


Viral Hijacking of BET Proteins


Abstract
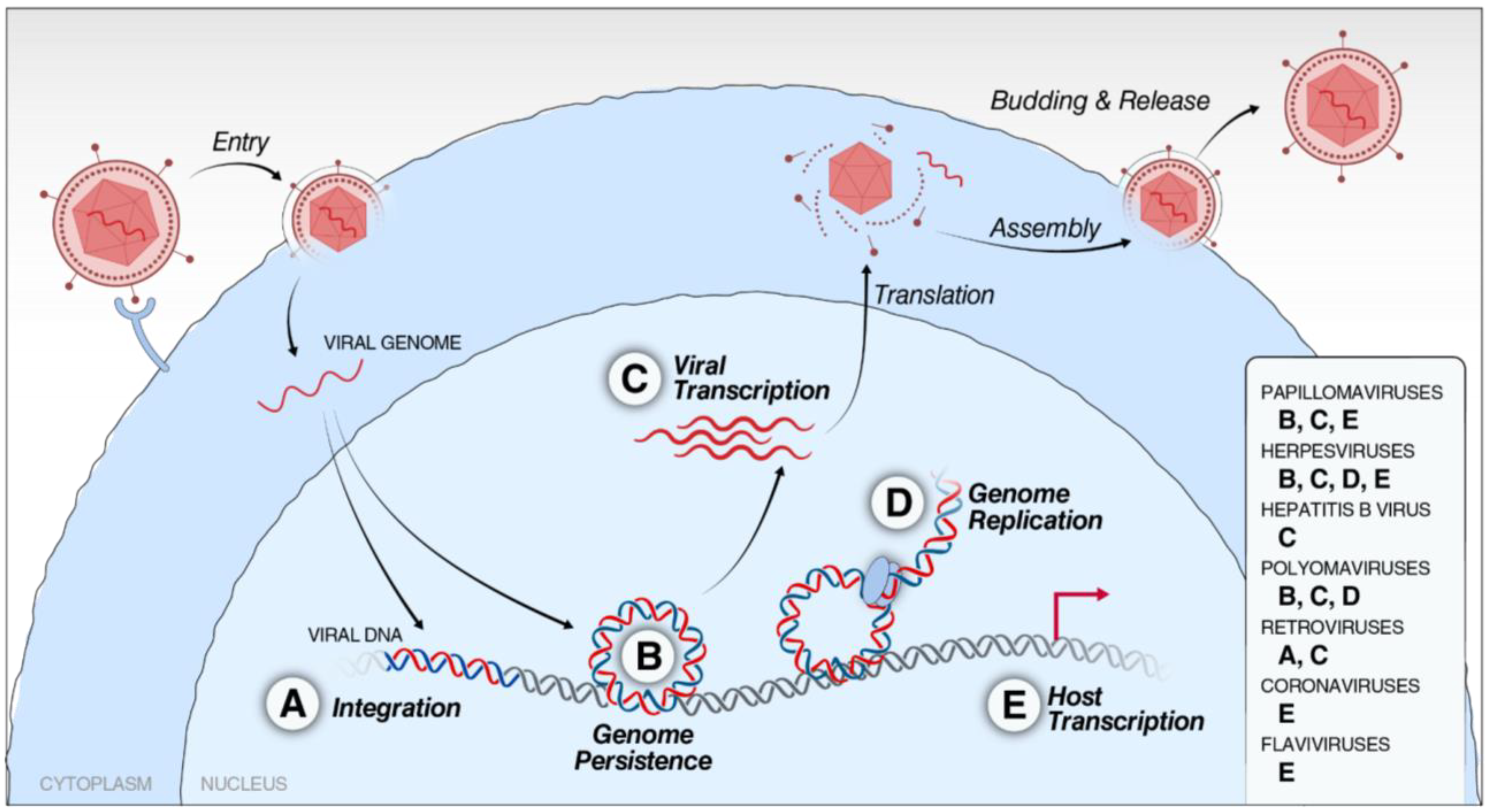
:1. Introduction
2. Domain Architecture of BET Proteins
3. Overlapping and Distinct Functions of BET Proteins
3.1. BRD2
3.2. BRD3
3.3. BRD4
4. Current BET Inhibitors Target All Family Members
5. The Role of BET Proteins in the Viral Life Cycle
5.1. Coordinators of Viral Genome Integration
5.2. Drivers of Viral Genome Persistence
5.3. Organizers of Viral Genome Replication
5.4. Accomplices in Viral Transcription
5.4.1. Recruitment of the BRD4:P-TEFb Complex
5.4.2. Competitive Binding to P-TEFb
5.5. Unwilling Disruptors of Host Transcription
6. Conclusions

{kind=link}
{kind=link}
| Virus | Viral Protein/Genome | BET Protein | BET Domain | Functions | References |
|---|---|---|---|---|---|
| MLV | IN | BRD2, BRD3, BRD4 | ET | Integration site selection | [54,55,56] |
| PERV A/C | IN | BRD2, BRD3, BRD4 | ET | Integration cofactor | [57] |
| Papillomavirus (HPV, CRPV, BPV) | E2 | BRD4 | C-terminus, NPS, BID | E2 stability, E2-mediated viral and host transcription, tethering viral genome to host chromatin | [9,59,63,64,65,66] |
| KSHV | LANA | BRD2, BRD3, BRD4 | ET | Tethering viral genome to host chromatin, LANA-mediated viral and host transcription | [6,71,130] |
| MHV-68 | orf73 | BRD2, BRD3, BRD4 | ET | Tethering viral genome to chromatin, orf73-mediated host transcription | [112] |
| EBV | EBNA1 OriLyt * | BRD2, BRD3, BRD4 | (C-terminus) | EBNA1-mediated viral transcription, tethering viral genome to host chromatin | [74,85] |
| HCMV | HCMV promoters * | BRD4 | PID | Competence for P-TEFb | [97,98] |
| MCPyV | LT | BRD4 | ET | Viral genome replication | [77] |
| JCPyV | NCRR * | BRD4 | (BDs, PID) | Viral transcription and genome replication | [78] |
| RacPyV | RacPyV genome * | BRD4 | (BDs, PID) | Viral transcription and genome replication, tether genome to host chromatin | [75] |
| HBV | HBV genome * | BRD4 | BD-independent | Viral transcription | [93] |
| HTLV-1 | Tax ** | BRD4 | PID | Competence for P-TEFb | [57,99] |
| HIV | Tat ** LTR * | BRD4 | PID BDs | Competence for P-TEFb Tat-mediated transcription, enforce viral latency | [103,105,106,107,131] |
| YFV | Capsid | BRD2, BRD3, BRD4 | BDs | Host transcription | [108] |
| SARS-CoV-2 | E | BRD2, BRD4 | BDs, ET | Host transcription | [109,110] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kulikowski, E.; Rakai, B.D.; Wong, N.C.W. Inhibitors of Bromodomain and Extra-Terminal Proteins for Treating Multiple Human Diseases. Med. Res. Rev. 2021, 41, 223–245. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Solomon, P.D.; Walshe, J.L.; Ford, D.J.; Wilkinson-White, L.; Payne, R.J.; Low, J.K.K.; Mackay, J.P. BET-Family Bromodomains Can Recognize Diacetylated Sequences from Transcription Factors Using a Conserved Mechanism. Biochemistry 2021, 60, 648–662. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.-P.; Picaud, S.; Fujisawa, T.; Hou, H.; Savitsky, P.; Uusküla-Reimand, L.; Gupta, G.D.; Abdouni, H.; Lin, Z.-Y.; Tucholska, M.; et al. Interactome Rewiring Following Pharmacological Targeting of BET Bromodomains. Mol. Cell 2019, 73, 621–638.e17. [Google Scholar] [CrossRef] [Green Version]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and Ligand of a Histone Acetyltransferase Bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [CrossRef]
- Josling, G.A.; Selvarajah, S.A.; Petter, M.; Duffy, M.F. The Role of Bromodomain Proteins in Regulating Gene Expression. Genes 2012, 3, 320–343. [Google Scholar] [CrossRef] [Green Version]
- Wai, D.C.C.; Szyszka, T.N.; Campbell, A.E.; Kwong, C.; Wilkinson-White, L.E.; Silva, A.P.G.; Low, J.K.K.; Kwan, A.H.; Gamsjaeger, R.; Chalmers, J.D.; et al. The BRD3 ET Domain Recognizes a Short Peptide Motif through a Mechanism That Is Conserved across Chromatin Remodelers and Transcriptional Regulators. J. Biol. Chem. 2018, 293, 7160–7175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, S.; Cho, S.; Zeng, L.; Zhang, Q.; Kaehlcke, K.; Mak, L.; Lau, J.; Bisgrove, D.; Schnölzer, M.; Verdin, E.; et al. Two-Pronged Binding with Bromodomain-Containing Protein 4 Liberates Positive Transcription Elongation Factor B from Inactive Ribonucleoprotein Complexes. J. Biol. Chem. 2012, 287, 1090–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawa, C.; Nedea, E.; Krogan, N.; Wada, T.; Handa, H.; Greenblatt, J.; Buratowski, S. Bromodomain Factor 1 (Bdf1) Is Phosphorylated by Protein Kinase CK2. Mol. Cell. Biol. 2004, 24, 4734–4742. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.-Y.; Nin, D.S.; Lee, A.-Y.; Simanski, S.; Kodadek, T.; Chiang, C.-M. BRD4 Phosphorylation Regulates HPV E2-Mediated Viral Transcription, Origin Replication, and Cellular MMP-9 Expression. Cell Rep. 2016, 16, 1733–1748. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.-Y.; Lee, A.-Y.; Lai, H.-T.; Zhang, H.; Chiang, C.-M. Phospho Switch Triggers Brd4 Chromatin Binding and Activator Recruitment for Gene-Specific Targeting. Mol. Cell 2013, 49, 843–857. [Google Scholar] [CrossRef]
- Malvezzi, F.; Stubbs, C.J.; Jowitt, T.A.; Dale, I.L.; Guo, X.; DeGnore, J.P.; Degliesposti, G.; Skehel, J.M.; Bannister, A.J.; McAlister, M.S. Phosphorylation-Dependent BRD4 Dimerization and Implications for Therapeutic Inhibition of BET Family Proteins. Commun. Biol. 2021, 4, 1273. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gutierrez, P.; Mundi, M.; Garcia-Dominguez, M. Association of Bromodomain BET Proteins with Chromatin Requires Dimerization through the Conserved Motif B. J. Cell Sci. 2012, 125, 3671–3680. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.L.; Kim, C.; Zhou, M.-M. The Functions of BET Proteins in Gene Transcription of Biology and Diseases. Front. Mol. Biosci. 2021, 8, 728777. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wu, R.; Tang, D.; Kang, R. The BET Family in Immunity and Disease. Signal Transduct. Target Ther. 2021, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Denis, G.V.; Green, M.R. A Novel, Mitogen-Activated Nuclear Kinase Is Related to a Drosophila Developmental Regulator. Genes Dev. 1996, 10, 261–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denis, G.V.; Vaziri, C.; Guo, N.; Faller, D.V. RING3 Kinase Transactivates Promoters of Cell Cycle Regulatory Genes through E2F. Cell Growth Differ. 2000, 11, 417–424. [Google Scholar]
- Xu, L.; Chen, Y.; Mayakonda, A.; Koh, L.; Chong, Y.K.; Buckley, D.L.; Sandanaraj, E.; Lim, S.W.; Lin, R.Y.-T.; Ke, X.-Y.; et al. Targetable BET Proteins- and E2F1-Dependent Transcriptional Program Maintains the Malignancy of Glioblastoma. Proc. Natl. Acad. Sci. USA 2018, 115, E5086–E5095. [Google Scholar] [CrossRef] [Green Version]
- LeRoy, G.; Rickards, B.; Flint, S.J. The Double Bromodomain Proteins Brd2 and Brd3 Couple Histone Acetylation to Transcription. Mol. Cell 2008, 30, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Denis, G.V.; McComb, M.E.; Faller, D.V.; Sinha, A.; Romesser, P.B.; Costello, C.E. Identification of Transcription Complexes That Contain the Double Bromodomain Protein Brd2 and Chromatin Remodeling Machines. J. Proteome Res. 2006, 5, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Tadeo, X.; Hou, H.; Tu, P.G.; Thompson, J.; Yates, J.R., 3rd; Jia, S. Epe1 Recruits BET Family Bromodomain Protein Bdf2 to Establish Heterochromatin Boundaries. Genes Dev. 2013, 27, 1886–1902. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.C.; Gilgenast, T.G.; Bartman, C.R.; Edwards, C.R.; Stonestrom, A.J.; Huang, P.; Emerson, D.J.; Evans, P.; Werner, M.T.; Keller, C.A.; et al. The BET Protein BRD2 Cooperates with CTCF to Enforce Transcriptional and Architectural Boundaries. Mol. Cell 2017, 66, 102–116.e7. [Google Scholar] [CrossRef] [PubMed]
- Lamonica, J.M.; Deng, W.; Kadauke, S.; Campbell, A.E.; Gamsjaeger, R.; Wang, H.; Cheng, Y.; Billin, A.N.; Hardison, R.C.; Mackay, J.P.; et al. Bromodomain Protein Brd3 Associates with Acetylated GATA1 to Promote Its Chromatin Occupancy at Erythroid Target Genes. Proc. Natl. Acad. Sci. USA 2011, 108, E159–E168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamsjaeger, R.; Webb, S.R.; Lamonica, J.M.; Billin, A.; Blobel, G.A.; Mackay, J.P. Structural Basis and Specificity of Acetylated Transcription Factor GATA1 Recognition by BET Family Bromodomain Protein Brd3. Mol. Cell. Biol. 2011, 31, 2632–2640. [Google Scholar] [CrossRef] [Green Version]
- Stonestrom, A.J.; Hsu, S.C.; Jahn, K.S.; Huang, P.; Keller, C.A.; Giardine, B.M.; Kadauke, S.; Campbell, A.E.; Evans, P.; Hardison, R.C.; et al. Functions of BET Proteins in Erythroid Gene Expression. Blood 2015, 125, 2825–2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneshvar, K.; Ardehali, M.B.; Klein, I.A.; Hsieh, F.-K.; Kratkiewicz, A.J.; Mahpour, A.; Cancelliere, S.O.L.; Zhou, C.; Cook, B.M.; Li, W.; et al. lncRNA DIGIT and BRD3 Protein Form Phase-Separated Condensates to Regulate Endoderm Differentiation. Nat. Cell Biol. 2020, 22, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Nakamura, T.; Fu, Y.; Lazar, Z.; Spector, D.L. Gene Bookmarking Accelerates the Kinetics of Post-Mitotic Transcriptional Re-Activation. Nat. Cell Biol. 2011, 13, 1295–1304. [Google Scholar] [CrossRef]
- Dey, A.; Ellenberg, J.; Farina, A.; Coleman, A.E.; Maruyama, T.; Sciortino, S.; Lippincott-Schwartz, J.; Ozato, K. A Bromodomain Protein, MCAP, Associates with Mitotic Chromosomes and Affects G(2)-to-M Transition. Mol. Cell. Biol. 2000, 20, 6537–6549. [Google Scholar] [CrossRef]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.-S.; Brady, J.N.; Ozato, K. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Devaiah, B.N.; Lewis, B.A.; Cherman, N.; Hewitt, M.C.; Albrecht, B.K.; Robey, P.G.; Ozato, K.; Sims, R.J., 3rd; Singer, D.S. BRD4 Is an Atypical Kinase That Phosphorylates serine2 of the RNA Polymerase II Carboxy-Terminal Domain. Proc. Natl. Acad. Sci. USA 2012, 109, 6927–6932. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 Extraterminal Domain Confers Transcription Activation Independent of pTEFb by Recruiting Multiple Proteins, Including NSD3. Mol. Cell. Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [Green Version]
- Sakamaki, J.-I.; Wilkinson, S.; Hahn, M.; Tasdemir, N.; O’Prey, J.; Clark, W.; Hedley, A.; Nixon, C.; Long, J.S.; New, M.; et al. Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol. Cell 2017, 66, 517–532.e9. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Case-Borden, C.; Gegonne, A.; Hsu, C.H.; Chen, Q.; Meerzaman, D.; Dey, A.; Ozato, K.; Singer, D.S. BRD4 Is a Histone Acetyltransferase That Evicts Nucleosomes from Chromatin. Nat. Struct. Mol. Biol. 2016, 23, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vershinin, Z.; Feldman, M.; Werner, T.; Weil, L.E.; Kublanovsky, M.; Abaev-Schneiderman, E.; Sklarz, M.; Lam, E.Y.N.; Alasad, K.; Picaud, S.; et al. BRD4 Methylation by the Methyltransferase SETD6 Regulates Selective Transcription to Control mRNA Translation. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Erber, L.; Luo, A.; Chen, Y. Targeted and Interactome Proteomics Revealed the Role of PHD2 in Regulating BRD4 Proline Hydroxylation. Mol. Cell. Proteom. 2019, 18, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yan, Y.; Wang, D.; Ding, D.; Ma, T.; Ye, Z.; Jimenez, R.; Wang, L.; Wu, H.; Huang, H. DUB3 Promotes BET Inhibitor Resistance and Cancer Progression by Deubiquitinating BRD4. Mol. Cell 2018, 71, 592–605.e4. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Baek, G.; Ramanand, S.G.; Sharp, A.; Gao, Y.; Yuan, W.; Welti, J.; Rodrigues, D.N.; Dolling, D.; Figueiredo, I.; et al. BRD4 Promotes DNA Repair and Mediates the Formation of TMPRSS2-ERG Gene Rearrangements in Prostate Cancer. Cell Rep. 2018, 22, 796–808. [Google Scholar] [CrossRef] [Green Version]
- Stanlie, A.; Yousif, A.S.; Akiyama, H.; Honjo, T.; Begum, N.A. Chromatin Reader Brd4 Functions in Ig Class Switching as a Repair Complex Adaptor of Nonhomologous End-Joining. Mol. Cell 2014, 55, 97–110. [Google Scholar] [CrossRef] [Green Version]
- Floyd, S.R.; Pacold, M.E.; Huang, Q.; Clarke, S.M.; Lam, F.C.; Cannell, I.G.; Bryson, B.D.; Rameseder, J.; Lee, M.J.; Blake, E.J.; et al. The Bromodomain Protein Brd4 Insulates Chromatin from DNA Damage Signalling. Nature 2013, 498, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Tang, P.; Zhang, J.; Liu, J.; Chiang, C.-M.; Ouyang, L. Targeting Bromodomain and Extraterminal Proteins for Drug Discovery: From Current Progress to Technological Development. J. Med. Chem. 2021, 64, 2419–2435. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective Inhibition of BET Bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-Cell Tumor Models and Synergizes with Targeted Drugs. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Baldan, F.; Allegri, L.; Lazarevic, M.; Catia, M.; Milosevic, M.; Damante, G.; Milasin, J. Biological and Molecular Effects of Bromodomain and Extra-Terminal (BET) Inhibitors JQ1, IBET-151, and IBET-762 in OSCC Cells. J. Oral Pathol. Med. 2019, 48, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhou, M.M. Bromodomain: An Acetyl-Lysine Binding Domain. FEBS Lett. 2002, 513, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Meslamani, J.; Smith, S.G.; Sanchez, R.; Zhou, M.-M. Structural Features and Inhibitors of Bromodomains. Drug Discov. Today Technol. 2016, 19, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, Y.; Gerona-Navarro, G.; Osman, R.; Zhou, M.-M. In Silico Design and Molecular Basis for the Selectivity of Olinone toward the First over the Second Bromodomain of BRD4. Proteins 2020, 88, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Gacias, M.; Gerona-Navarro, G.; Plotnikov, A.N.; Zhang, G.; Zeng, L.; Kaur, J.; Moy, G.; Rusinova, E.; Rodriguez, Y.; Matikainen, B.; et al. Selective Chemical Modulation of Gene Transcription Favors Oligodendrocyte Lineage Progression. Chem. Biol. 2014, 21, 841–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.-W.; Bamborough, P.; Petretich, M.; et al. Selective Targeting of BD1 and BD2 of the BET Proteins in Cancer and Immunoinflammation. Science 2020, 368, 387–394. [Google Scholar] [CrossRef]
- Lin, X.; Huang, X.; Bellin, R.; Faivre, E.; Hessler, P.; Lam, L.; Bui, M.H.; Wilcox, D.; Uziel, T.; Ferguson, D.C.; et al. Abstract 800: ABBV-744, a First-in-Class and Highly Selective Inhibitor of the Second Bromodomain of BET Family Proteins, Displays Robust Activities in Preclinical Models of Acute Myelogenous Leukemia. Cancer Res. 2018, 78, 800. [Google Scholar] [CrossRef]
- McLure, K.G.; Gesner, E.M.; Tsujikawa, L.; Kharenko, O.A.; Attwell, S.; Campeau, E.; Wasiak, S.; Stein, A.; White, A.; Fontano, E.; et al. RVX-208, an Inducer of ApoA-I in Humans, Is a BET Bromodomain Antagonist. PLoS ONE 2013, 8, e83190. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.E.; Mayer, A.; Buckley, D.L.; Erb, M.A.; Roderick, J.E.; Vittori, S.; Reyes, J.M.; di Iulio, J.; Souza, A.; Ott, C.J.; et al. BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol. Cell 2017, 67, 5–18.e19. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Hu, J.; Xu, F.; Chen, Z.; Bai, L.; Fernandez-Salas, E.; Lin, M.; Liu, L.; Yang, C.-Y.; Zhao, Y.; et al. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J. Med. Chem. 2018, 61, 462–481. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M. Transcription Start Regions in the Human Genome Are Favored Targets for MLV Integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.S.; Maetzig, T.; Maertens, G.N.; Sharif, A.; Rothe, M.; Weidner-Glunde, M.; Galla, M.; Schambach, A.; Cherepanov, P.; Schulz, T.F. Bromo- and Extraterminal Domain Chromatin Regulators Serve as Cofactors for Murine Leukemia Virus Integration. J. Virol. 2013, 87, 12721–12736. [Google Scholar] [CrossRef] [Green Version]
- De Rijck, J.; de Kogel, C.; Demeulemeester, J.; Vets, S.; El Ashkar, S.; Malani, N.; Bushman, F.D.; Landuyt, B.; Husson, S.J.; Busschots, K.; et al. The BET Family of Proteins Targets Moloney Murine Leukemia Virus Integration near Transcription Start Sites. Cell Rep. 2013, 5, 886–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Larue, R.C.; Plumb, M.R.; Malani, N.; Male, F.; Slaughter, A.; Kessl, J.J.; Shkriabai, N.; Coward, E.; Aiyer, S.S.; et al. BET Proteins Promote Efficient Murine Leukemia Virus Integration at Transcription Start Sites. Proc. Natl. Acad. Sci. USA 2013, 110, 12036–12041. [Google Scholar] [CrossRef] [Green Version]
- Gallay, K.; Blot, G.; Chahpazoff, M.; Yajjou-Hamalian, H.; Confort, M.-P.; De Boisséson, C.; Leroux, A.; Luengo, C.; Fiorini, F.; Lavigne, M.; et al. In Vitro, in Cellulo and Structural Characterizations of the Interaction between the Integrase of Porcine Endogenous Retrovirus A/C and Proteins of the BET Family. Virology 2019, 532, 69–81. [Google Scholar] [CrossRef]
- Serrao, E.; Ballandras-Colas, A.; Cherepanov, P.; Maertens, G.N.; Engelman, A.N. Key Determinants of Target DNA Recognition by Retroviral Intasomes. Retrovirology 2015, 12, 39. [Google Scholar] [CrossRef] [Green Version]
- Iftner, T.; Haedicke-Jarboui, J.; Wu, S.-Y.; Chiang, C.-M. Involvement of Brd4 in Different Steps of the Papillomavirus Life Cycle. Virus Res. 2017, 231, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Abbate, E.A.; Voitenleitner, C.; Botchan, M.R. Structure of the Papillomavirus DNA-Tethering Complex E2:Brd4 and a Peptide That Ablates HPV Chromosomal Association. Mol. Cell 2006, 24, 877–889. [Google Scholar] [CrossRef]
- Muller, M.; Jacob, Y.; Jones, L.; Weiss, A.; Brino, L.; Chantier, T.; Lotteau, V.; Favre, M.; Demeret, C. Large Scale Genotype Comparison of Human Papillomavirus E2-Host Interaction Networks Provides New Insights for e2 Molecular Functions. PLoS Pathog. 2012, 8, e1002761. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.G.; Colf, L.A.; McBride, A.A. Variations in the Association of Papillomavirus E2 Proteins with Mitotic Chromosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 1047–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, M.K.; McPhillips, M.G.; Ozato, K.; McBride, A.A. The Mitotic Chromosome Binding Activity of the Papillomavirus E2 Protein Correlates with Interaction with the Cellular Chromosomal Protein, Brd4. J. Virol. 2005, 79, 4806–4818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, M.A.; Balogh, K.K.; Brendle, S.A.; Campbell, C.A.; Chen, M.X.; Furze, R.C.; Harada, I.L.; Holyer, I.D.; Kumar, U.; Lee, K.; et al. BET Bromodomain Inhibitors Show Anti-Papillomavirus Activity In Vitro and Block CRPV Wart Growth In Vivo. Antivir. Res. 2018, 154, 158–165. [Google Scholar] [CrossRef]
- Ilves, I.; Mäemets, K.; Silla, T.; Janikson, K.; Ustav, M. Brd4 Is Involved in Multiple Processes of the Bovine Papillomavirus Type 1 Life Cycle. J. Virol. 2006, 80, 3660–3665. [Google Scholar] [CrossRef] [Green Version]
- You, J.; Schweiger, M.-R.; Howley, P.M. Inhibition of E2 Binding to Brd4 Enhances Viral Genome Loss and Phenotypic Reversion of Bovine Papillomavirus-Transformed Cells. J. Virol. 2005, 79, 14956–14961. [Google Scholar] [CrossRef] [Green Version]
- McPhillips, M.G.; Oliveira, J.G.; Spindler, J.E.; Mitra, R.; McBride, A.A. Brd4 Is Required for e2-Mediated Transcriptional Activation but Not Genome Partitioning of All Papillomaviruses. J. Virol. 2006, 80, 9530–9543. [Google Scholar] [CrossRef] [Green Version]
- McBride, A.A.; Jang, M.K. Current Understanding of the Role of the Brd4 Protein in the Papillomavirus Lifecycle. Viruses 2013, 5, 1374–1394. [Google Scholar] [CrossRef] [Green Version]
- Poddar, A.; Reed, S.C.; McPhillips, M.G.; Spindler, J.E.; McBride, A.A. The Human Papillomavirus Type 8 E2 Tethering Protein Targets the Ribosomal DNA Loci of Host Mitotic Chromosomes. J. Virol. 2009, 83, 640–650. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, M.M.; Boner, W.; Morgan, I.M. TopBP1 Regulates Human Papillomavirus Type 16 E2 Interaction with Chromatin. J. Virol. 2007, 81, 4338–4342. [Google Scholar] [CrossRef] [Green Version]
- You, J.; Srinivasan, V.; Denis, G.V.; Harrington, W.J., Jr.; Ballestas, M.E.; Kaye, K.M.; Howley, P.M. Kaposi’s Sarcoma-Associated Herpesvirus Latency-Associated Nuclear Antigen Interacts with Bromodomain Protein Brd4 on Host Mitotic Chromosomes. J. Virol. 2006, 80, 8909–8919. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, K.; Kiss, C.; Platt, G.M.; Simpson, G.R.; Kashuba, E.; Klein, G.; Schulz, T.F.; Szekely, L. Latent Nuclear Antigen of Kaposi’s Sarcoma Herpesvirus/human Herpesvirus-8 Induces and Relocates RING3 to Nuclear Heterochromatin Regions. J. Gen. Virol. 2002, 83, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Szekely, L.; Kiss, C.; Mattsson, K.; Kashuba, E.; Pokrovskaja, K.; Juhasz, A.; Holmvall, P.; Klein, G. Human Herpesvirus-8-Encoded LNA-1 Accumulates in Heterochromatin- Associated Nuclear Bodies. J. Gen. Virol. 1999, 80 Pt 11, 2889–2900. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Wang, S.; Nguyen, T.; Shire, K.; Frappier, L. The EBNA1 Protein of Epstein-Barr Virus Functionally Interacts with Brd4. J. Virol. 2008, 82, 12009–12019. [Google Scholar] [CrossRef] [Green Version]
- Church, M.E.; Estrada, M.; Leutenegger, C.M.; Dela Cruz, F.N.; Pesavento, P.A.; Woolard, K.D. BRD4 Is Associated with Raccoon Polyomavirus Genome and Mediates Viral Gene Transcription and Maintenance of a Stem Cell State in Neuroglial Tumour Cells. J. Gen. Virol. 2016, 97, 2939–2948. [Google Scholar] [CrossRef]
- Decaprio, J.A.; Imperiale, M.J.; Major, E.O. Polyomaviruses. In Fields Virology; Howley, P.M., Knipe, D.M., Cohen, J.L., Damania, B.A., Eds.; Wolters Kluwer: Singapore, 2013; Volume 2, pp. 1633–1661. ISBN 9781451105636. [Google Scholar]
- Wang, X.; Li, J.; Schowalter, R.M.; Jiao, J.; Buck, C.B.; You, J. Bromodomain Protein Brd4 Plays a Key Role in Merkel Cell Polyomavirus DNA Replication. PLoS Pathog. 2012, 8, e1003021. [Google Scholar] [CrossRef] [Green Version]
- Wollebo, H.S.; Bellizzi, A.; Cossari, D.H.; Salkind, J.; Safak, M.; White, M.K. The Brd4 Acetyllysine-Binding Protein Is Involved in Activation of Polyomavirus JC. J. Neurovirol. 2016, 22, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, N.; Chen, D.; McBride, A.A. Papillomaviruses Use Recombination-Dependent Replication to Vegetatively Amplify Their Genomes in Differentiated Cells. PLoS Pathog. 2013, 9, e1003321. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human Papillomaviruses Recruit Cellular DNA Repair and Homologous Recombination Factors to Viral Replication Centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [Green Version]
- Moody, C.A.; Laimins, L.A. Human Papillomaviruses Activate the ATM DNA Damage Pathway for Viral Genome Amplification upon Differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Helfer, C.M.; Pancholi, N.; Bradner, J.E.; You, J. Recruitment of Brd4 to the Human Papillomavirus Type 16 DNA Replication Complex Is Essential for Replication of Viral DNA. J. Virol. 2013, 87, 3871–3884. [Google Scholar] [CrossRef] [PubMed]
- DeSmet, M.; Jose, L.; Isaq, N.; Androphy, E.J. Phosphorylation of a Conserved Tyrosine in the Papillomavirus E2 Protein Regulates Brd4 Binding and Viral Replication. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakakibara, N.; Chen, D.; Jang, M.K.; Kang, D.W.; Luecke, H.F.; Wu, S.-Y.; Chiang, C.-M.; McBride, A.A. Brd4 Is Displaced from HPV Replication Factories as They Expand and Amplify Viral DNA. PLoS Pathog. 2013, 9, e1003777. [Google Scholar] [CrossRef] [PubMed]
- Keck, K.M.; Moquin, S.A.; He, A.; Fernandez, S.G.; Somberg, J.J.; Liu, S.M.; Martinez, D.M.; Miranda, J.L. Bromodomain and Extraterminal Inhibitors Block the Epstein-Barr Virus Lytic Cycle at Two Distinct Steps. J. Biol. Chem. 2017, 292, 13284–13295. [Google Scholar] [CrossRef] [Green Version]
- Chin, M.T.; Hirochika, R.; Hirochika, H.; Broker, T.R.; Chow, L.T. Regulation of Human Papillomavirus Type 11 Enhancer and E6 Promoter by Activating and Repressing Proteins from the E2 Open Reading Frame: Functional and Biochemical Studies. J. Virol. 1988, 62, 2994–3002. [Google Scholar] [CrossRef] [Green Version]
- Cripe, T.P.; Haugen, T.H.; Turk, J.P.; Tabatabai, F.; Schmid, P.G., 3rd; Dürst, M.; Gissmann, L.; Roman, A.; Turek, L.P. Transcriptional Regulation of the Human Papillomavirus-16 E6-E7 Promoter by a Keratinocyte-Dependent Enhancer, and by Viral E2 Trans-Activator and Repressor Gene Products: Implications for Cervical Carcinogenesis. EMBO J. 1987, 6, 3745–3753. [Google Scholar] [CrossRef]
- Spalholz, B.A.; Yang, Y.C.; Howley, P.M. Transactivation of a Bovine Papilloma Virus Transcriptional Regulatory Element by the E2 Gene Product. Cell 1985, 42, 183–191. [Google Scholar] [CrossRef]
- Helfer, C.M.; Yan, J.; You, J. The Cellular Bromodomain Protein Brd4 Has Multiple Functions in E2-Mediated Papillomavirus Transcription Activation. Viruses 2014, 6, 3228–3249. [Google Scholar] [CrossRef] [Green Version]
- Sénéchal, H.; Poirier, G.G.; Coulombe, B.; Laimins, L.A.; Archambault, J. Amino Acid Substitutions That Specifically Impair the Transcriptional Activity of Papillomavirus E2 Affect Binding to the Long Isoform of Brd4. Virology 2007, 358, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Schweiger, M.-R.; You, J.; Howley, P.M. Bromodomain Protein 4 Mediates the Papillomavirus E2 Transcriptional Activation Function. J. Virol. 2006, 80, 4276–4285. [Google Scholar] [CrossRef] [Green Version]
- Schweiger, M.-R.; Ottinger, M.; You, J.; Howley, P.M. Brd4-Independent Transcriptional Repression Function of the Papillomavirus e2 Proteins. J. Virol. 2007, 81, 9612–9622. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.C.; Dai, Q.; Luo, Z.; Wang, Y.; Chong, R.H.-H.; Tan, Y.J.; Xie, W.; Lee, G.-H.; Lin, C. Transcriptional Elongation Control of Hepatitis B Virus Covalently Closed Circular DNA Transcription by Super Elongation Complex and BRD4. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groves, I.J.; Sinclair, J.H.; Wills, M.R. Bromodomain Inhibitors as Therapeutics for Herpesvirus-Related Disease: All BETs Are Off? Front. Cell. Infect. Microbiol. 2020, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Zhang, W.; Chen, X.; Ma, Y.; Dai, Y.; Fan, Y.; Hou, Y.; Tan, R.X.; Li, E. An Epigenetic Compound Library Screen Identifies BET Inhibitors That Promote HSV-1 and -2 Replication by Bridging P-TEFb to Viral Gene Promoters through BRD4. PLoS Pathog. 2016, 12, e1005950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfonso-Dunn, R.; Turner, A.-M.W.; Jean Beltran, P.M.; Arbuckle, J.H.; Budayeva, H.G.; Cristea, I.M.; Kristie, T.M. Transcriptional Elongation of HSV Immediate Early Genes by the Super Elongation Complex Drives Lytic Infection and Reactivation from Latency. Cell Host Microbe 2017, 21, 507–517.e5. [Google Scholar] [CrossRef] [Green Version]
- Groves, I.J.; Jackson, S.E.; Poole, E.L.; Nachshon, A.; Rozman, B.; Schwartz, M.; Prinjha, R.K.; Tough, D.F.; Sinclair, J.H.; Wills, M.R. Bromodomain Proteins Regulate Human Cytomegalovirus Latency and Reactivation Allowing Epigenetic Therapeutic Intervention. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Kapasi, A.J.; Spector, D.H. Inhibition of the Cyclin-Dependent Kinases at the Beginning of Human Cytomegalovirus Infection Specifically Alters the Levels and Localization of the RNA Polymerase II Carboxyl-Terminal Domain Kinases cdk9 and cdk7 at the Viral Transcriptosome. J. Virol. 2008, 82, 394–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Lu, H.; Park, H.; Wilson-Chiru, J.; Linton, R.; Brady, J.N. Tax Interacts with P-TEFb in a Novel Manner to Stimulate Human T-Lymphotropic Virus Type 1 Transcription. J. Virol. 2006, 80, 4781–4791. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Yik, J.H.N.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for Stimulation of Transcriptional Elongation by the Bromodomain Protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Huo, L.; Li, D.; Sun, X.; Shi, X.; Karna, P.; Yang, W.; Liu, M.; Qiao, W.; Aneja, R.; Zhou, J. Regulation of Tat Acetylation and Transactivation Activity by the Microtubule-Associated Deacetylase HDAC6. J. Biol. Chem. 2011, 286, 9280–9286. [Google Scholar] [CrossRef] [Green Version]
- Kiernan, R.E.; Vanhulle, C.; Schiltz, L.; Adam, E.; Xiao, H.; Maudoux, F.; Calomme, C.; Burny, A.; Nakatani, Y.; Jeang, K.-T.; et al. HIV-1 Tat Transcriptional Activity Is Regulated by Acetylation. EMBO J. 1999, 18, 6106–6118. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Schnölzer, M.; Garnica, J.; Fischle, W.; Emiliani, S.; Rackwitz, H.R.; Verdin, E. Acetylation of the HIV-1 Tat Protein by p300 Is Important for Its Transcriptional Activity. Curr. Biol. 1999, 9, 1489–1492. [Google Scholar] [CrossRef] [Green Version]
- Pagans, S.; Pedal, A.; North, B.J.; Kaehlcke, K.; Marshall, B.L.; Dorr, A.; Hetzer-Egger, C.; Henklein, P.; Frye, R.; McBurney, M.W.; et al. SIRT1 Regulates HIV Transcription via Tat Deacetylation. PLoS Biol. 2005, 3, e41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-Interacting Domain of BRD4 Inhibits HIV Transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef] [Green Version]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.-C.; Planelles, V.; Bradner, J.E.; et al. BET Bromodomain-Targeting Compounds Reactivate HIV from Latency via a Tat-Independent Mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef]
- Conrad, R.J.; Fozouni, P.; Thomas, S.; Sy, H.; Zhang, Q.; Zhou, M.-M.; Ott, M. The Short Isoform of BRD4 Promotes HIV-1 Latency by Engaging Repressive SWI/SNF Chromatin-Remodeling Complexes. Mol. Cell 2017, 67, 1001–1012.e6. [Google Scholar] [CrossRef] [Green Version]
- Mourão, D.; Chen, S.; Schaefer, U.; Bozzacco, L.; Carneiro, L.A.; Gerber, A.; Adura, C.; Dill, B.D.; Molina, H.; Carroll, T.; et al. A Histone-like Motif in Yellow Fever Virus Contributes to Viral Replication. bioRxiv 2020, 2020.05.05.078782. [Google Scholar]
- Chen, I.P.; Longbotham, J.E.; McMahon, S.; Suryawanshi, R.K.; Khalid, M.M.; Taha, T.Y.; Tabata, T.; Hayashi, J.M.; Soveg, F.W.; Carlson-Stevermer, J.; et al. Viral E Protein Neutralizes BET Protein-Mediated Post-Entry Antagonism of SARS-CoV-2. Cell Rep. 2022, 40, 111088. [Google Scholar] [CrossRef]
- Vann, K.R.; Acharya, A.; Jang, S.M.; Lachance, C.; Zandian, M.; Holt, T.A.; Smith, A.L.; Pandey, K.; Durden, D.L.; El-Gamal, D.; et al. Binding of the SARS-CoV-2 Envelope E Protein to Human BRD4 Is Essential for Infection. Structure 2022, 30, 1224–1232. [Google Scholar] [CrossRef]
- de Wilde, J.; De-Castro Arce, J.; Snijders, P.J.F.; Meijer, C.J.L.M.; Rösl, F.; Steenbergen, R.D.M. Alterations in AP-1 and AP-1 Regulatory Genes during HPV-Induced Carcinogenesis. Cell. Oncol. 2008, 30, 77–87. [Google Scholar] [CrossRef]
- Ottinger, M.; Pliquet, D.; Christalla, T.; Frank, R.; Stewart, J.P.; Schulz, T.F. The Interaction of the Gammaherpesvirus 68 orf73 Protein with Cellular BET Proteins Affects the Activation of Cell Cycle Promoters. J. Virol. 2009, 83, 4423–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehm, D.; Conrad, R.J.; Ott, M. Bromodomain Proteins in HIV Infection. Viruses 2013, 5, 1571–1586. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; MacKenzie, K.R.; Jain, P.; Santini, C.; Young, D.W.; Matzuk, M.M. Metabolism of JQ1, an Inhibitor of Bromodomain and Extra Terminal Bromodomain Proteins, in Human and Mouse Liver Microsomes. Biol. Reprod. 2020, 103, 427–436. [Google Scholar] [CrossRef]
- Zhu, J.; Gaiha, G.D.; John, S.P.; Pertel, T.; Chin, C.R.; Gao, G.; Qu, H.; Walker, B.D.; Elledge, S.J.; Brass, A.L. Reactivation of Latent HIV-1 by Inhibition of BRD4. Cell Rep. 2012, 2, 807–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET Bromodomain Inhibition as a Novel Strategy for Reactivation of HIV-1. J. Leukoc. Biol. 2012, 92, 1147–1154. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Shen, Y.; Yang, H.; Wang, Y.; Jiang, Z.; Yang, X.; Zhong, Y.; Pan, H.; Xu, J.; Lu, H.; et al. BET Inhibitors RVX-208 and PFI-1 Reactivate HIV-1 from Latency. Sci. Rep. 2017, 7, 16646. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-X.; Lin, J.; Liang, T.-Z.; Duan, H.; Tan, X.-H.; Xi, B.-M.; Li, L.; Liu, S.-W. The BET Bromodomain Inhibitor Apabetalone Induces Apoptosis of Latent HIV-1 Reservoir Cells Following Viral Reactivation. Acta Pharmacol. Sin. 2019, 40, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Qu, X.; Shen, Y.; Jiang, Z.; Wang, P.; Zeng, H.; Ji, H.; Deng, J.; Yang, X.; Li, X.; et al. The BET Inhibitor OTX015 Reactivates Latent HIV-1 through P-TEFb. Sci. Rep. 2016, 6, 24100. [Google Scholar] [CrossRef]
- Zhao, M.; De Crignis, E.; Rokx, C.; Verbon, A.; van Gelder, T.; Mahmoudi, T.; Katsikis, P.D.; Mueller, Y.M. T Cell Toxicity of HIV Latency Reversing Agents. Pharmacol. Res. 2019, 139, 524–534. [Google Scholar] [CrossRef]
- Canarte, V.; Munger, K. BRD4 Downregulation Inhibits the Viability of Cervical Cancer Cells without Affecting Viral Oncoprotein Expression. Curr. Res. Virol. Sci. 2021, 2, 100010. [Google Scholar] [CrossRef]
- Gilham, D.; Smith, A.L.; Fu, L.; Moore, D.Y.; Muralidharan, A.; Reid, S.P.M.; Stotz, S.C.; Johansson, J.O.; Sweeney, M.; Wong, N.C.W.; et al. Bromodomain and Extraterminal Protein Inhibitor, Apabetalone (RVX-208), Reduces ACE2 Expression and Attenuates SARS-Cov-2 Infection In Vitro. Biomedicines 2021, 9, 437. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.J.; Humphrey, S.J.; Fortuna, P.R.J.; Lor, M.; Foster, S.R.; Quaife-Ryan, G.A.; Johnston, R.L.; Dumenil, T.; Bishop, C.; Rudraraju, R.; et al. BET Inhibition Blocks Inflammation-Induced Cardiac Dysfunction and SARS-CoV-2 Infection. Cell 2021, 184, 2167–2182.e22. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Wang, X.-M.; Mannan, R.; Pitchiaya, S.; Zhang, Y.; Wotring, J.W.; Xiao, L.; Robinson, D.R.; Wu, Y.-M.; Tien, J.C.-Y.; et al. Targeting Transcriptional Regulation of SARS-CoV-2 Entry Factors ACE2 and TMPRSS2. Proc. Natl. Acad. Sci. USA 2020, 118, e2021450118. [Google Scholar] [CrossRef] [PubMed]
- Samelson, A.J.; Tran, Q.D.; Robinot, R.; Carrau, L.; Rezelj, V.V.; Kain, A.M.; Chen, M.; Ramadoss, G.N.; Guo, X.; Lim, S.A.; et al. BRD2 Inhibition Blocks SARS-CoV-2 Infection by Reducing Transcription of the Host Cell Receptor ACE2. Nat. Cell Biol. 2022, 24, 24–34. [Google Scholar] [CrossRef]
- Acharya, A.; Pathania, A.S.; Pandey, K.; Thurman, M.; Vann, K.R.; Kutateladze, T.G.; Challagundala, K.B.; Durden, D.L.; Byrareddy, S.N. PI3K-α/mTOR/BRD4 Inhibitor Alone or in Combination with Other Anti-Virals Blocks Replication of SARS-CoV-2 and Its Variants of Concern Including Delta and Omicron. Clin. Transl. Med. 2022, 12, e806. [Google Scholar] [CrossRef]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an Inhibitor of BET Transcriptional Regulators with Selectivity for the Second Bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef] [Green Version]
- Cai, D.; Lee, A.-Y.; Chiang, C.-M.; Kodadek, T. Peptoid Ligands That Bind Selectively to Phosphoproteins. Bioorg. Med. Chem. Lett. 2011, 21, 4960–4964. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.-M. Phospho-BRD4: Transcription Plasticity and Drug Targeting. Drug Discov. Today Technol. 2016, 19, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Viejo-Borbolla, A.; Ottinger, M.; Brüning, E.; Bürger, A.; König, R.; Kati, E.; Sheldon, J.A.; Schulz, T.F. Brd2/RING3 Interacts with a Chromatin-Binding Domain in the Kaposi’s Sarcoma-Associated Herpesvirus Latency-Associated Nuclear Antigen 1 (LANA-1) That Is Required for Multiple Functions of LANA-1. J. Virol. 2005, 79, 13618–13629. [Google Scholar] [CrossRef] [Green Version]
- Barboric, M.; Peterlin, B.M. A New Paradigm in Eukaryotic Biology: HIV Tat and the Control of Transcriptional Elongation. PLoS Biol. 2005, 3, e76. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, I.P.; Ott, M. Viral Hijacking of BET Proteins. Viruses 2022, 14, 2274. https://doi.org/10.3390/v14102274
Chen IP, Ott M. Viral Hijacking of BET Proteins. Viruses. 2022; 14(10):2274. https://doi.org/10.3390/v14102274
Chicago/Turabian StyleChen, Irene P., and Melanie Ott. 2022. "Viral Hijacking of BET Proteins" Viruses 14, no. 10: 2274. https://doi.org/10.3390/v14102274
APA StyleChen, I. P., & Ott, M. (2022). Viral Hijacking of BET Proteins. Viruses, 14(10), 2274. https://doi.org/10.3390/v14102274