
Identification and Characterization of Critical Processing Parameters in the Fabrication of Double-Emulsion Poly(lactic-co-glycolic) Acid Microparticles

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Design
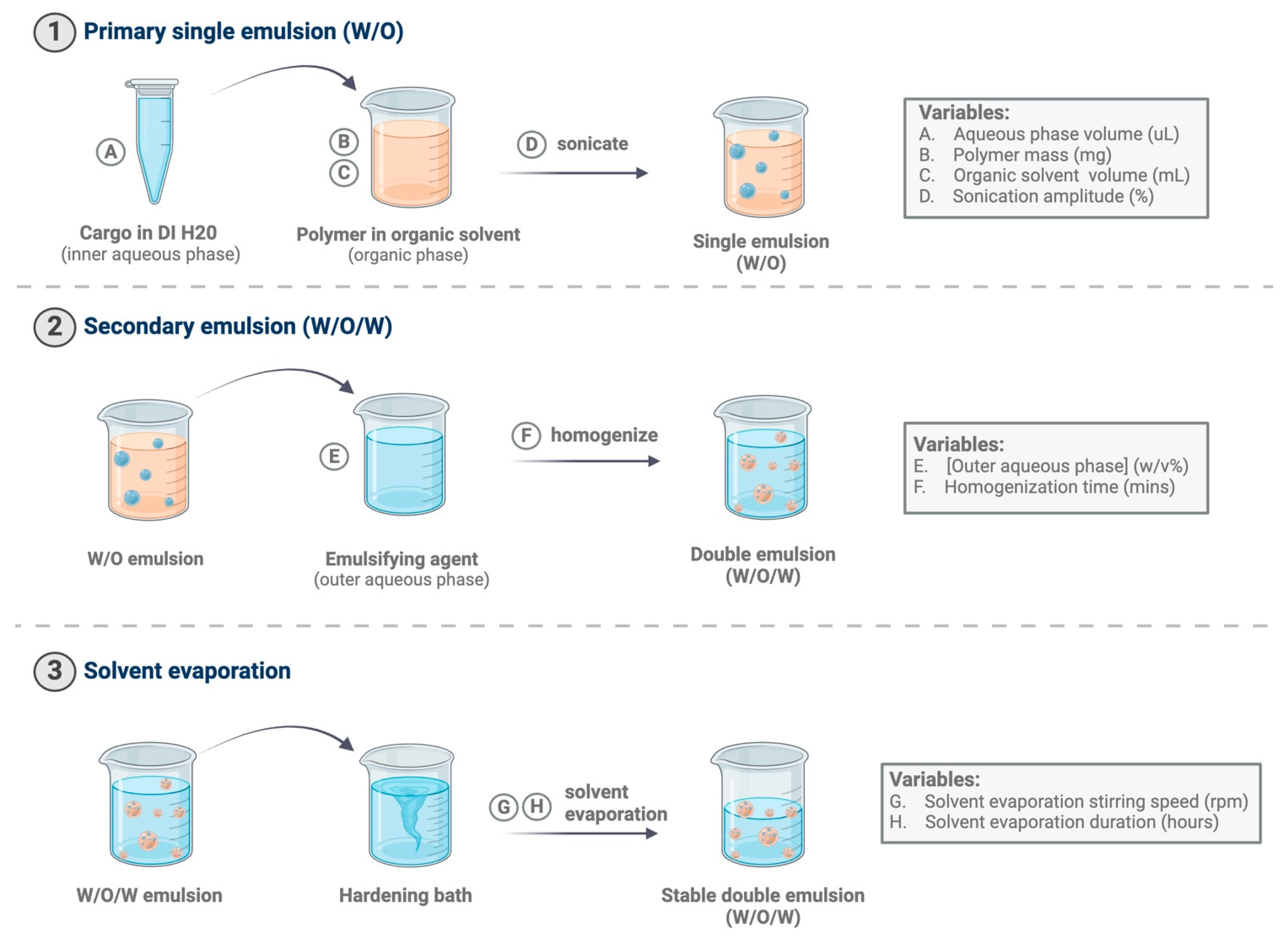
2.3. Fabrication of PLGA Microparticles
2.3.1. Blank MPs
2.3.2. CCL22 MPs
2.4. Characterization of PLGA Microparticles
2.4.1. Blank MPs
2.4.2. CCL22-MPs
3. Results and Discussion
3.1. Analysis of Design of Experiments Model
3.2. Identification of Critical Processing Parameters in Double-Emulsion Microparticle Fabrication
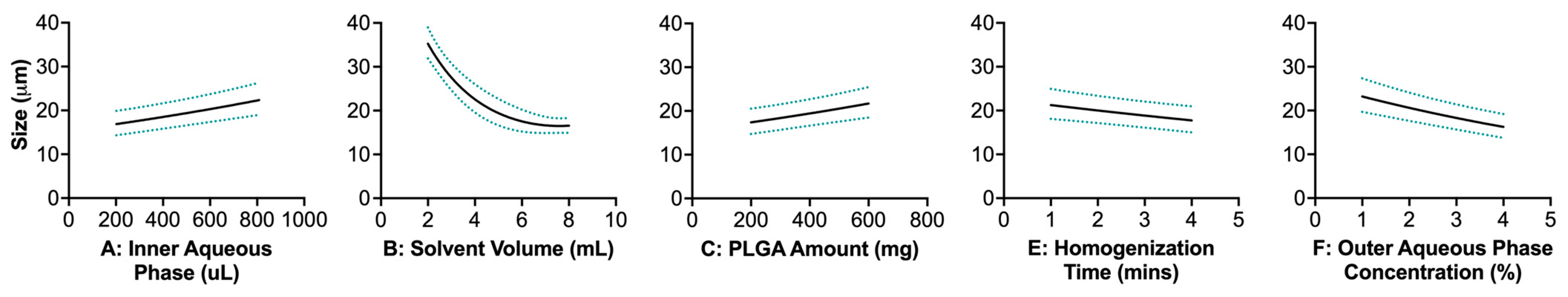
3.3. Effect of Single-Factor and Interaction Variables on Unloaded Double-Emulsion Microparticle Size
3.3.1. Single-Factor Effects
Inner Aqueous Phase Volume
Solvent Volume
Amount of Poly(lactic-co-glycolic) Acid
Homogenization Time
Outer Aqueous Phase Concentration
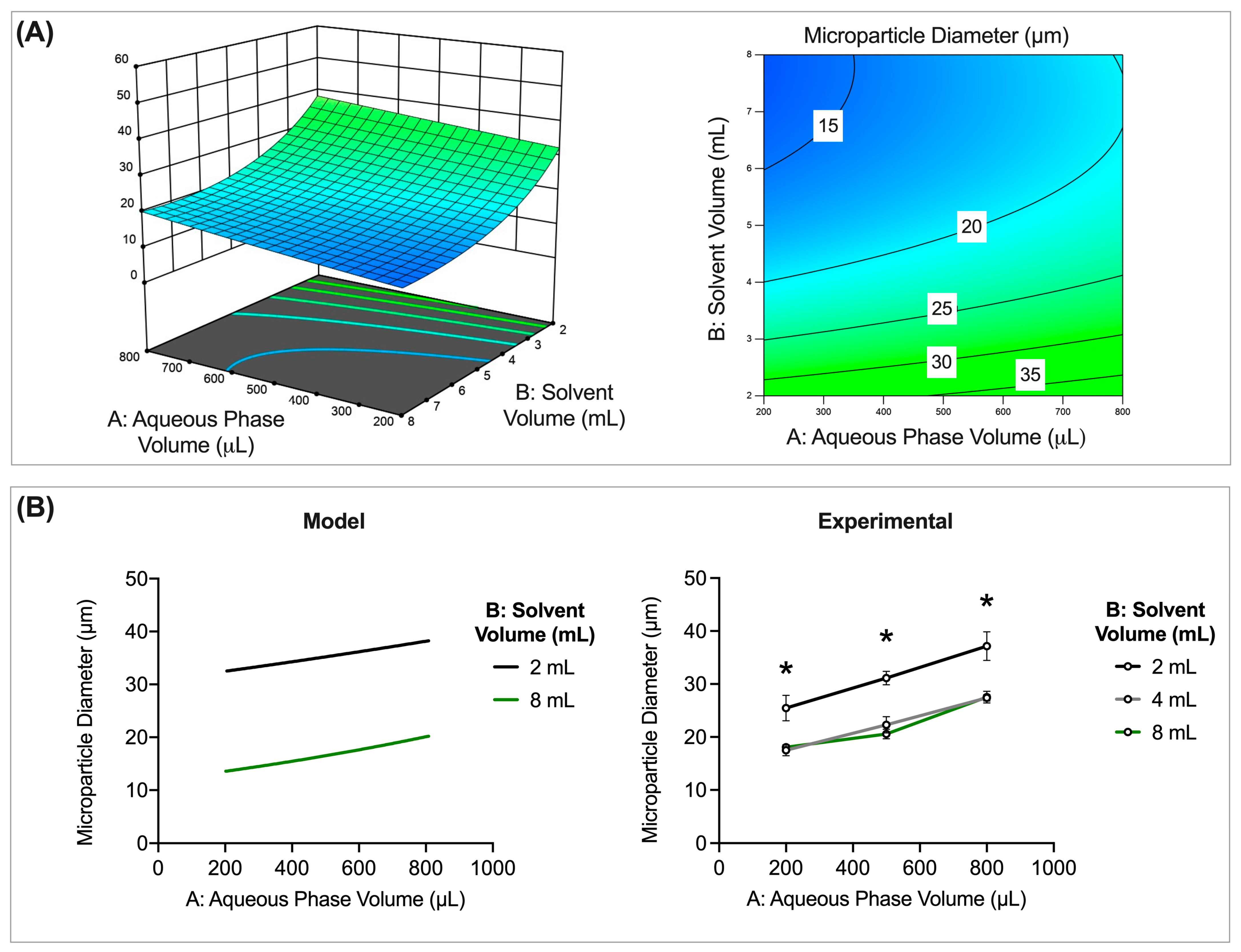
3.3.2. Interaction Effects
Inner Aqueous Phase Volume and Solvent Volume
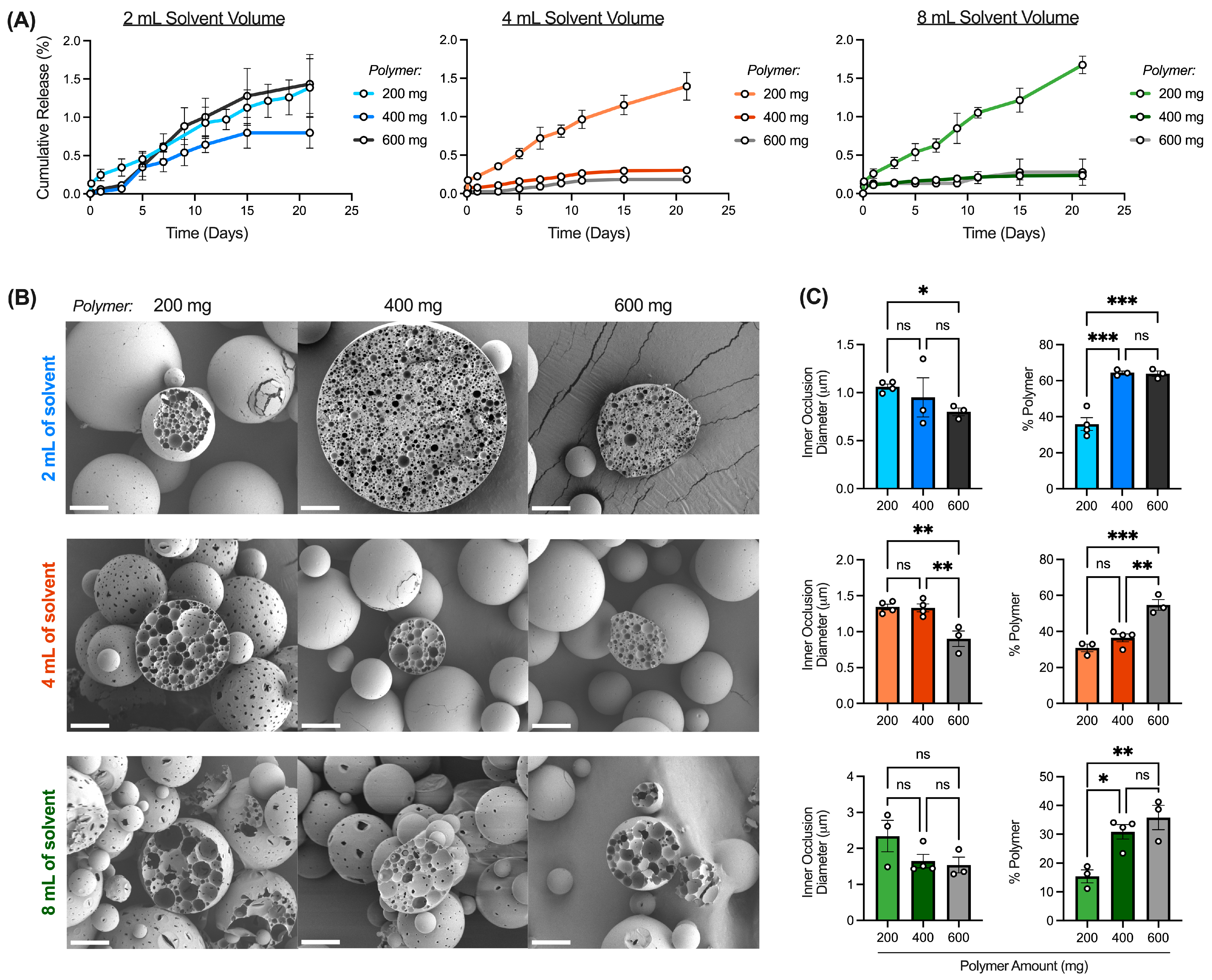
Solvent Volume and Polymer Amount
3.4. Effect of Critical Processing Parameters on CQAs of rhCCL22-Loaded Microparticles
3.4.1. Interaction between Inner Aqueous Phase Volume and Solvent Volume
3.4.2. Interaction between Solvent Volume and Polymer Amount
3.5. Applying Principles of DOE to Scale-Up Batch Manufacturing
3.5.1. Interaction between Inner Aqueous Phase Volume and Polymer Amount
3.5.2. Identifying a Formulation of CCL22-MPs with Increased Batch Size
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BSA | Bovine serum albumin |
| CMAs | Critical material attributes |
| CPPs | Critical processing parameters |
| CQAs | Critical quality attributes |
| DOE | Design of experiments |
| IA | Inner aqueous phase |
| MP(s) | Microparticle(s) |
| NaCl | Sodium chloride |
| O/W | Oil-in-water (single) emulsion |
| PLGA | Poly(lactic-co-glycolic acid) |
| PVA | Polyvinyl alcohol |
| QbD | Quality by design |
| QTPP | Quality target product profile |
| SEM | Scanning electron microscopy |
| W/O/W | Water–oil–water (double) emulsion |
References
- Tibbitt, M.W.; Dahlman, J.E.; Langer, R. Emerging Frontiers in Drug Delivery. J. Am. Chem. Soc. 2016, 138, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric systems for controlled drug release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef] [PubMed]
- Bentley, E.R.; Little, S.R. Local delivery strategies to restore immune homeostasis in the context of inflammation. Adv. Drug Deliv. Rev. 2021, 178, 113971. [Google Scholar] [CrossRef] [PubMed]
- Han, F.Y.; Thurecht, K.J.; Whittaker, A.K.; Smith, M.T. Bioerodable PLGA-Based Microparticles for Producing Sustained-Release Drug Formulations and Strategies for Improving Drug Loading. Front. Pharmacol. 2016, 7, 185. [Google Scholar] [CrossRef] [PubMed]
- Lagreca, E.; Onesto, V.; Di Natale, C.; La Manna, S.; Netti, P.A.; Vecchione, R. Recent advances in the formulation of PLGA microparticles for controlled drug delivery. Prog. Biomater. 2020, 9, 153–174. [Google Scholar] [CrossRef] [PubMed]
- Rosca, I.D.; Watari, F.; Uo, M. Microparticle formation and its mechanism in single and double emulsion solvent evaporation. J. Control. Release 2004, 99, 271–280. [Google Scholar] [CrossRef] [PubMed]
- McGinity, J.W.; O’Donnell, P.B. Preparation of microspheres by the solvent evaporation technique. Adv. Drug Deliv. Rev. 1997, 28, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.; Zafar, N.; Fessi, H.; Elaissari, A. Double emulsion solvent evaporation techniques used for drug encapsulation. Int. J. Pharm. 2015, 496, 173–190. [Google Scholar] [CrossRef] [PubMed]
- Wischke, C.; Schwendeman, S.P. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int. J. Pharm. 2008, 364, 298–327. [Google Scholar] [CrossRef]
- Fu, K.; Klibanov, A.M.; Langer, R. Protein stability in controlled-release systems. Nat. Biotechnol. 2000, 18, 24–25. [Google Scholar] [CrossRef]
- Pagels, R.F.; Prud’homme, R.K. Polymeric nanoparticles and microparticles for the delivery of peptides, biologics, and soluble therapeutics. J. Control. Release 2015, 219, 519–535. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qin, B.; Xia, G.; Choi, S.H. FDA’s Poly (Lactic-Co-Glycolic Acid) Research Program and Regulatory Outcomes. AAPS J. 2021, 23, 92. [Google Scholar] [CrossRef]
- Park, K.; Skidmore, S.; Hadar, J.; Garner, J.; Park, H.; Otte, A.; Soh, B.K.; Yoon, G.; Yu, D.; Yun, Y.; et al. Injectable, long-acting PLGA formulations: Analyzing PLGA and understanding microparticle formation. J. Control. Release 2019, 304, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Amidon, G.; Khan, M.A.; Hoag, S.W.; Polli, J.; Raju, G.K.; Woodcock, J. Understanding pharmaceutical quality by design. AAPS J. 2014, 16, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, L.; Wan, F.; Bera, H.; Cun, D.; Rantanen, J.; Yang, M. Quality by design thinking in the development of long-acting injectable PLGA/PLA-based microspheres for peptide and protein drug delivery. Int. J. Pharm. 2020, 585, 119441. [Google Scholar] [CrossRef] [PubMed]
- Pramod, K.; Tahir, M.A.; Charoo, N.A.; Ansari, S.H.; Ali, J. Pharmaceutical product development: A quality by design approach. Int. J. Pharm. Investig. 2016, 6, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Politis, S.N.; Colombo, P.; Colombo, G.; Rekkas, D.M. Design of experiments (DoE) in pharmaceutical development. Drug Dev. Ind. Pharm. 2017, 43, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Su, Y.; Zhang, H.; Liu, N.; Wang, Z.; Gao, X.; Gao, J.; Zheng, A. Poly(lactic-co-glycolic acid) microsphere production based on quality by design: A review. Drug Deliv. 2021, 28, 1342–1355. [Google Scholar] [CrossRef]
- Singh, B.; Bhatowa, R.; Tripathi, C.B.; Kapil, R. Developing micro-/nanoparticulate drug delivery systems using “design of experiments”. Int. J. Pharm. Investig. 2011, 1, 75–87. [Google Scholar] [CrossRef]
- Tavares Luiz, M.; Santos Rosa Viegas, J.; Palma Abriata, J.; Viegas, F.; Testa Moura de Carvalho Vicentini, F.; Lopes Badra Bentley, M.V.; Chorilli, M.; Maldonado Marchetti, J.; Tapia-Blacido, D.R. Design of experiments (DoE) to develop and to optimize nanoparticles as drug delivery systems. Eur. J. Pharm. Biopharm. 2021, 165, 127–148. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.D.; Zhang, W.; Balmert, S.C.; Aral, A.M.; Acharya, A.P.; Kulahci, Y.; Li, J.; Turnquist, H.R.; Thomson, A.W.; Solari, M.G.; et al. In situ recruitment of regulatory T cells promotes donor-specific tolerance in vascularized composite allotransplantation. Sci. Adv. 2020, 6, eaax8429. [Google Scholar] [CrossRef] [PubMed]
- Glowacki, A.J.; Yoshizawa, S.; Jhunjhunwala, S.; Vieira, A.E.; Garlet, G.P.; Sfeir, C.; Little, S.R. Prevention of inflammation-mediated bone loss in murine and canine periodontal disease via recruitment of regulatory lymphocytes. Proc. Natl. Acad. Sci. USA 2013, 110, 18525–18530. [Google Scholar] [CrossRef] [PubMed]
- Jhunjhunwala, S.; Raimondi, G.; Glowacki, A.J.; Hall, S.J.; Maskarinec, D.; Thorne, S.H.; Thomson, A.W.; Little, S.R. Bioinspired controlled release of CCL22 recruits regulatory T cells in vivo. Adv. Mater. 2012, 24, 4735–4738. [Google Scholar] [CrossRef] [PubMed]
- Sediq, A.S.; Waasdorp, S.K.D.; Nejadnik, M.R.; van Beers, M.M.C.; Meulenaar, J.; Verrijk, R.; Jiskoot, W. Determination of the Porosity of PLGA Microparticles by Tracking Their Sedimentation Velocity Using a Flow Imaging Microscope (FlowCAM). Pharm. Res. 2017, 34, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Nair, M.; Lusignan, C.P.; Boris, D.C. Preparation of uniform micrometer-sized polymer particles with closed-cell porous architecture made by limited coalescence of a double emulsion. Colloids Surf. A Physicochem. Eng. Asp. 2014, 443, 583–595, Erratum in Colloids Surf. A Physicochem. Eng. Asp. 2014, 450, 166. [Google Scholar] [CrossRef]
- Zandstra, J.; Hiemstra, C.; Petersen, A.H.; Zuidema, J.; van Beuge, M.M.; Rodriguez, S.; Lathuile, A.A.; Veldhuis, G.J.; Steendam, R.; Bank, R.A.; et al. Microsphere size influences the foreign body reaction. Eur. Cell Mater. 2014, 28, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Kohane, D.S. Microparticles and nanoparticles for drug delivery. Biotechnol. Bioeng. 2007, 96, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Gaignaux, A.; Reeff, J.; Siepmann, F.; Siepmann, J.; De Vriese, C.; Goole, J.; Amighi, K. Development and evaluation of sustained-release clonidine-loaded PLGA microparticles. Int. J. Pharm. 2012, 437, 20–28. [Google Scholar] [CrossRef]
- Ding, S.; Serra, C.A.; Vandamme, T.F.; Yu, W.; Anton, N. Double emulsions prepared by two-step emulsification: History, state-of-the-art and perspective. J. Control. Release 2019, 295, 31–49. [Google Scholar] [CrossRef]
- Taghipour, B.; Yakhchali, M.; Haririan, I.; Tamaddon, A.M.; Samani, S.M. The effects of technical and compositional variables on the size and release profile of bovine serum albumin from PLGA based particulate systems. Res. Pharm. Sci. 2014, 9, 407–420. [Google Scholar] [PubMed]
- Bullard, J.W.; Pauli, A.T.; Garboczi, E.J.; Martys, N.S. A comparison of viscosity-concentration relationships for emulsions. J. Colloid Interface Sci. 2009, 330, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Strivens, T.A. An Introduction to Rheology; Woodhead Publishing: Sawston, UK, 1999; pp. 550–574. [Google Scholar]
- Rajeev, M.; Helms, C.C.; Rajeev, M.; Helms, C.C. A Study of the Relationship between Polymer Solution Entanglement and Electrospun PCL Fiber Mechanics. Polymers 2023, 15, 4555. [Google Scholar] [CrossRef] [PubMed]
- Zambaux, M.F.; Bonneaux, F.; Gref, R.; Maincent, P.; Dellacherie, E.; Alonso, M.J.; Labrude, P.; Vigneron, C. Influence of experimental parameters on the characteristics of poly(lactic acid) nanoparticles prepared by a double emulsion method. J. Control. Release 1998, 50, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Yeo, Y.; Park, K. Control of encapsulation efficiency and initial burst in polymeric microparticle systems. Arch. Pharm. Res. 2004, 27, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Klose, D.; Siepmann, F.; Elkharraz, K.; Krenzlin, S.; Siepmann, J. How porosity and size affect the drug release mechanisms from PLGA-based microparticles. Int. J. Pharm. 2006, 314, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, S.N.; Federspiel, W.J.; Little, S.R. A simple model framework for the prediction of controlled release from bulk eroding polymer matrices. J. Mater. Chem. 2008, 18, 1873–1880. [Google Scholar] [CrossRef]
- Rothstein, S.N.; Federspiel, W.J.; Little, S.R. A unified mathematical model for the prediction of controlled release from surface and bulk eroding polymer matrices. Biomaterials 2009, 30, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Balmert, S.C.; Zmolek, A.C.; Glowacki, A.J.; Knab, T.D.; Rothstein, S.N.; Wokpetah, J.M.; Fedorchak, M.V.; Little, S.R. Positive Charge of “Sticky” Peptides and Proteins Impedes Release From Negatively Charged PLGA Matrices. J. Mater. Chem. B 2015, 3, 4723–4734. [Google Scholar] [CrossRef]
- Sophocleous, A.M.; Desai, K.G.; Mazzara, J.M.; Tong, L.; Cheng, J.X.; Olsen, K.F.; Schwendeman, S.P. The nature of peptide interactions with acid end-group PLGAs and facile aqueous-based microencapsulation of therapeutic peptides. J. Control. Release 2013, 172, 662–670. [Google Scholar] [CrossRef]
- Vlachopoulos, A.; Karlioti, G.; Balla, E.; Daniilidis, V.; Kalamas, T.; Stefanidou, M.; Bikiaris, N.D.; Christodoulou, E.; Koumentakou, I.; Karavas, E.; et al. Poly(Lactic Acid)-Based Microparticles for Drug Delivery Applications: An Overview of Recent Advances. Pharmaceutics 2022, 14, 359. [Google Scholar] [CrossRef] [PubMed]
- Jiang, E.Y.; Desroches, S.T.; Mikos, A.G. Particle carriers for controlled release of peptides. J. Control. Release 2023, 360, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Won, Y.Y. Phenomenology of the Initial Burst Release of Drugs from PLGA Microparticles. ACS Biomater. Sci. Eng. 2020, 6, 6053–6062. [Google Scholar] [CrossRef] [PubMed]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Tamada, J.A.; Langer, R. Erosion kinetics of hydrolytically degradable polymers. Proc. Natl. Acad. Sci. USA 1993, 90, 552–556. [Google Scholar] [CrossRef]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors | Name | Units | Minimum | Maximum |
|---|---|---|---|---|
| A | aqueous phase volume | μL | 200 | 800 |
| B | solvent volume | mL | 2 | 8 |
| C | PLGA amount | mg | 200 | 600 |
| D | sonication amplitude | % | 25 | 100 |
| E | homogenization time | mins | 1 | 4 |
| F | outer aqueous phase concentration | % | 1 | 4 |
| G | solvent evaporation duration | hrs | 2 | 8 |
| H | stirring speed | rpm | 100 | 600 |
| ANOVA Results | p-Value | |
|---|---|---|
| Terms | ||
| Whole plot | D—sonication amplitude | 0.8315 |
| Subplot | A—aqueous phase volume | <0.0001 |
| Single-Factor | B—solvent volume | <0.0001 |
| C—PLGA amount | 0.0001 | |
| E—homogenization time | 0.0016 | |
| F—outer aqueous phase concentration | <0.0001 | |
| G—solvent evaporation duration | 0.7189 | |
| Interactions | AB | 0.0479 |
| BC | 0.0326 | |
| CD | 0.0784 | |
| EG | 0.0431 | |
| FG | 0.0243 | |
| Quadratic | B2 | 0.0069 |
| Formulation | Solvent Volume (mL) | Polymer Amount (mg) | Inner Aqueous Phase (μL) | D21 Cumulative Release (ng rhCCL22/mg MP) | |
|---|---|---|---|---|---|
| Mean | SD | ||||
| A | 4 | 200 | 200 | 0.564 | 0.043 |
| B | 2 | 400 | 500 | 0.598 | 0.23 |
| C | 2 | 400 | 800 | 1.031 | 0.028 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bentley, E.R.; Subick, S.; Pezzillo, M.; Balmert, S.C.; Herbert, A.; Little, S.R. Identification and Characterization of Critical Processing Parameters in the Fabrication of Double-Emulsion Poly(lactic-co-glycolic) Acid Microparticles. Pharmaceutics 2024, 16, 796. https://doi.org/10.3390/pharmaceutics16060796
Bentley ER, Subick S, Pezzillo M, Balmert SC, Herbert A, Little SR. Identification and Characterization of Critical Processing Parameters in the Fabrication of Double-Emulsion Poly(lactic-co-glycolic) Acid Microparticles. Pharmaceutics. 2024; 16(6):796. https://doi.org/10.3390/pharmaceutics16060796
Chicago/Turabian StyleBentley, Elizabeth R., Stacia Subick, Michael Pezzillo, Stephen C. Balmert, Aidan Herbert, and Steven R. Little. 2024. "Identification and Characterization of Critical Processing Parameters in the Fabrication of Double-Emulsion Poly(lactic-co-glycolic) Acid Microparticles" Pharmaceutics 16, no. 6: 796. https://doi.org/10.3390/pharmaceutics16060796
APA StyleBentley, E. R., Subick, S., Pezzillo, M., Balmert, S. C., Herbert, A., & Little, S. R. (2024). Identification and Characterization of Critical Processing Parameters in the Fabrication of Double-Emulsion Poly(lactic-co-glycolic) Acid Microparticles. Pharmaceutics, 16(6), 796. https://doi.org/10.3390/pharmaceutics16060796