
Non-Random Pattern of Integration for Epstein-Barr Virus with Preference for Gene-Poor Genomic Chromosomal Regions into the Genome of Burkitt Lymphoma Cell Lines

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Amplification and Detection of EBV DNA by PCR
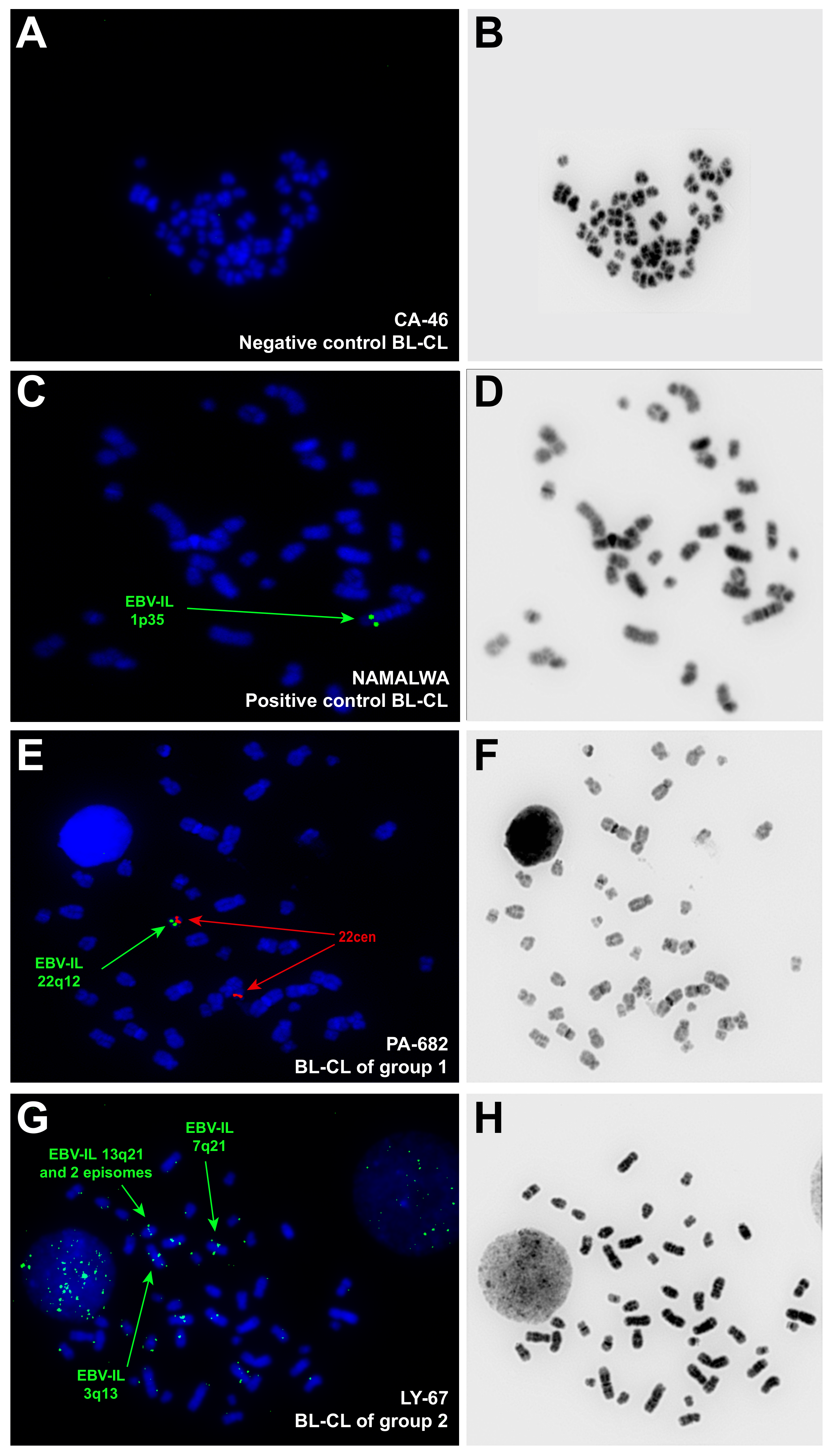
2.3. Assessment of the EBV-IS by FISH and Data Analysis
2.4. Statistical Analyses
3. Results
3.1. Distribution of EBV-IS in the Whole Collective of BL-CL
3.2. Comparison between EBV-IS in BL-CLs and LCLs
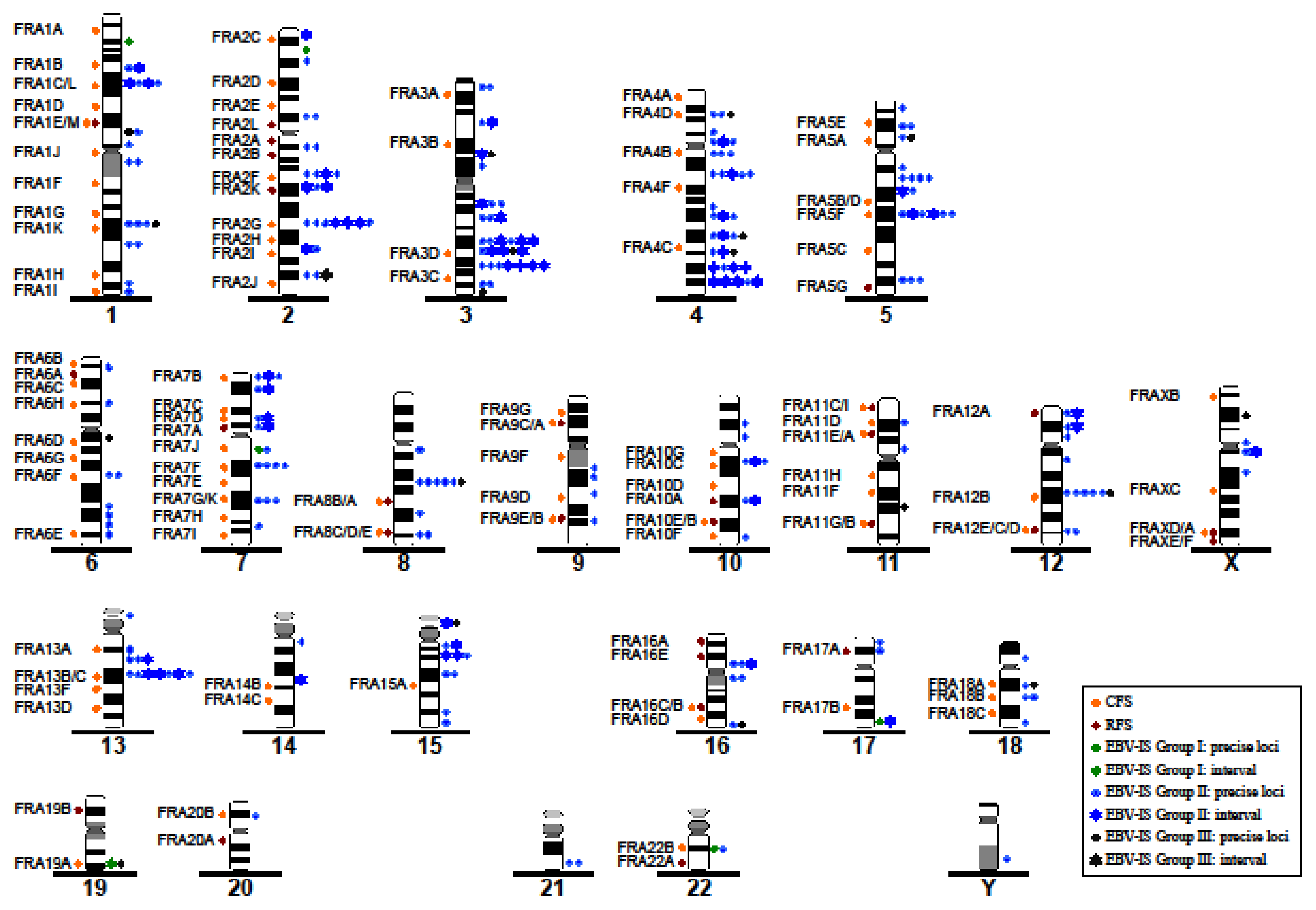
3.3. Correlation between EBV-IS and Genetically Unstable Regions
3.4. Pattern of Integration of Statistically Significant EBV in the Chromosomal DNA Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Shannon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein-Barr virus-associated lymphomas. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160271. [Google Scholar] [CrossRef]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef]
- Boerma, E.G.; Siebert, R.; Kluin, P.M.; Baudis, M. Translocations involving 8q24 in Burkitt lymphoma and other malignant lymphomas: A historical review of cytogenetics in the light of todays knowledge. Leukemia 2009, 23, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Molyneux, E.M.; Rochford, R.; Griffin, B.; Newton, R.; Jackson, G.; Menon, G.; Harrison, C.J.; Israels, T.; Bailey, S. Burkitt’s lymphoma. Lancet 2012, 379, 1234–1244. [Google Scholar] [CrossRef] [Green Version]
- Brady, G.; MacArthur, G.J.; Farrell, P.J. Epstein–Barr virus and Burkitt lymphoma. J. Clin. Pathol. 2007, 60, 1397–1402. [Google Scholar] [CrossRef]
- Cader, F.Z.; Kearns, P.; Young, L.; Murray, P.; Vockerodt, M. The contribution of the Epstein-Barr virus to the pathogenesis of childhood lymphomas. Cancer Treat. Rev. 2010, 36, 348–353. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Allday, M.J. The curious case of the tumour virus: 50 years of Burkitt’s lymphoma. Nat. Rev. Microbiol. 2008, 6, 913–924. [Google Scholar] [CrossRef]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 23, e131. [Google Scholar] [CrossRef] [Green Version]
- Xiao, K.; Yu, Z.; Li, X.; Li, X.; Tang, K.; Tu, C.; Qi, P.; Liao, Q.; Chen, P.; Zeng, Z.; et al. Genome-wide Analysis of Epstein-Barr Virus (EBV) Integration and Strain in C666-1 and Raji Cells. J. Cancer 2016, 7, 214–224. [Google Scholar] [CrossRef] [Green Version]
- Delecluse, H.J.; Bartnizke, S.; Hammerschmidt, W.; Bullerdiek, J.; Bornkamm, G.W. Episomal and integrated copies of Epstein-Barr virus coexist in Burkitt lymphoma cell lines. J. Virol. 1993, 67, 1292–1299. [Google Scholar] [CrossRef] [Green Version]
- Wolf, J.; Pawlita, M.; Jox, A.; Kohls, S.; Bartnitzke, S.; Diehl, V.; Bullerdiek, J. Integration of Epstein Barr virus near the breakpoint of a translocation 11;19 in a Burkitt’s lymphoma cell line. Cancer Genet. Cytogenet. 1993, 67, 90–94. [Google Scholar] [CrossRef]
- Lestou, V.S.; De Braekeleer, M.; Strehl, S.; Ott, G.; Gadner, H.; Ambros, P.F. Non-random integration of Epstein-Barr virus in lymphoblastoid cell lines. Genes Chromosomes Cancer 1993, 8, 38–48. [Google Scholar] [CrossRef]
- Ohshima, K.; Suzumiya, J.; Kanda, M.; Kato, A.; Kikuchi, M. Integrated and episomal forms of Epstein-Barr virus (EBV) in EBV associated disease. Cancer Lett. 1998, 122, 43–50. [Google Scholar] [CrossRef]
- Hurley, E.A.; Agger, S.; McNeil, J.A.; Lawrence, J.B.; Calendar, A.; Lenoir, G.; Thorley-Lawson, D.A. When Epstein-Barr virus persistently infects B-cell lines, it frequently integrates. J. Virol. 1991, 65, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, J.B.; Villnave, C.A.; Singer, R.H. Sensitive, high-resolution chromatin and chromosome mapping in situ: Presence and orientation of two closely integrated copies of EBV in a lymphoma line. Cell 1988, 52, 51–61. [Google Scholar]
- Popescu, N.C.; Chen, M.C.; Simpson, S.; Solinas, S.; DiPaolo, J.A. A Burkitt lymphoma cell line with integrated Epstein-Barr virus at a stable chromosome modification site. Virology 1993, 195, 248–251. [Google Scholar] [CrossRef]
- Luo, W.J.; Takakuwa, T.; Ham, M.F.; Wada, N.; Liu, A.; Fujita, S.; Sakane-Ishikawa, E.; Aozasa, K. Epstein-Barr virus is integrated between REL and BCL-11A in American Burkitt lymphoma cell line (NAB-2). Lab. Investig. 2004, 84, 1193–1199. [Google Scholar] [CrossRef]
- Henderson, A.; Ripley, S.; Heller, M.; Kieff, E. Chromosome site for Epstein- Barr virus DNA in a Burkitt tumor cell line and in lymphocytes growth- transformed in vitro. Proc. Natl. Acad. Sci. USA 1983, 80, 1987–1991. [Google Scholar] [CrossRef] [Green Version]
- Trescol-Biémont, M.C.; Biémont, C.; Daillie, J. Localization polymorphism of EBV DNA genomes in the chromosomes of Burkitt lymphoma cell lines. Chromosoma 1987, 95, 144–147. [Google Scholar] [CrossRef]
- Takakuwa, T.; Luo, W.J.; Ham, M.F.; Wada, N.; Aozasa, K. Identification of Epstein-Barr virus integrated sites in lymphoblastoid cell line (IB4). Virus Res. 2005, 108, 133–138. [Google Scholar] [CrossRef]
- Gao, J.; Luo, X.; Tang, K.; Li, X.; Li, G. Epstein-Barr virus integrates frequently into chromosome 4q, 2q, 1q and 7q of Burkitt’s lymphoma cell line (Raji). J. Virol. Methods 2006, 136, 193–199. [Google Scholar] [CrossRef]
- Wuu, K.D.; Chen, Y.J.; Wuu, S.W. Frequency and distribution of chromosomal integration sites of the Epstein Barr virus genome. J. Formos Med. Assoc. 1996, 95, 911–916. [Google Scholar]
- Shiraishi, Y.; Taguchi, T.; Ohta, Y.; Hirai, K. Chromosomal localization of the Epstein-Barr virus (EBV) genome in Bloom’s syndrome B-lymphoblastoid cell lines transformed with EBV. Chromosoma 1985, 93, 157–164. [Google Scholar] [CrossRef]
- Caporossi, D.; Vernole, P.; Porfirio, B.; Tedeschi, B.; Frezza, D.; Nicoletti, B.; Calef, E. Specific sites for EBV association in the Namalwa Burkitt lymphoma cell line and in a lymphoblastoid line transformed in vitro with EBV. Cytogenet. Cell Genet. 1988, 48, 220–223. [Google Scholar] [CrossRef]
- Hayashida, M.; Daibata, M.; Tagami, E.; Taguchi, T.; Maekawa, F.; Takeoka, K.; Fukutsuka, K.; Shimomura, D.; Hayashi, T.; Iwatani, Y.; et al. Establishment and characterization of a novel Hodgkin lymphoma cell line, AM-HLH, carrying the Epstein-Barr virus genome integrated into the host chromosome. Hematol. Oncol. 2017, 35, 567–575. [Google Scholar] [CrossRef]
- Peng, R.J.; Han, B.W.; Cai, Q.Q.; Zuo, X.Y.; Xia, T.; Chen, J.R.; Feng, L.-N.; Lim, J.Q.; Chen, S.-W.; Zeng, M.; et al. Genomic and transcriptomic landscapes of Epstein-Barr virus in extranodal natural killer T-cell lymphoma. Leukemia 2019, 33, 1451–1462. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, W.-L.; Zhu, Q.; Zhang, S.; Yao, Y.-Y.; Xiang, T.; Feng, Q.-S.; Zhang, Z.; Peng, R.-J.; Jia, W.-H.; et al. Genome-wide profiling of Epstein-Barr virus integration by targeted sequencing in Epstein-Barr virus associated malignancies. Theranostic 2019, 9, 1115–1124. [Google Scholar] [CrossRef]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef]
- Zhao, L.H.; Liu, X.; Yan, H.X.; Li, W.Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.P.; Zhuang, X.H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992. [Google Scholar] [CrossRef]
- Murga Penas, E.M.; Schilling, G.; Behrmann, P.; Klokow, M.; Vettorazzi, E.; Bokemeyer, C.; Dierlamm, J. Comprehensive cytogenetic and molecular cytogenetic analysis of 44 Burkitt lymphoma cell lines: Secondary chromosomal changes characterization, karyotypic evolution, and comparison with primary samples. Genes Chromosomes Cancer 2014, 53, 497–515. [Google Scholar] [CrossRef]
- Ciuffi, A.; Ronen, K.; Brady, T.; Malani, N.; Wang, G.; Berry, C.C.; Bushman, F.D. Methods for integration site distribution analyses in animal cell genomes. Methods 2009, 47, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Lukusa, T.; Fryns, J.P. Human chromosome fragility. Biochim. Biophys. Acta 2008, 1779, 3–16. [Google Scholar] [CrossRef]
- Bickmore, W.A. Patterns in the genome. Heredity 2019, 123, 50–57. [Google Scholar] [CrossRef] [Green Version]
- Federico, C.; Andreozzi, L.; Saccone, S.; Bernardi, G. Gene Density in the Giemsa Bands of Human Chromosomes. Chromosome Res. 2000, 8, 737–746. [Google Scholar] [CrossRef]
- Chang, Y.; Cheng, S.D.; Tsai, C.H. Chromosomal integration of Epstein-Barr virus genomes in nasopharyngeal carcinoma cells. Head Neck 2002, 24, 143–150. [Google Scholar] [CrossRef]
- Leenman, E.E.; Panzer-Gruemayer, R.E.; Fischer, S.; Leitch, H.A.; Horsman, D.E.; Lion, T.; Gadner, H.; Ambros, P.F.; Lestou, V.S. Rapid determination of Epstein-Barr virus latent or lytic infection in single human cells using in situ hybridization. Mod. Pathol. 2004, 17, 1564–1572. [Google Scholar] [CrossRef] [Green Version]
- Kamranvar, S.A.; Gruhne, B.; Szeles, A.; Masucci, M.G. Epstein-Barr Virus Promotes Genomic Instability in Burkitt’s Lymphoma. Oncogene 2007, 26, 5115–5123. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.; Qiu, L.; Mrasek, K.; Zhang, J.; Liehr, T.; Quintana, L.G.; Li, Z. Common fragile sites: Genomic hotspots of DNA damage and carcinogenesis. Int. J. Mol. Sci. 2012, 13, 11974–11999. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Li, B.; Xu, T.; Hu, R.; Tan, D.; Song, X.; Jia, P.; Zhao, Z. VISDB: A manually curated database of viral integration sites in the human genome. Nucleic Acids Res. 2020, 48, D633–D641. [Google Scholar] [CrossRef]
- Gao, G.; Johnson, S.H.; Vasmatzis, G.; Pauley, C.E.; Tombers, N.M.; Kasperbauer, J.L.; Smith, D.I. Common fragile sites (CFS) and extremely large CFS genes are targets for human papillomavirus integrations and chromosome rearrangements in oropharyngeal squamous cell carcinoma. Genes Chromosomes Cancer 2017, 56, 59–74. [Google Scholar] [CrossRef]
- Moquin, S.A.; Thomas, S.; Whalen, S.; Warburton, A.; Fernandez, S.G.; McBride, A.A.; Pollard, K.S.; Miranda, J.L. The Epstein-Barr Virus Episome Maneuvers Between Nuclear Chromatin Compartments During Reactivation. J. Virol. 2018, 92, e01413-17. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Cell Line | Origin | Chromosomal Location | Gene Involved | Reference |
|---|---|---|---|---|
| BL36 | BL * | 11p15 + Episomal form | Unknown | [12] |
| BL60 | BL | der (19)t(11;19) + 1 Episomal form | Near the breakpoint | [13] |
| BL137 | BL * | 1p34 + Episomal form | Unknown | [12] |
| EB-2 | BL | 1p, 1q, 3p, 3q, 4p, 4q, 5p, 5q, 6p, 6q, 8p, 8q, 11q, 12q, 13q, 14q, 19p, 10q, 21p + Episomal form | None | [14] |
| [15] | ||||
| [12] | ||||
| [16] | ||||
| [17] | ||||
| NAB-2 | BL | 2p13 | Between REL and BCL11A | [18] |
| [19] | ||||
| Namalwa | BL | 1p35 | MACF-1 | [20] |
| Raji | BL ** | Preferential: 1q, 2q, 4q, and 7q Further: 1p, 3p, 3q, 5q, 6q, 7p, 9q, 11p, 14q, 15q + Episomal form 1q31, 3q13, 7q21, 7q3, 8q23, 21q | BACH2/6q15 | [21] |
| [22] | ||||
| [23] | ||||
| Jijoye | BL ** | Preferential: 13q2 Further: 3p, 3q, 4p, 4q, 5p, 6p, 6q, 7q, 8q, 11p, 18q, 21q + Episomal form | Unknown | [21] |
| P3HR-1 | LCL (derived from Jijoye) | 1q31, 5q2, 13q2, 21q1 + Episomal form | Unknown | [21] |
| IB-4 | LCL | 4q25 + Episomal form | RP11-119H12 | [20] |
| [22] | ||||
| n = 3 | LCL ** | 1p31, 1q31, 2q32, 3q13, 6q24, 7q31 (in all 3 LCL) + Episomal form | Unknown | [24] |
| n = 14 | LCL ** | Preferential: 5p14 (in all 14 LCLs); 2p22 (in 13); 1p31, 1q43, 3q28, 4q13, 5q12, 11p15 (in 12); 1q31, 2q32, 4p15, 5q34, 10q26, 12p12 (in 11); 1p36, 3p24, 4q26, 6q24, 10q21, 13q13 (in 10) Further: up to 60 integration sites in total + Episomal form | Unknown | [14] |
| n = 4 | LCLs *** | 1p31, 1q31, 4q22~25, 5q21, 13q21 and 14q21 (in all); 2q22~24, 3q11, 3q22~24, 3q26, 5q32~34 and 14q31 (presumably in all) + Episomal form | Unknown | [25] |
| ATL9/g | LCL | 1p31~32, 1q31, 2p12, 4p11, 5q21, 11p13, 11q22, 13q2, 16p13, 18q12 + Episomal form | Unknown | [26] |
| AM-HLH | HL | Multiple copies on chromosome 20 of a der(21)t(19;20;21) | Unknown | [27] |
| BL-CL | Frequency: Recurrent IS | Frequency: Non-Recurrent IS | |
|---|---|---|---|
| Group 1 | BL60 | 100%: 19 on der (19)t(17,19) (near the breakpoint) | None. Episomal. |
| Naliaka * | 100%: 17q25 75%: 7q11 | None. Episomal. | |
| Namalwa | 100%: 1p35 | None. | |
| PA-682 | 100%: 22q12 | None. Episomal. | |
| Seraphine | 100%: 2p23 | None. Episomal. | |
| Group 2 | Maku | 100%: 13p11 on der (13)t(3;13) ins (3;13) | 2q31, 3p25, 3q24, 4p12, 4q31, 5p14, 8q21. Episomal. |
| AG876 | 80%: 7p22 53%: 13q21 | 1q12, 3q12~13, 3q24, 3q28, 4q33~35, 5q13, 7q31, 8q11, 10p11, 10q23, 10q26, 13q14, 16p11. Episomal. | |
| LY-67 | 80%: 13q21 67%: 7q21 20%: 12q21 13%: 3q26 | 1q43, 3p, 3q13, 4p13, 4q21, 5q13, 5q21, 6q26, 10q21, 15p. Episomal. | |
| Switzer | 67%: 2q21 33%: 2q31 27%: 1q31, 3q26 20%: 4q31~34 13%: 1p32, 13q21~31 | 2p12, 2q36, 4q12, 5q14~23, 5q34, 7p13, 7q11, 7q34, 8q21, 15q21, 18p11.2. Episomal. | |
| BL16 | 40%: 4q21 33%: 4q32 20%: 3q28, 10q11~21 13%: 1p11, 2p22, 2q12, 3q24~26, 7q31, 15q21, 16q11 | 2q21, 2q31, 3p25, 3q21, 4p15, 4q12, 4q26, 5q21, 5q34, 9q13, 9q22, 9q33, 10p12, 12q21, 14q11, 15q11, 16p11, 18q23. Episomal. | |
| JBL-2 | 33%: 15q13~15 20%: 13q21~31 13%: 1q31, 8q24 on der (8)t(2;8), 11p14, 17p12 | Yq12, 1q44, 4p15, 4q26, 5q12, 5q13, 6q23, 7p12, 7q21, 8q21 on t(2;8), 8q23, 11p11, 12p12, 12q21, 13q13, 15q25. Episomal. | |
| Rael | 33%: 4q32~34 20%: 2p11~12, 4q21~24, 4q26~28 13%: 4p12 | Xq10~11, 1p32~34, 2q22~24, 2q31, 5q13, 6q21, 6q24, 7p22, 12q13, 13q14, 14q22~23, 16q10~11, 18q21. Episomal. | |
| Akuba | 27%: 4p11~12 20%: 13q21 13%: 1q32, 4q12, 8q21, 15q13~14 | Xq21, 1p31, 2q22, 3q21, 3q24, 3q26, 4q21, 5p13, 6p21, 6q12, 6q21, 7q21, 12p13, 12q24, 16p11~12. Episomal. | |
| JI | 27%: 15q11~14 13%: 2q21~31, 12p12~13, 12q24 | 1q13 on der(6)t(1;6), 3q25, 4q25, 5q21~23, 7p21, 6p24 on der(6)t(1;6), 8q24, 12q21, 15q26, 17p13, 21q22. Episomal. | |
| Silfere | 20%: 2q31~33, 17q22~25 13%: 3q21~26 | 1p13, 1p21~31, 3p21, 4q28, 5q14, 5q21, 5q34, 7q31, 10q21, 12p11, 12q21, 16q24, 18q21, 20p12, 21q22. Episomal. | |
| Solubo | 20%: 13q14~22 13%: 4q34, 7p11~13 | 1p21~31, 2pter, 2q21, 2q33, 5p15, 9q21. Episomal. | |
| BL18 | 13%: 2q24~32, 3p13~21 | Xp10, Xq10, 1p31, 1q31, 2q36, 3q24~26, 4q21, 6q25, 7q21, 8q21, 10q23~25, 22q12. Episomal. | |
| LY-91 | 13%: 5q21, 7p21~22, 13q21~31, 15p11~13 | 1q32, 2p24~25, 2q12, 3p12, 3q13, 3q26~27, 4q28, 4q32~35, 5p14, 9p, 12p, 18q12. Episomal. | |
| Group 3 | EB-1 | none | 2q34~36, 3q29, 4q28, 5p13, 12q21, 16q24, 19q13. Episomal. |
| LY-47 | none | 1p13, 3p13, 3q25, 4p15, 11q22, 15p11, 18q12. Episomal. | |
| Salim Mwalim | none | Xp21, 1q31, 4q31, 7p on dup(7)(q11q22.3), 8q21. Episomal. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janjetovic, S.; Hinke, J.; Balachandran, S.; Akyüz, N.; Behrmann, P.; Bokemeyer, C.; Dierlamm, J.; Murga Penas, E.M. Non-Random Pattern of Integration for Epstein-Barr Virus with Preference for Gene-Poor Genomic Chromosomal Regions into the Genome of Burkitt Lymphoma Cell Lines. Viruses 2022, 14, 86. https://doi.org/10.3390/v14010086
Janjetovic S, Hinke J, Balachandran S, Akyüz N, Behrmann P, Bokemeyer C, Dierlamm J, Murga Penas EM. Non-Random Pattern of Integration for Epstein-Barr Virus with Preference for Gene-Poor Genomic Chromosomal Regions into the Genome of Burkitt Lymphoma Cell Lines. Viruses. 2022; 14(1):86. https://doi.org/10.3390/v14010086
Chicago/Turabian StyleJanjetovic, Snjezana, Juliane Hinke, Saranya Balachandran, Nuray Akyüz, Petra Behrmann, Carsten Bokemeyer, Judith Dierlamm, and Eva Maria Murga Penas. 2022. "Non-Random Pattern of Integration for Epstein-Barr Virus with Preference for Gene-Poor Genomic Chromosomal Regions into the Genome of Burkitt Lymphoma Cell Lines" Viruses 14, no. 1: 86. https://doi.org/10.3390/v14010086