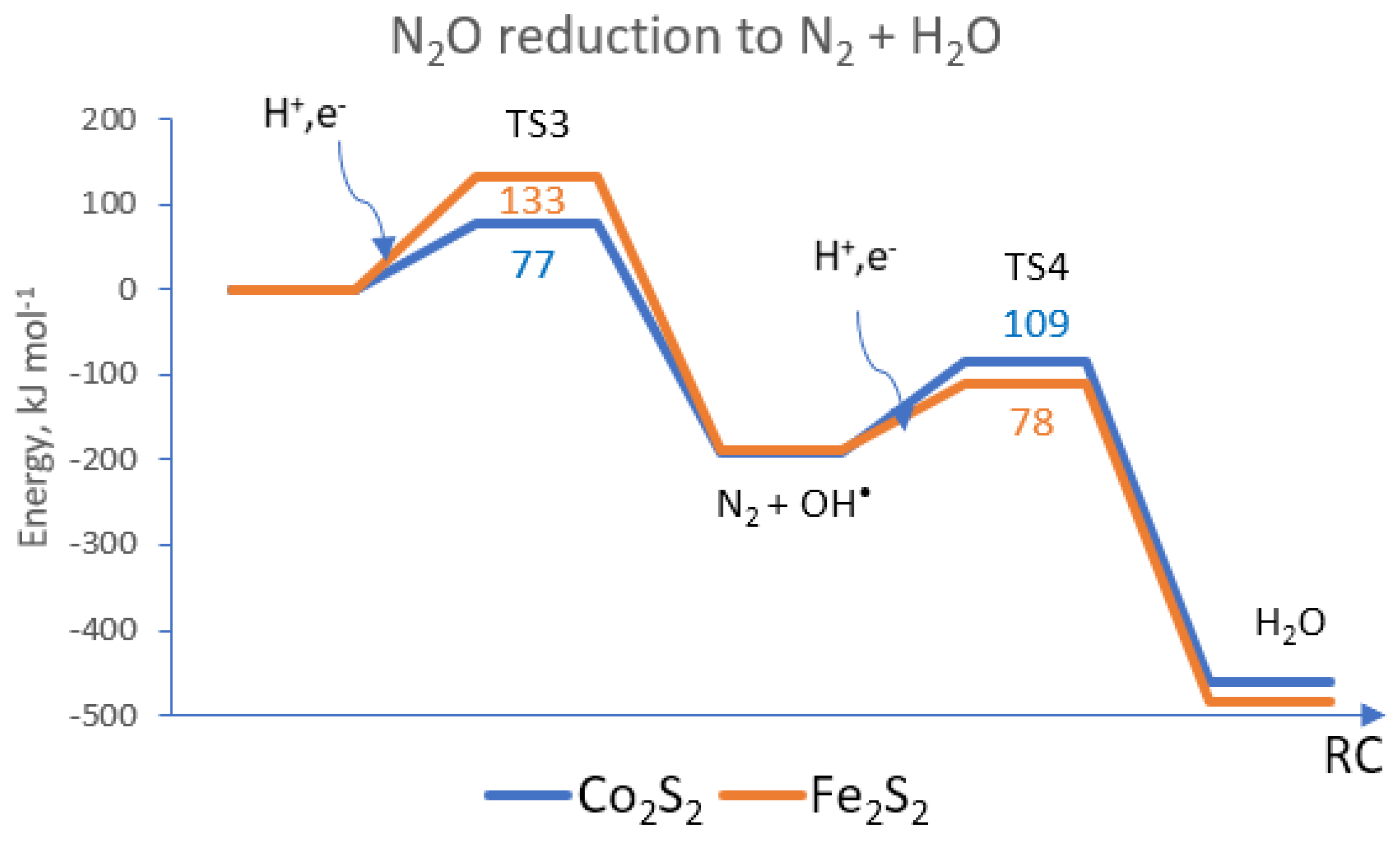



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Bond Length N-O, Å | Bond Length N-N, Å | PA, kJ mol−1 | H+,e− Affinity, kJ mol−1 |
|---|---|---|---|---|
| NO | 1.1456 | 529 | 233 | |
| Exp, Ref. [18] | 1.1508 | 531 | ||
| N2O | 1.1840 | 1.1210 | 554 at N 582 at O | |
| Exp, Ref. [19] | 1.1860 | 1.1260 | 549.8 at N 575.2 at O | |
| (NO)2 cis, N2O2 | 1.1471 | 1.9730 | 638 | 286 |
| (NO)2 trans-ONNO | 1.2118 | 1.1564 | 682 | 311 |
| Molecule | Bond Length M-S, Å | Bond Length M-N, Å | Bond Length S-N, Å | NO BE, kJ mol−1 |
|---|---|---|---|---|
| Co2S2(CO)5(NO) Co-NO bond | 2.236 | 1.964 | 17.4 | |
| Co2S2(CO)4(NO)2 2 Co-NO bonds | 2.245 | 1.821 | 30.1 | |
| Co2S2(NO)4 | 2.317 | 1.641 | 76.1 | |
| Co2S2(CO)6(NO) S-NO bond | 2.297; 2.373 | 1.864 | 22.4 | |
| Fe2S2(CO)5(NO) Fe-NO bond | 2.236 | 1.726 | 50.4 | |
| Fe2S2(CO)4(NO)2 2 Fe-NO bonds | 2.275 | 1.722 | 58.6 | |
| Fe2S2(NO)4 | 2.168 | 1.636 | 88.5 | |
| Fe2S2(CO)6(NO) S-NO bond | 2.231; 2.273 | 1.939 | 32.1 |
| Complex, Bonds | Light Absorption, nm | Oscillator Strength |
|---|---|---|
| Co2S2(CO)5(NO) Co-NO bond | 663 | 0.0025 |
| Fe2S2(CO)5(NO) Fe-NO bond | 739 | 0.0065 |
| Co2S2(NO)4 | 524 | 0.0174 |
| Fe2S2(NO)4 | 535 1057 | 0.0011 0.0087 |
| Co2S2(CO)6(NO) S-NO bond | 820 | 0.0133 |
| Fe2S2(CO)6(NO) S-NO bond | 739 | 0.0065 |
| Co2S2(CO)6(OH) S-OH bond | 874 | 0.0282 |
| Fe2S2(CO)6(OH) S-OH bond | 965 | 0.0205 |
| Fe2S2(CO)6(OH) Fe-OH-Fe bond | 679 | 0.0091 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uzunova, E.L.; Georgieva, I.M. Adsorption and Catalytic Reduction of Nitrogen Oxides (NO, N2O) on Disulfide Cluster Complexes of Cobalt and Iron—A Density Functional Study. Materials 2024, 17, 4764. https://doi.org/10.3390/ma17194764
Uzunova EL, Georgieva IM. Adsorption and Catalytic Reduction of Nitrogen Oxides (NO, N2O) on Disulfide Cluster Complexes of Cobalt and Iron—A Density Functional Study. Materials. 2024; 17(19):4764. https://doi.org/10.3390/ma17194764
Chicago/Turabian StyleUzunova, Ellie L., and Ivelina M. Georgieva. 2024. "Adsorption and Catalytic Reduction of Nitrogen Oxides (NO, N2O) on Disulfide Cluster Complexes of Cobalt and Iron—A Density Functional Study" Materials 17, no. 19: 4764. https://doi.org/10.3390/ma17194764