A Novel Anphevirus in Aedes albopictus Mosquitoes Is Distributed Worldwide and Interacts with the Host RNA Interference Pathway



Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Discovery
2.2. Virus Genome Annotation
2.3. Phylogenetic Analysis
2.4. Virus-Derived Small RNA Profile
3. Results
3.1. Identification and Assembly of AealbAV Genome
3.2. Genomic Organization, Gene Content and RNA Motifs of AealbAV
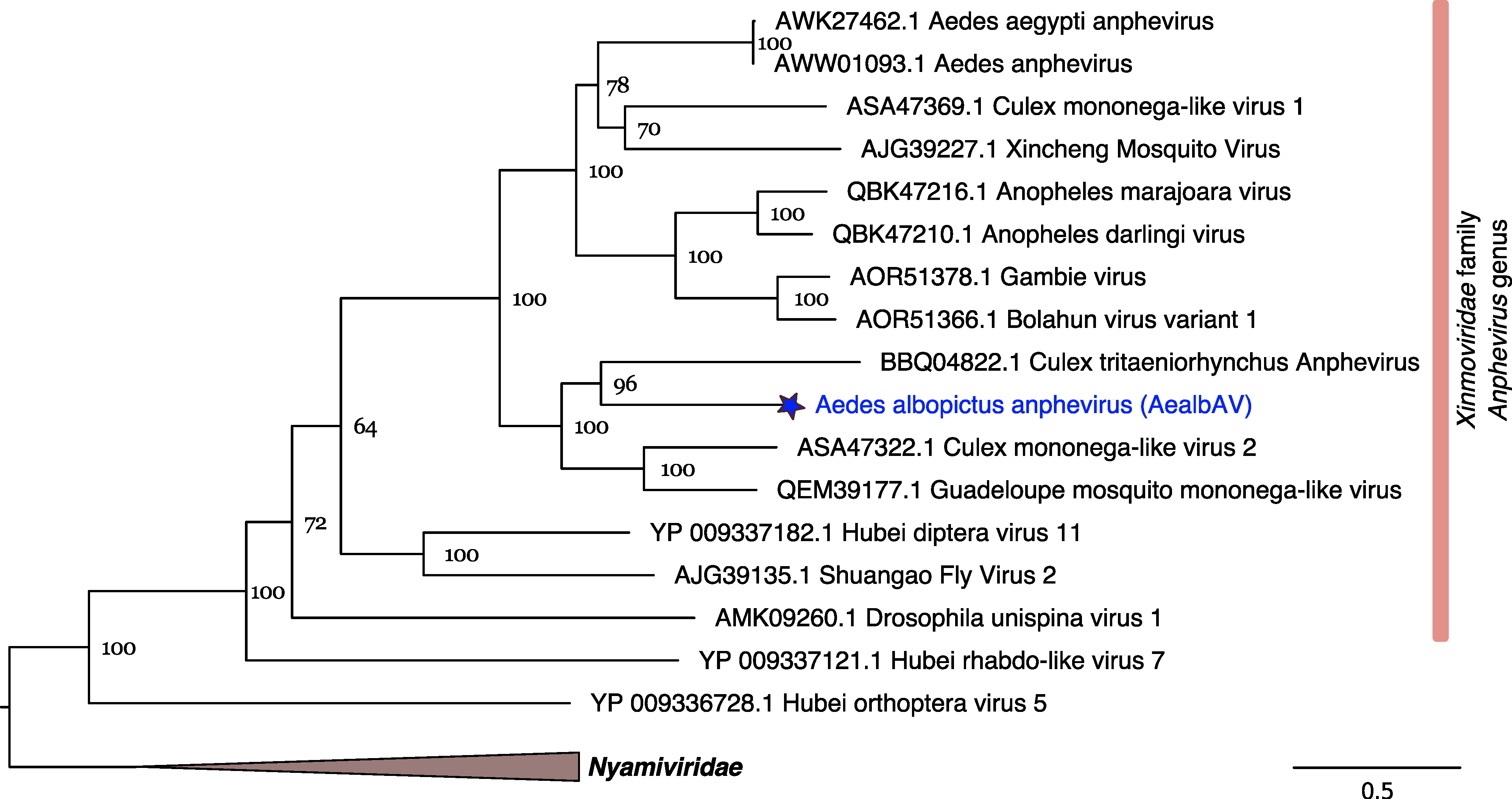
3.3. AealbAV Is Highly Divergent from Currently Known Anpheviruses
3.4. AealbAV Is Distributed Worldwide in Ae. albopictus Laboratory Colonies and Wild Populations
3.5. Ae. albopictus Mosquitoes Worldwide Carry Highly Similar AealbAV Strains
3.6. Eggs and Various Tissues of Ae. albopictus Harbor AealbAV
3.7. Ae. albopictus RNAi Response Targets AealbAV
3.8. Putative Anphevirus-Like NIRVs in Ae. albopictus Genomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Carneiro, L.A.; Travassos, L.H. Autophagy and viral diseases transmitted by Aedes aegypti and Aedes albopictus. Microbes Infect. 2016, 18, 169–171. [Google Scholar] [CrossRef]
- Lounibos, L.P.; Kramer, L.D. Invasiveness of Aedes aegypti and Aedes albopictus and Vectorial Capacity for Chikungunya Virus. J. Infect. Dis. 2016, 214, S453–S458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, B.A.; Wilson, A.E.; Zohdy, S. Aedes albopictus is a competent vector of Zika virus: A meta-analysis. PLoS ONE 2019, 14, e0216794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolling, B.G.; Weaver, S.C.; Tesh, R.B.; Vasilakis, N. Insect-Specific Virus Discovery: Significance for the Arbovirus Community. Viruses 2015, 7, 4911–4928. [Google Scholar] [CrossRef] [Green Version]
- Elrefaey, A.M.; Abdelnabi, R.; Rosas, A.L.R.; Wang, L.; Basu, S.; Delang, L. Understanding the Mechanisms Underlying Host Restriction of Insect-Specific Viruses. Viruses 2020, 12, 964. [Google Scholar] [CrossRef]
- Goenaga, S.; Kenney, J.L.; Duggal, N.K.; DeLorey, M.; Ebel, G.D.; Zhang, B.; Levis, S.C.; Enria, D.A.; Brault, A.C. Potential for Co-Infection of a Mosquito-Specific Flavivirus, Nhumirim Virus, to Block West Nile Virus Transmission in Mosquitoes. Viruses 2015, 7, 5801–5812. [Google Scholar] [CrossRef] [Green Version]
- Romo, H.; Kenney, J.L.; Blitvich, B.J.; Brault, A.C. Restriction of Zika virus infection and transmission in Aedes aegypti mediated by an insect-specific flavivirus. Emerg. Microbes Infect. 2018, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baidaliuk, A.; Miot, E.F.; Lequime, S.; Moltini-Conclois, I.; Delaigue, F.; Dabo, S.; Dickson, L.B.; Aubry, F.; Merkling, S.H.; Cao-Lormeau, V.-M.; et al. Cell-Fusing Agent Virus Reduces Arbovirus Dissemination in Aedes aegypti Mosquitoes In Vivo. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, M.J.; Frydman, H.M.; Connor, J.H. Dual Insect specific virus infection limits Arbovirus replication in Aedes mosquito cells. Virology 2018, 518, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Amuzu, H.E.; Tsyganov, K.; Koh, C.; Herbert, R.I.; Powell, D.R.; McGraw, E.A. Wolbachiaenhances insect-specific flavivirus infection inAedes aegyptimosquitoes. Ecol. Evol. 2018, 8, 5441–5454. [Google Scholar] [CrossRef] [PubMed]
- McLean, B.J.; Dainty, K.R.; Flores, H.; Neill, S.L.O. Differential suppression of persistent insect specific viruses in trans-infected wMel and wMelPop-CLA Aedes-derived mosquito lines. Virology 2018, 527, 141–145. [Google Scholar] [CrossRef]
- Schnettler, E.; Sreenu, V.B.; Mottram, T.; McFarlane, M. Wolbachia restricts insect-specific flavivirus infection in Aedes aegypti cells. J. Gen. Virol. 2016, 97, 3024–3029. [Google Scholar] [CrossRef]
- I Patterson, E.; Villinger, J.; Muthoni, J.N.; Dobel-Ober, L.; Hughes, G.L. Exploiting insect-specific viruses as a novel strategy to control vector-borne disease. Curr. Opin. Insect Sci. 2020, 39, 50–56. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, D.; Li, Y.; Yang, C.; Wu, Y.; Liang, X.; Liang, Y.; Pan, X.; Hu, L.; Sun, Q.; et al. Incompatible and sterile insect techniques combined eliminate mosquitoes. Nature 2019, 572, 56–61. [Google Scholar] [CrossRef]
- Amarasinghe, G.K.; Aréchiga-Ceballos, N.; Banyard, A.C.; Basler, C.F.; Bavari, S.; Bennett, A.J.; Blasdell, K.R.; Briese, T.; Bukreyev, A.; Cai, Y.; et al. Taxonomy of the order Mononegavirales: Update 2018. Arch. Virol. 2018, 163, 2283–2294. [Google Scholar] [CrossRef] [Green Version]
- Whelan, S.P.J.; Barr, J.N.; Wertz, G.W. Transcription and Replication of Nonsegmented Negative-Strand RNA Viruses. In Current Topics in Microbiology and Immunology; Springer Science and Business Media LLC: New York, NY, USA, 2004; Volume 283, pp. 61–119. [Google Scholar]
- Varjak, M.; Leggewie, M.; Schnettler, E. The antiviral piRNA response in mosquitoes? J. Gen. Virol. 2018, 99, 1551–1562. [Google Scholar] [CrossRef]
- Asgari, S. Chapter Two—microRNAs as Regulators of Insect Host–Pathogen Interactions and Immunity. In Advances in Insect Physiology; Smagghe, G., Ed.; Crop Protection; Academic Press: Cambridge, MA, USA, 2018; Volume 55, pp. 19–45. [Google Scholar]
- Kodama, Y.; Shumway, M.; Leinonen, R. On behalf of the International Nucleotide Sequence Database Collaboration The sequence read archive: Explosive growth of sequencing data. Nucleic Acids Res. 2011, 40, D54–D56. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-G.; Jiang, X.; Gu, J.; Xu, M.; Wu, Y.; Deng, Y.; Zhang, C.; Bonizzoni, M.; Dermauw, W.; Vontas, J.; et al. Genome sequence of the Asian Tiger mosquito, Aedes albopictus, reveals insights into its biology, genetics, and evolution. Proc. Natl. Acad. Sci. USA 2015, 112, E5907–E5915. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.R.; Koren, S.; A Dilley, K.; Puri, V.; Brown, D.M.; Harkins, D.M.; Thibaud-Nissen, F.; Rosen, B.; Chen, X.-G.; Tu, Z.; et al. Analysis of the Aedes albopictus C6/36 genome provides insight into cell line utility for viral propagation. GigaScience 2018, 7, 1–13. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.S.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2012, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Hahne, F.; Ivanek, R. Visualizing Genomic Data Using Gviz and Bioconductor. In Methods in Molecular Biology; Springer Science and Business Media LLC: New York, NY, USA, 2016; Volume 1418, pp. 335–351. [Google Scholar]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.-S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; I Hurwitz, D.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [Green Version]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes11Edited by F. Cohen. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Käll, L.; Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Uhrig, S.; Klein, H. PingPongPro: A tool for the detection of piRNA-mediated transposon-silencing in small RNA-Seq data. Bioinformatics 2018, 35, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Gomulski, L.M.; Manni, M.; Carraretto, D.; Nolan, T.; Lawson, D.; Ribeiro, J.M.; Malacrida, A.R.; Gasperi, G. Transcriptional variation of sensory-related genes in natural populations of Aedes albopictus. BMC Genom. 2020, 21, 1–22. [Google Scholar] [CrossRef]
- Kubacki, J.; Flacio, E.; Qi, W.; Guidi, V.; Tonolla, M.; Fraefel, C. Viral Metagenomic Analysis of Aedes albopictus Mosquitos from Southern Switzerland. Viruses 2020, 12, 929. [Google Scholar] [CrossRef]
- Stanojević, M.; Li, K.; Stamenković, G.; Ilić, B.; Paunović, M.; Pešić, B.; Maslovara, I.Đ.; Šiljić, M.; Ćirković, V.; Zhang, Y.-Z. Depicting the RNA Virome of Hematophagous Arthropods from Belgrade, Serbia. Viruses 2020, 12, 975. [Google Scholar] [CrossRef]
- Poelchau, M.; Reynolds, J.A.; Denlinger, D.L.; Elsik, C.G.; Armbruster, P.A. A de novo transcriptome of the Asian tiger mosquito, Aedes albopictus, to identify candidate transcripts for diapause preparation. BMC Genom. 2011, 12, 619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, F.; Salvemini, M.; Fiorillo, C.; Nolan, T.; Zwiebel, L.J.; Ribeiro, J.M.; Arcà, B. Deciphering the olfactory repertoire of the tiger mosquito Aedes albopictus. BMC Genom. 2017, 18, 1–23. [Google Scholar] [CrossRef]
- Poelchau, M.; Reynolds, J.A.; Elsik, C.G.; Denlinger, D.L.; Armbruster, P.A. Deep sequencing reveals complex mechanisms of diapause preparation in the invasive mosquito, Aedes albopictus. Proc. R. Soc. B Biol. Sci. 2013, 280, 20130143. [Google Scholar] [CrossRef] [Green Version]
- Parry, R.; Asgari, S. Aedes Anphevirus: An Insect-Specific Virus Distributed Worldwide in Aedes aegypti Mosquitoes That Has Complex Interplays with Wolbachia and Dengue Virus Infection in Cells. J. Virol. 2018, 92, 92. [Google Scholar] [CrossRef] [Green Version]
- Amarasinghe, G.K.; Ayllón, M.A.; Bào, Y.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the order Mononegavirales: Update 2019. Arch. Virol. 2019, 164, 1967–1980. [Google Scholar] [CrossRef] [Green Version]
- Liang-Jun, C.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Kang, Y.-J.; Chen, L.-J.; Qin, X.-C.; Xu, J.; Holmes, E.C.; Zhang, Y.-Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef]
- Fauver, J.R.; Grubaugh, N.D.; Krajacich, B.J.; Weger-Lucarelli, J.; Lakin, S.M.; Fakoli, L.S.; Bolay, F.K.; DiclaroII, J.W.; Dabiré, K.R.; Foy, B.D.; et al. West African Anopheles gambiae mosquitoes harbor a taxonomically diverse virome including new insect-specific flaviviruses, mononegaviruses, and totiviruses. Virology 2016, 498, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.-S.; Imrie, A.; Holmes, E.C. High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Scarpassa, V.M.; Debat, H.J.; Alencar, R.B.; Saraiva, J.F.; Calvo, E.; Arcà, B.; Ribeiro, J.M.C. An insight into the sialotranscriptome and virome of Amazonian anophelines. BMC Genom. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Faizah, A.N.; Kobayashi, D.; Isawa, H.; Amoa-Bosompem, M.; Murota, K.; Higa, Y.; Futami, K.; Shimada, S.; Kim, K.S.; Itokawa, K.; et al. Deciphering the Virome of Culex vishnui Subgroup Mosquitoes, the Major Vectors of Japanese Encephalitis, in Japan. Viruses 2020, 12, 264. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Poelchau, M.F.; Armbruster, P.A. Global Transcriptional Dynamics of Diapause Induction in Non-Blood-Fed and Blood-Fed Aedes albopictus. PLOS Neglected Trop. Dis. 2015, 9, e0003724. [Google Scholar] [CrossRef]
- Batz, Z.A.; Goff, A.C.; Armbruster, P.A. MicroRNAs are differentially abundant during Aedes albopictus diapause maintenance but not diapause induction. Insect Mol. Biol. 2017, 26, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Poelchau, M.F.; Reynolds, J.A.; Denlinger, D.L.; Elsik, C.G.; Armbruster, P.A. Transcriptome sequencing as a platform to elucidate molecular components of the diapause response in the Asian tiger mosquitoAedes albopictus. Physiol. Èntomol. 2013, 38, 173–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morazzani, E.M.; Wiley, M.R.; Murreddu, M.G.; Adelman, Z.N.; Myles, K.M. Production of Virus-Derived Ping-Pong-Dependent piRNA-like Small RNAs in the Mosquito Soma. PLoS Pathog. 2012, 8, e1002470. [Google Scholar] [CrossRef]
- Frangeul, L.; Blanc, H.; Saleh, M.-C.; Suzuki, Y. Differential Small RNA Responses against Co-Infecting Insect-Specific Viruses in Aedes albopictus Mosquitoes. Viruses 2020, 12, 468. [Google Scholar] [CrossRef] [Green Version]
- Esquivel, C.J.; Cassone, B.J.; Piermarini, P.M. Ade novotranscriptome of the Malpighian tubules in non-blood-fed and blood-fed Asian tiger mosquitoesAedes albopictus: Insights into diuresis, detoxification, and blood meal processing. PeerJ 2016, 4, e1784. [Google Scholar] [CrossRef] [Green Version]
- Esquivel, C.J.; Cassone, B.J.; Piermarini, P.M. Transcriptomic Evidence for a Dramatic Functional Transition of the Malpighian Tubules after a Blood Meal in the Asian Tiger Mosquito Aedes albopictus. PLoS Negl. Trop. Dis. 2014, 8, e2929. [Google Scholar] [CrossRef] [Green Version]
- Manni, M.; Guglielmino, C.R.; Scolari, F.; Vega-Rúa, A.; Failloux, A.-B.; Somboon, P.; Lisa, A.; Savini, G.; Bonizzoni, M.; Gomulski, L.M.; et al. Genetic evidence for a worldwide chaotic dispersion pattern of the arbovirus vector, Aedes albopictus. PLoS Negl. Trop. Dis. 2017, 11, e0005332. [Google Scholar] [CrossRef]
- Kraemer, M.U.G.; E Sinka, M.; A Duda, K.; Mylne, A.Q.N.; Shearer, F.M.; Barker, C.M.; Moore, C.G.; Carvalho, R.G.; E Coelho, G.; Van Bortel, W.; et al. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. eLife 2015, 4, e08347. [Google Scholar] [CrossRef]
- Pischedda, E.; Scolari, F.; Valerio, F.; Carballar-Lejarazú, R.; Catapano, P.L.; Waterhouse, R.M.; Bonizzoni, M. Insights Into an Unexplored Component of the Mosquito Repeatome: Distribution and Variability of Viral Sequences Integrated Into the Genome of the Arboviral Vector Aedes albopictus. Front. Genet. 2019, 10, 93. [Google Scholar] [CrossRef] [Green Version]
- Houé, V.; Gabiane, G.; Dauga, C.; Suez, M.; Madec, Y.; Mousson, L.; Marconcini, M.; Yen, P.-S.; De Lamballerie, X.; Bonizzoni, M.; et al. Evolution and biological significance of flaviviral elements in the genome of the arboviral vector Aedes albopictus. Emerg. Microbes Infect. 2019, 8, 1265–1279. [Google Scholar] [CrossRef] [Green Version]
- Palatini, U.; Miesen, P.; Carballar-Lejarazu, R.; Ometto, L.; Rizzo, E.; Tu, Z.; Van Rij, R.P.; Bonizzoni, M. Comparative genomics shows that viral integrations are abundant and express piRNAs in the arboviral vectors Aedes aegypti and Aedes albopictus. BMC Genom. 2017, 18, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaller, C.K.; Cattaneo, R.; Schnell, M.J. Reverse genetics of Mononegavirales: How they work, new vaccines, and new cancer therapeutics. Virology 2015, 331–344. [Google Scholar] [CrossRef] [Green Version]
- Haddow, A.D.; Guzman, H.; Popov, V.L.; Wood, T.G.; Widen, S.G.; Haddow, A.D.; Tesh, R.B.; Weaver, S.C. First isolation of Aedes flavivirus in the Western Hemisphere and evidence of vertical transmission in the mosquito Aedes (Stegomyia) albopictus (Diptera: Culicidae). Virology 2013, 440, 134–139. [Google Scholar] [CrossRef] [Green Version]
- Bolling, B.G.; Eisen, L.; Moore, C.G.; Blair, C.D. Insect-Specific Flaviviruses from Culex Mosquitoes in Colorado, with Evidence of Vertical Transmission. Am. J. Trop. Med. Hyg. 2011, 85, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Vasilakis, N.; Tesh, R.B. Insect-specific viruses and their potential impact on arbovirus transmission. Curr. Opin. Virol. 2015, 15, 69–74. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SRA Study | Number of Libraries | Origin | Lab Colony Location | Sampling Year for Colony Establishment | Collection Site | Sampling Time/(Lab Colony Generation) | Library Type | Library Layout | Evidence | Max Depth of Coverage (% Breadth of Coverage) | Tissue | Sex |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SRP012105 | 12 | Lab colony | Georgetown University (USA) | 2008 | Manassas (USA) | -/(F10) | RNA-Seq | Paired (Illumina) | Assembled genomes | 361.5 (100) | Embryos | Mixed |
| SRP018112 | 17 | Lab colony | Georgetown University (USA) | 2008 | Manassas (USA) | -/(F13) | RNA-Seq | Paired (Illumina) | Assembled genomes | 57.4 (100) | 1st instar pharate larvae (whole eggs) | Mixed |
| SRP050258 | 16 | Lab colony | Georgetown University (USA) | 2013 | Manassas (USA) | 2013/(F3) | RNA-Seq | Paired (Illumina) | Assembled genomes | 27.7 (100) | Adult whole body | Female |
| SRP096579 | 8 | Lab colony | Georgetown University (USA) | 2010 | Manassas (USA) | -/(F12) | miRNA-Seq | Single (Illumina) | Mapped reads | 2.6 (73) | Whole Eggs (Pharate Larvae) | Mixed |
| SRP007714 | 2 | Lab colony | Georgetown University (USA) | 2008 | Manassas (USA) | 2010/(F5) | RNA-Seq | Single (454) | Mapped reads | 4.0 (64) | Oocytes | Female |
| SRP228299 * | 2 | Lab colony | Institut Pasteur (FRA) | 2008 | Kawasaki (JPN) | 2015/- | miRNA-Seq | Single (Illumina) | Mapped reads | 49.8 (99) | Whole body, ovaries | Female |
| SRP071220 | 8 | Lab colony | Sapienza University (ITA) | 2012 | Rome (ITA) | 2013/- | RNA-Seq | Paired (Illumina) | Assembled genomes | 169.3 (100) | Whole body, heads, antennae, maxillary palps | Male/Female |
| SRP056407 | 20 | Lab colony (MRA-804) | The Ohio State University (USA) | na | Florida (USA) | 2012/- | RNA-Seq | Paired (Illumina) | Assembled genomes | 33.6 (100) | Malpighian tubules | Female |
| SRP034701 | 18 | Lab colony (MRA-804) | The Ohio State University (USA) | na | Florida (USA) | 2012/- | RNA-Seq | Single (Illumina) | Mapped reads | 3.7 (85) | Malpighian tubules | Female |
| SRP008316 ** | 5 | Lab colony | Fralin Life Science Institute (USA) | na | na | 2012/- | miRNA-Seq | Single (Illumina) | Assembled genomes | 1228.7 (100) | Whole body, head and thorax | Female |
| SRP188743 | 9 | Wild-caught | - | - | Guangzhou (CHN) | 2017 | miRNA-Seq | Single (Illumina) | Mapped reads | 4.8 (77) | Adult Whole body, larvae | Mixed |
| PRJNA602498 | 1 | Wild-caught | - | - | Arco (ITA) | 2011 | RNA-Seq | Paired (Illumina) | Assembled genomes | 129.4261 (100) | Antennae | Mixed |
| PRJNA602498 | 1 | Wild-caught | - | - | Trento (ITA) | 2011 | RNA-Seq | Paired (Illumina) | Assembled genomes | 512.9825 (100) | Antennae | Mixed |
| PRJNA602498 | 1 | Wild-caught | - | - | Ban Rai (THA) | 2012 | RNA-Seq | Paired (Illumina) | Assembled genomes | 27.8252 (100) | Antennae | Mixed |
| SRP266553 | 14 | Wild-caught | - | - | Ticino (CHE) | 2019 | DNA-Seq | Single (Illumina) | Assembled genomes | 21.5 (100) | Adult whole body | Mixed |
| Serbia Mononega-Like Virus 1 | Culex Tritaeniorhynchus Anphevirus | Culex Mononega-Like Virus 2 | Guadeloupe Mosquito Mononega-Like Virus | Aedes Anphevirus | |
|---|---|---|---|---|---|
| ORF1 (N) | 67.8 (43)/55.3 (98) | -/29.9 (50) | -/- | -/- | -/- |
| ORF2 (STM) | -/- | -/- | -/- | -/- | -/- |
| ORF3 (G1) | -/39.6 (78) | -/- | -/- | -/- | -/- |
| ORF4 (G2) | 70.4 (91)/74.2 (99) | 74.9 (12)/41.9 (93) | 65.9 (16)/43.7 (96) | 65.6 (11)/43.7 (96) | -/42.0 (96) |
| ORF5 (ZnF) | -/- | -/- | -/- | -/- | -/- |
| ORF6 | -/- | -/- | -/- | -/- | -/- |
| ORF7 (RdRp) | 71.1 (70)/69.0 (99) | 64.9 (13)/39.2 (95) | 64.7 (12)/41.7 (96) | -/41.6 (97) | -/36.5 (96) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manni, M.; Zdobnov, E.M. A Novel Anphevirus in Aedes albopictus Mosquitoes Is Distributed Worldwide and Interacts with the Host RNA Interference Pathway. Viruses 2020, 12, 1264. https://doi.org/10.3390/v12111264
Manni M, Zdobnov EM. A Novel Anphevirus in Aedes albopictus Mosquitoes Is Distributed Worldwide and Interacts with the Host RNA Interference Pathway. Viruses. 2020; 12(11):1264. https://doi.org/10.3390/v12111264
Chicago/Turabian StyleManni, Mosè, and Evgeny M. Zdobnov. 2020. "A Novel Anphevirus in Aedes albopictus Mosquitoes Is Distributed Worldwide and Interacts with the Host RNA Interference Pathway" Viruses 12, no. 11: 1264. https://doi.org/10.3390/v12111264