

Biological Barriers for Drug Delivery and Development of Innovative Therapeutic Approaches in HIV, Pancreatic Cancer, and Hemophilia A/B
 , ,
, ,  ,
,
Abstract
:1. Introduction
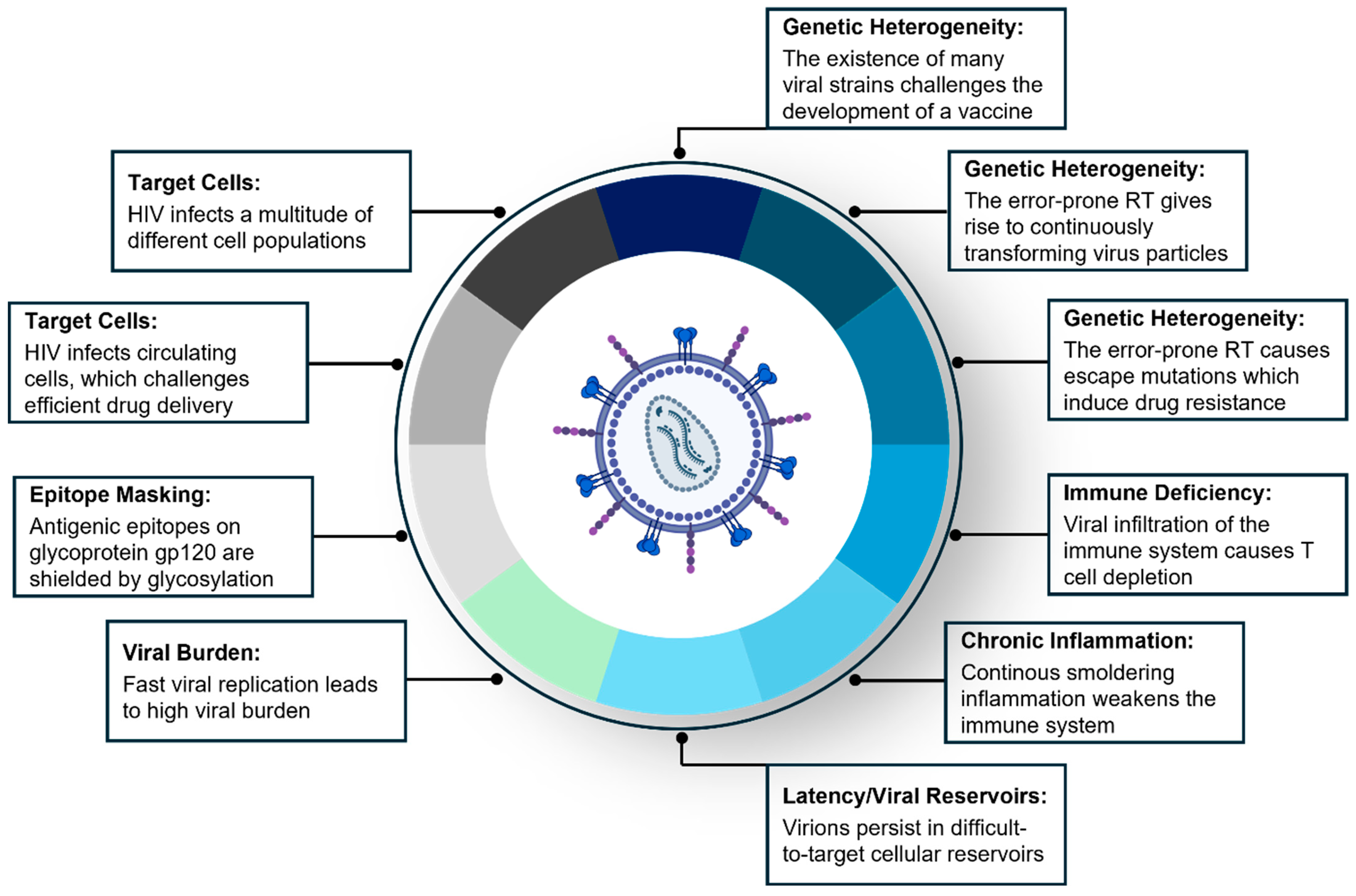
2. Chapter I: HIV/AIDS—Biological Barriers in HIV and Innovative Therapeutic Strategies to Prevent and Treat HIV Infection
2.1. Introduction to HIV/AIDS
2.2. ART/HAART
2.3. HIV Vaccine
2.4. HIV Microbicide
3. Chapter II: PANCREATIC CANCER—Biological Barriers and Novel Innovative Therapeutic Approaches to Treat Pancreatic Cancer
3.1. Introduction to Pancreatic Cancer
3.2. Tumor Microenvironment (TME) as a Biological Barrier
3.3. Genetic Diversity and Clonal Evolution as a Biological Barrier
3.4. Metastasis as a Biological Barrier
3.5. Therapeutic Strategies to Overcome Biological Barriers in Pancreatic Cancer
3.5.1. Basic Therapeutic Principles
3.5.2. Therapeutic Targeting of the TME
3.5.3. Therapeutic Targeting of Genetic Diversity
3.5.4. Therapeutic Targeting of Metastasis
- (a)
- the epithelial–mesenchymal transformation of tumor cells at the site of the primary tumor;
- (b)
- the migration of metastatic cells as circulating tumor cells (CTCs) in the blood stream and/or lymphatic tissues;
- (c)
- metastatic colonies in distant organs.
3.5.5. Summary of Therapeutic Approaches in Pancreatic Cancer
4. Chapter III: HEMOPHILIA—Biological Barriers in Rare Genetic Disorders and Gene Therapy as a Novel Therapeutic Approach in Hemophilia A and B
4.1. Introduction to Rare Genetic Diseases
4.2. Biology of AAV-Mediated Gene Therapy as Novel Therapeutic Approach
4.3. Short History of Gene Therapy
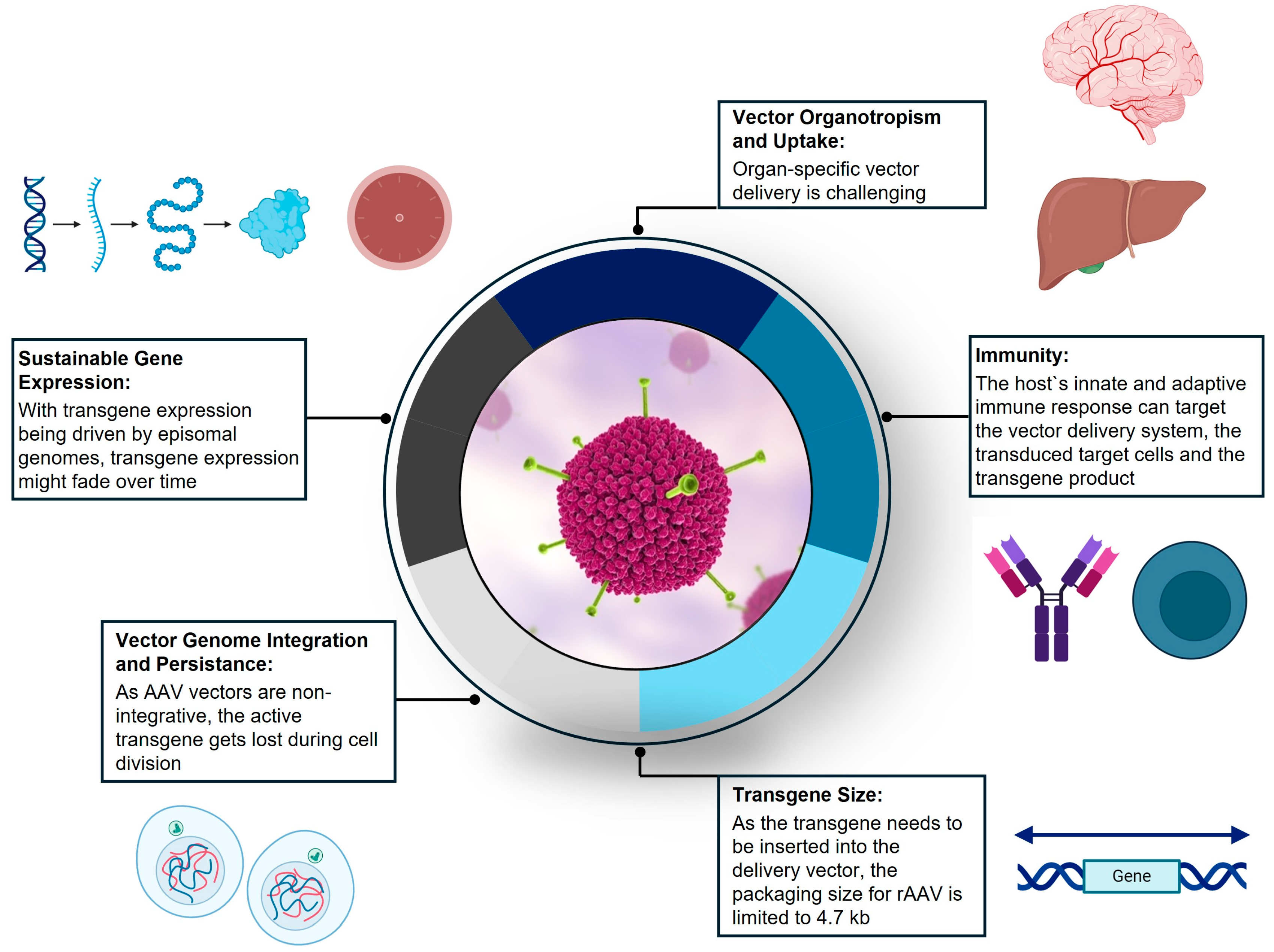
4.4. Biological Barriers for the Application of Gene Therapy
- As the AAV vector is engineered based on a wild-type parvovirus which naturally infects humans, it often elicits a B-cell response, resulting in neutralizing antibodies [145,166]. Immune responses in the GENEr8-1 trial (NCT03370913) for hemophilia A were predominantly directed toward the AAV5 capsid, and all study subjects developed a durable response against the AAV5 serotype used in the study. This high-titer antibody response will likely prevent redosing with the same serotype or other AAV serotypes due to the high degree of homology between serotypes.
- b.
- Transduced cells can elicit capsid T-cell responses in humans, which has been implicated in effecting the duration of the transgene expression [142]. When a T-cell response leads to the rejection and apoptosis of transduced cells, the transgene expression is abrogated, which renders gene therapy ineffective [145].
- c.

{kind=link}
{kind=link}
{kind=link}
| rAAV-Mediated Gene Therapy: Challenges for Drug Delivery | References | |
|---|---|---|
| Transgene Size | Vector packaging size limited to 4.7 kb Transgene engineering of FVIII cDNA | [162,163] |
| Vector Uptake and Organotropism | Vector uptake is organ-/cell-dependent Capsid modifications aiming at retargeting AAV tropism permit tissue-specific gene transfer Selection of tissue-/cell-specific promoters can direct transgene expression to cells of interest | [142,150,162,164,167] |
| Vector Genome Integration and Persistence | rAAV delivers transgenes as non-integrative episomes, which are lost upon cell division Lowering vector doses reduces the risk of insertional mutagenesis | [155,164,165] |
| Gene Expression Sustainability | Transgene expression, driven by circular episomes, depends on episomal persistence, epigenetic modulation and choice of promoters Transgene expression is challenged by adaptive immune responses to the transduced cells and/or transgene itself | [143,163,164,166,167] |
| Immunological Barrier | ||
| Humoral antibody-mediated immune response to viral vector | [146,162,164,167] | |
| Cellular CTL-mediated immune response to transduced host cells | [143,146,162,167] | |
| Humoral and cellular immune response to transgene | [146,162,164,167] | |
4.5. Therapeutic Application of Gene Therapy in Hemophilia A and B
5. Concluding Remarks
- The relevance of the host immune system as a predominant biological barrier;
- The importance of a targeted, personalized, and combinational therapy approach;
- The emergence of new druggable targets in HIV, cancer, and rare diseases;
- The development of new cutting-edge therapy modalities (such as RNAi-based therapeutics, mRNA-based cancer vaccines, and AAV-based gene therapy) in all three disease areas we visited today.
Author Contributions
Funding
Conflicts of Interest
References
- Agarwal-Jans, S. Timeline: HIV. Cell 2020, 183, 550. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Arias, L.; Delgado, R. Update and latest advances in antiretroviral therapy. Trends Pharmacol. Sci. 2022, 43, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS JUNP on H (o. J.). UNAIDS Global AIDS Update 2023. Available online: https://thepath.unaids.org/wp-content/themes/unaids2023/assets/files/2023_report.pdf (accessed on 1 August 2024).
- Bekker, L.-G.; Beyrer, C.; Mgodi, N.; Lewin, S.R.; Delany-Moretlwe, S.; Taiwo, B.; Masters, M.C.; Lazarus, J.V. HIV infection. Nat. Rev. Dis. Primer 2023, 9, 42. [Google Scholar] [CrossRef]
- Prabhu, V.M.; Padwal, V.; Velhal, S.; Salwe, S.; Nagar, V.; Patil, P.; Bandivdekar, A.H.; Patel, V. Vaginal Epithelium Transiently Harbours HIV-1 Facilitating Transmission. Front. Cell. Infect. Microbiol. 2021, 11, 634647. [Google Scholar] [CrossRef]
- Zila, V.; Margiotta, E.; Turoňová, B.; Müller, T.G.; Zimmerli, C.E.; Mattei, S.; Allegretti, M.; Börner, K.; Rada, J.; Müller, B.; et al. Cone-shaped HIV-1 capsids are transported through intact nuclear pores. Cell 2021, 184, 1032–1046.e18. [Google Scholar] [CrossRef]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef]
- Mourez, T.; Simon, F.; Plantier, J.-C. Non-M variants of human immunodeficiency virus type 1. Clin. Microbiol. Rev. 2013, 26, 448–461. [Google Scholar] [CrossRef]
- Delviks-Frankenberry, K.; Galli, A.; Nikolaitchik, O.; Mens, H.; Pathak, V.K.; Hu, W.-S. Mechanisms and factors that influence high frequency retroviral recombination. Viruses 2011, 3, 1650–1680. [Google Scholar] [CrossRef]
- Hemelaar, J. The origin and diversity of the HIV-1 pandemic. Trends Mol. Med. 2012, 18, 182–192. [Google Scholar] [CrossRef]
- Rossi, E.; Meuser, M.E.; Cunanan, C.J.; Cocklin, S. Structure, Function, and Interactions of the HIV-1 Capsid Protein. Life 2021, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Connell, B.; Lortat-Jacob, H. Human Immunodeficiency Virus and Heparan Sulfate: From Attachment to Entry Inhibition. Front. Immunol. 2013, 4, 385. [Google Scholar] [CrossRef]
- Zhuang, S.; Torbett, B.E. Interactions of HIV-1 Capsid with Host Factors and Their Implications for Developing Novel Therapeutics. Viruses 2021, 13, 417. [Google Scholar] [CrossRef] [PubMed]
- Grandgenett, D.P.; Engelman, A.N. Brief Histories of Retroviral Integration Research and Associated International Conferences. Viruses 2024, 16, 604. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H. Challenges in the development of an HIV-1 vaccine. Nature 2008, 455, 613–619. [Google Scholar] [CrossRef]
- Ng’uni, T.; Chasara, C.; Ndhlovu, Z.M. Major Scientific Hurdles in HIV Vaccine Development: Historical Perspective and Future Directions. Front. Immunol. 2020, 11, 590780. [Google Scholar] [CrossRef]
- Haynes, B.F.; Wiehe, K.; Borrow, P.; Saunders, K.O.; Korber, B.; Wagh, K.; McMichael, A.J.; Kelsoe, G.; Hahn, B.H.; Alt, F.; et al. Strategies for HIV-1 vaccines that induce broadly neutralizing antibodies. Nat. Rev. Immunol. 2023, 23, 142–158. [Google Scholar] [CrossRef]
- Abram, M.E.; Ferris, A.L.; Shao, W.; Alvord, W.G.; Hughes, S.H. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 2010, 84, 9864–9878. [Google Scholar] [CrossRef]
- Taylor, B.S.; Sobieszczyk, M.E.; McCutchan, F.E.; Hammer, S.M. The challenge of HIV-1 subtype diversity. N. Engl. J. Med. 2008, 358, 1590–1602. [Google Scholar] [CrossRef]
- Obare, L.M.; Temu, T.; Mallal, S.A.; Wanjalla, C.N. Inflammation in HIV and Its Impact on Atherosclerotic Cardiovascular Disease. Circ. Res. 2024, 134, 1515–1545. [Google Scholar] [CrossRef]
- McArthur, J.C.; Johnson, T.P. Chronic inflammation mediates brain injury in HIV infection: Relevance for cure strategies. Curr. Opin. Neurol. 2020, 33, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Tang, H. HIV-1 Infection and Glucose Metabolism Reprogramming of T Cells: Another Approach Toward Functional Cure and Reservoir Eradication. Front. Immunol. 2020, 11, 572677. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Maeda, K. HIV Reservoirs and Treatment Strategies toward Curing HIV Infection. Int. J. Mol. Sci. 2024, 25, 2621. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, D.; Badar, U.; Javaid, M.; Farooqui, N.; Shah, S.A.; Iftikhar, A.; Sultan, F.; Mir, F.; Furqan, S.; Mahmood, S.F.; et al. Genetic and antiretroviral drug resistance mutations analysis of reverse transcriptase and protease gene from Pakistani people living with HIV-1. PLoS ONE 2023, 18, e0290425. [Google Scholar] [CrossRef]
- Khan, T.; Mayuresh Patkar, M.; Momin, M.; Omri, A. Macrophage targeted nanocarrier delivery systems in HIV therapeutics. Expert Opin. Drug Deliv. 2020, 17, 903–918. [Google Scholar] [CrossRef]
- Ahaus, P.; Potthoff, A.; Kayser, A.; Wach, J.; Brockmeyer, N.H.; Skaletz-Rorowski, A. HIV-Präexpositionsprophylaxe-Versorgung in intersektoraler Zusammenarbeit. Hautarzt 2020, 71, 211–218. [Google Scholar] [CrossRef]
- Ahaus, P.; Schmidt, A.J.; Skaletz-Rorowski, A.; Uhrmacher, M.; Serova, K.; Kayser, A.; Wach, J.; Nambiar, S.; Brockmeyer, N.H.; Potthoff, A. Changes in the user profiles of HIV pre-exposure prophylaxis (PrEP) before and after PrEP reimbursement. J. Infect. Public Health 2022, 15, 955–960. [Google Scholar] [CrossRef]
- Agrahari, V.; Anderson, S.M.; Peet, M.M.; Wong, A.P.; Singh, O.N.; Doncel, G.F.; Clark, M.R. Long-acting HIV pre-exposure prophylaxis (PrEP) approaches: Recent advances, emerging technologies, and development challenges. Expert Opin. Drug Deliv. 2022, 19, 1365–1380. [Google Scholar] [CrossRef]
- Marrazzo, J.; Tao, L.; Becker, M.; Leech, A.A.; Taylor, A.W.; Ussery, F.; Kiragu, M.; Reza-Paul, S.; Myers, J.; Bekker, L.-G.; et al. HIV Preexposure Prophylaxis With Emtricitabine and Tenofovir Disoproxil Fumarate Among Cisgender Women. JAMA 2024, 331, 930–937. [Google Scholar] [CrossRef]
- Mayer, K.H.; Allan-Blitz, L.-T. Post-exposure prophylaxis to prevent HIV: New drugs, new approaches, and more questions. Lancet HIV 2023, 10, e816–e824. [Google Scholar] [CrossRef]
- Burton, D.R. Advancing an HIV vaccine; advancing vaccinology. Nat. Rev. Immunol. 2019, 19, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Giese, S.; Pelchen-Matthews, A.; Marsh, M. HIV–The Cell Biology of Virus Infection and Replication. In Encyclopedia of Cell Biology; Bradshaw, R.A., Stahl, P.D., Eds.; Academic Press: Waltham, MA, USA, 2016; pp. 828–838. [Google Scholar]
- Schiffner, T.; Phung, I.; Ray, R.; Irimia, A.; Tian, M.; Swanson, O.; Lee, J.H.; Lee, C.-C.D.; Marina-Zárate, E.; Cho, S.Y.; et al. Vaccination induces broadly neutralizing antibody precursors to HIV gp41. Nat. Immunol. 2024, 25, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Basar, E.; Shum, B.; Skaletz-Rorowski, A.; Wu, Y.; Nambiar, S.; Brockmeyer, N.H. Cholesterol-conjugated siRNAs silence gene expression in mucosal dendritic cells in cervicovaginal tissue in mice. J. Eur. Acad. Dermatol. Venereol. 2023, 37, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Carias, A.M.; McCoombe, S.; McRaven, M.; Anderson, M.; Galloway, N.; Vandergrift, N.; Fought, A.J.; Lurain, J.; Duplantis, M.; Veazey, R.S.; et al. Defining the interaction of HIV-1 with the mucosal barriers of the female reproductive tract. J. Virol. 2013, 87, 11388–11400. [Google Scholar] [CrossRef] [PubMed]
- Hladik, F.; McElrath, M.J. Setting the stage: Host invasion by HIV. Nat. Rev. Immunol. 2008, 8, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Ayele, H.; Perner, M.; McKinnon, L.R.; Birse, K.; Farr Zuend, C.; Burgener, A. An updated review on the effects of depot medroxyprogesterone acetate on the mucosal biology of the female genital tract. Am. J. Reprod. Immunol. 2021, 86, e13455. [Google Scholar] [CrossRef] [PubMed]
- Amabebe, E.; Anumba, D.O.C. The Vaginal Microenvironment: The Physiologic Role of Lactobacilli. Front. Med. 2018, 5, 181. [Google Scholar] [CrossRef] [PubMed]
- Mirmonsef, P.; Modur, S.; Burgad, D.; Gilbert, D.; Golub, E.T.; French, A.L.; McCotter, K.; Landay, A.L.; Spear, G.T. Exploratory comparison of vaginal glycogen and Lactobacillus levels in premenopausal and postmenopausal women. Menopause 2015, 22, 702–709. [Google Scholar] [CrossRef]
- Zhou, J.Z.; Way, S.S.; Chen, K. Immunology of Uterine and Vaginal Mucosae: (Trends in Immunology 39, 302-314, 2018). Trends Immunol. 2018, 39, 355. [Google Scholar] [CrossRef]
- Shen, R.; Richter, H.E.; Smith, P.D. Early HIV-1 target cells in human vaginal and ectocervical mucosa. Am. J. Reprod. Immunol. 2011, 65, 261–267. [Google Scholar] [CrossRef]
- Sibeko, S.; Sanderson, M.; Moyo, S.; Botha, M.H. Role of the epithelium in human papillomavirus and human immunodeficiency virus infections in the female genital tract. Front. Reprod. Health 2024, 6, 1408198. [Google Scholar] [CrossRef] [PubMed]
- Yasen, A.; Herrera, R.; Rosbe, K.; Lien, K.; Tugizov, S.M. HIV internalization into oral and genital epithelial cells by endocytosis and macropinocytosis leads to viral sequestration in the vesicles. Virology 2018, 515, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Day, C.J.; Hardison, R.L.; Spillings, B.L.; Poole, J.; Jurcisek, J.A.; Mak, J.; Jennings, M.P.; Edwards, J.L. Complement Receptor 3 Mediates HIV-1 Transcytosis across an Intact Cervical Epithelial Cell Barrier: New Insight into HIV Transmission in Women. mBio 2022, 13, e0217721. [Google Scholar] [CrossRef] [PubMed]
- Pena-Cruz, V.; Agosto, L.M.; Akiyama, H.; Olson, A.; Moreau, Y.; Larrieux, J.-R.; Henderson, A.; Gummuluru, S.; Sagar, M. HIV-1 replicates and persists in vaginal epithelial dendritic cells. J. Clin. Investig. 2018, 128, 3439–3444. [Google Scholar] [CrossRef]
- Yasen, A.; Herrera, R.; Rosbe, K.; Lien, K.; Tugizov, S.M. Release of HIV-1 sequestered in the vesicles of oral and genital mucosal epithelial cells by epithelial-lymphocyte interaction. PLoS Pathog. 2017, 13, e1006247. [Google Scholar] [CrossRef]
- Tugizov, S.M. Human immunodeficiency virus interaction with oral and genital mucosal epithelia may lead to epithelial-mesenchymal transition and sequestration of virions in the endosomal compartments. Oral Dis. 2020, 26 (Suppl. 1), 40–46. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; Kwon, D.S.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Middel, J.; Cornelissen, I.L.; Nottet, H.S.; KewalRamani, V.N.; Littman, D.R.; et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef]
- Sewald, X.; Ladinsky, M.S.; Uchil, P.D.; Beloor, J.; Pi, R.; Herrmann, C.; Motamedi, N.; Murooka, T.T.; Brehm, M.A.; Greiner, D.L.; et al. Retroviruses use CD169-mediated trans-infection of permissive lymphocytes to establish infection. Science 2015, 350, 563–567. [Google Scholar] [CrossRef]
- Gummuluru, S.; Pina Ramirez, N.-G.; Akiyama, H. CD169-dependent cell-associated HIV-1 transmission: A driver of virus dissemination. J. Infect. Dis. 2014, 210 (Suppl. 3), S641–S647. [Google Scholar] [CrossRef] [PubMed]
- Ménager, M.M.; Littman, D.R. Actin Dynamics Regulates Dendritic Cell-Mediated Transfer of HIV-1 to T Cells. Cell 2016, 164, 695–709. [Google Scholar] [CrossRef]
- Nikolic, D.S.; Lehmann, M.; Felts, R.; Garcia, E.; Blanchet, F.P.; Subramaniam, S.; Piguet, V. HIV-1 activates Cdc42 and induces membrane extensions in immature dendritic cells to facilitate cell-to-cell virus propagation. Blood 2011, 118, 4841–4852. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Alcover, A.; Schwartz, O. Modulation of the immunological synapse: A key to HIV-1 pathogenesis? Nat. Rev. Immunol. 2007, 7, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Hannon, G.J. RNA interference. Nature 2002, 418, 244–251. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Dykxhoorn, D.M.; Lieberman, J. The silent revolution: RNA interference as basic biology, research tool, and therapeutic. Annu. Rev. Med. 2005, 56, 401–423. [Google Scholar] [CrossRef] [PubMed]
- Dykxhoorn, D.M. P-bodies and RNAi: The missing link? J. RNAi Gene Silenc. Int. J. RNA Gene Target. Res. 2005, 2, 105–106. [Google Scholar]
- Beckham, C.J.; Parker, R. P Bodies, Stress Granules, and Viral Life Cycles. Cell Host Microbe 2008, 3, 206–212. [Google Scholar] [CrossRef]
- Dykxhoorn, D.M.; Lieberman, J. Running interference: Prospects and obstacles to using small interfering RNAs as small molecule drugs. Annu. Rev. Biomed. Eng. 2006, 8, 377–402. [Google Scholar] [CrossRef]
- Kang, H.; Ga, Y.J.; Kim, S.H.; Cho, Y.H.; Kim, J.W.; Kim, C.; Yeh, J.-Y. Small interfering RNA (siRNA)-based therapeutic applications against viruses: Principles, potential, and challenges. J. Biomed. Sci. 2023, 30, 88. [Google Scholar] [CrossRef]
- Palliser, D.; Chowdhury, D.; Wang, Q.-Y.; Lee, S.J.; Bronson, R.T.; Knipe, D.M.; Lieberman, J. An siRNA-based microbicide protects mice from lethal herpes simplex virus 2 infection. Nature 2006, 439, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Navarro, F.; Lal, A.; Basar, E.; Pandey, R.K.; Manoharan, M.; Feng, Y.; Lee, S.J.; Lieberman, J.; Palliser, D. Durable protection from Herpes Simplex Virus-2 transmission following intravaginal application of siRNAs targeting both a viral and host gene. Cell Host Microbe 2009, 5, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, L.A. Silencing sexually transmitted infections: Topical siRNA-based interventions for the prevention of HIV and HSV. Infect. Dis. Obstet. Gynecol. 2014, 2014, 125087. [Google Scholar] [CrossRef]
- Hibma, M.H. Silencing genes in the vaginal mucosa by topical application of a cholesterol-modified siRNA to inhibit HIV transmission. J. Eur. Acad. Dermatol. Venereol. JEADV 2023, 37, 467–468. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Ukidve, A.; Cu, K.; Kumbhojkar, N.; Lahann, J.; Mitragotri, S. Overcoming biological barriers to improve solid tumor immunotherapy. Drug Deliv. Transl. Res. 2021, 11, 2276–2301. [Google Scholar] [CrossRef]
- Afghani, E.; Klein, A.P. Pancreatic Adenocarcinoma: Trends in Epidemiology, Risk Factors, and Outcomes. Hematol. Oncol. Clin. N. Am. 2022, 36, 879–895. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. Available online: https://gco.iarc.fr/en (accessed on 1 August 2024).
- Tarannum, M.; Vivero-Escoto, J.L. Nanoparticle-based therapeutic strategies targeting major clinical challenges in pancreatic cancer treatment. Adv. Drug Deliv. Rev. 2022, 187, 114357. [Google Scholar] [CrossRef]
- Sturgeon, R.; Goel, P.; Singh, R.K. Tumor-associated neutrophils in pancreatic cancer progression and metastasis. Am. J. Cancer Res. 2023, 13, 6176–6189. [Google Scholar]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Ilic, I.; Ilic, M. International patterns in incidence and mortality trends of pancreatic cancer in the last three decades: A joinpoint regression analysis. World J. Gastroenterol. 2022, 28, 4698–4715. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Lyssiotis, C.A.; Pasca di Magliano, M.; Maitra, A. Pancreatic cancer: Advances and challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Urbanova, M.; Cihova, M.; Buocikova, V.; Slopovsky, J.; Dubovan, P.; Pindak, D.; Tomas, M.; García-Bermejo, L.; Rodríguez-Garrote, M.; Earl, J.; et al. Nanomedicine and epigenetics: New alliances to increase the odds in pancreatic cancer survival. Biomed. Pharmacother. 2023, 165, 115179. [Google Scholar] [CrossRef] [PubMed]
- Hartupee, C.; Nagalo, B.M.; Chabu, C.Y.; Tesfay, M.Z.; Coleman-Barnett, J.; West, J.T.; Moaven, O. Pancreatic cancer tumor microenvironment is a major therapeutic barrier and target. Front. Immunol. 2024, 15, 1287459. [Google Scholar] [CrossRef]
- Joseph, A.M.; Al Aiyan, A.; Al-Ramadi, B.; Singh, S.K.; Kishore, U. Innate and adaptive immune-directed tumour microenvironment in pancreatic ductal adenocarcinoma. Front. Immunol. 2024, 15, 1323198. [Google Scholar] [CrossRef]
- Xu, Z.; Pothula, S.; Wilson, J.; Apte, M. Pancreatic cancer and its stroma: A conspiracy theory. World J. Gastroenterol. WJG 2014, 20, 11216–11229. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.P. Pancreatic cancer epidemiology: Understanding the role of lifestyle and inherited risk factors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 493–502. [Google Scholar] [CrossRef]
- Xiang, Z.-J.; Hu, T.; Wang, Y.; Wang, H.; Xu, L.; Cui, N. Neutrophil-lymphocyte ratio (NLR) was associated with prognosis and immunomodulatory in patients with pancreatic ductal adenocarcinoma (PDAC). Biosci. Rep. 2020, 40, BSR20201190. [Google Scholar] [CrossRef]
- Li, Y.; Xiang, S.; Pan, W.; Wang, J.; Zhan, H.; Liu, S. Targeting tumor immunosuppressive microenvironment for pancreatic cancer immunotherapy: Current research and future perspective. Front. Oncol. 2023, 13, 1166860. [Google Scholar] [CrossRef]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef]
- Caronni, N.; La Terza, F.; Vittoria, F.M.; Barbiera, G.; Mezzanzanica, L.; Cuzzola, V.; Barresi, S.; Pellegatta, M.; Canevazzi, P.; Dunsmore, G.; et al. IL-1β+ macrophages fuel pathogenic inflammation in pancreatic cancer. Nature 2023, 623, 415–422. [Google Scholar] [CrossRef]
- Stanciu, S.; Ionita-Radu, F.; Stefani, C.; Miricescu, D.; Stanescu-Spinu, I.-I.; Greabu, M.; Ripszky Totan, A.; Jinga, M. Targeting PI3K/AKT/mTOR Signaling Pathway in Pancreatic Cancer: From Molecular to Clinical Aspects. Int. J. Mol. Sci. 2022, 23, 10132. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Li, L.; Ruan, J.; Zhang, N.; Dai, J.; Xu, X.; Tian, X.; Hu, J. Identification of prognostic and driver gene mutations in acute myeloid leukemia by a bioinformatics analysis. Transl. Cancer Res. 2023, 12, 1552–1564. [Google Scholar] [CrossRef]
- Regel, I.; Mayerle, J.; Ujjwal Mukund, M. Current Strategies and Future Perspectives for Precision Medicine in Pancreatic Cancer. Cancers 2020, 12, 1024. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Sahin, I.H.; Lowery, M.A.; Stadler, Z.K.; Salo-Mullen, E.; Iacobuzio-Donahue, C.A.; Kelsen, D.P.; O’Reilly, E.M. Genomic instability in pancreatic adenocarcinoma: A new step towards precision medicine and novel therapeutic approaches. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 893–905. [Google Scholar] [CrossRef]
- Hu, H.; Ye, Z.; Qin, Y.; Xu, X.; Yu, X.; Zhuo, Q.; Ji, S. Mutations in key driver genes of pancreatic cancer: Molecularly targeted therapies and other clinical implications. Acta Pharmacol. Sin. 2021, 42, 1725–1741. [Google Scholar] [CrossRef]
- Ho, I.-L.; Li, C.-Y.; Wang, F.; Zhao, L.; Liu, J.; Yen, E.-Y.; Dyke, C.A.; Shah, R.; Liu, Z.; Çetin, A.O.; et al. Clonal dominance defines metastatic dissemination in pancreatic cancer. Sci. Adv. 2024, 10, eadd9342. [Google Scholar] [CrossRef]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Luo, Q.; Hu, Z.; Zhao, H.; Fan, Y.; Tu, X.; Wang, Y.; Liu, X. The role of TGF-β in the tumor microenvironment of pancreatic cancer. Genes Dis. 2023, 10, 1513–1524. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Da, X.; Mo, J.; Li, Q.; Cao, B.; Huang, J.; Lu, Y.; Lu, L.; Fan, M.; Lu, H. Targeted co-delivery of PD-L1 monoclonal antibody and sorafenib to circulating tumor cells via platelet-functionalized nanocarriers. Biochem. Biophys. Res. Commun. 2023, 671, 335–342. [Google Scholar] [CrossRef]
- Cozzo, A.J.; Coleman, M.F.; Hursting, S.D. You complete me: Tumor cell-myeloid cell nuclear fusion as a facilitator of organ-specific metastasis. Front. Oncol. 2023, 13, 1191332. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Usman, S.; Waseem, N.H.; Nguyen, T.K.N.; Mohsin, S.; Jamal, A.; Teh, M.-T.; Waseem, A. Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis. Cancers 2021, 13, 4985. [Google Scholar] [CrossRef]
- Chen, T.; Jia, W.; Zhang, B.; Xie, H.; Wu, Q. EMT transcription factors activated circuits: A novel tool to study EMT dynamics and its therapeutic implications. Synth. Syst. Biotechnol. 2024, 9, 1–10. [Google Scholar] [CrossRef]
- Bulle, A.; Lim, K.-H. Beyond just a tight fortress: Contribution of stroma to epithelial-mesenchymal transition in pancreatic cancer. Signal Transduct. Target. Ther. 2020, 5, 249. [Google Scholar] [CrossRef]
- Valent, P.; Sadovnik, I.; Eisenwort, G.; Herrmann, H.; Bauer, K.; Mueller, N.; Sperr, W.R.; Wicklein, D.; Schumacher, U. Redistribution, homing and organ-invasion of neoplastic stem cells in myeloid neoplasms. Semin. Cancer Biol. 2020, 60, 191–201. [Google Scholar] [CrossRef]
- Le, M.T.N.; Hamar, P.; Guo, C.; Basar, E.; Perdigão-Henriques, R.; Balaj, L.; Lieberman, J. miR-200-containing extracellular vesicles promote breast cancer cell metastasis. J. Clin. Investig. 2014, 124, 5109–5128. [Google Scholar] [CrossRef]
- Dykxhoorn, D.M.; Wu, Y.; Xie, H.; Yu, F.; Lal, A.; Petrocca, F.; Martinvalet, D.; Song, E.; Lim, B.; Lieberman, J. miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS ONE 2009, 4, e7181. [Google Scholar] [CrossRef]
- Lemke, J.; Scheele, J.; Kapapa, T.; Wirtz, C.R.; Henne-Bruns, D.; Kornmann, M. Brain metastasis in pancreatic cancer. Int. J. Mol. Sci. 2013, 14, 4163–4173. [Google Scholar] [CrossRef]
- Ye, W.; Zhao, Y.; Li, H.; Na, R.; Li, F.; Mei, Q.; Zhao, M.; Zhou, S. Doxorubicin-poly (ethylene glycol)-alendronate self-assembled micelles for targeted therapy of bone metastatic cancer. Sci. Rep. 2015, 5, 14614. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Thota, R.; Pauff, J.M.; Berlin, J.D. Treatment of metastatic pancreatic adenocarcinoma: A review. Oncology 2014, 28, 70–74. [Google Scholar]
- De Dosso, S.; Siebenhüner, A.R.; Winder, T.; Meisel, A.; Fritsch, R.; Astaras, C.; Szturz, P.; Borner, M. Treatment landscape of metastatic pancreatic cancer. Cancer Treat. Rev. 2021, 96, 102180. [Google Scholar] [CrossRef]
- Lamb, Y.N.; Scott, L.J. Liposomal Irinotecan: A Review in Metastatic Pancreatic Adenocarcinoma. Drugs 2017, 77, 785–792. [Google Scholar] [CrossRef]
- Tempero, M.A.; Van Cutsem, E.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. HALO 109-301: A randomized, double-blind, placebo-controlled, phase 3 study of pegvorhyaluronidase alfa (PEGPH20) + nab-paclitaxel/gemcitabine (AG) in patients (pts) with previously untreated hyaluronan (HA)-high metastatic pancreatic ductal adenocarcinoma (mPDA). J. Clin. Oncol. 2020, 38, 638. [Google Scholar] [CrossRef]
- Liu, H.-C.; Davila Gonzalez, D.; Viswanath, D.I.; Vander Pol, R.S.; Saunders, S.Z.; Di Trani, N.; Xu, Y.; Zheng, J.; Chen, S.-H.; Chua, C.Y.X.; et al. Sustained Intratumoral Administration of Agonist CD40 Antibody Overcomes Immunosuppressive Tumor Microenvironment in Pancreatic Cancer. Adv. Sci. 2023, 10, 2206873. [Google Scholar] [CrossRef]
- Lim, C.Y.; Chang, J.H.; Lee, W.S.; Kim, J.; Park, I.Y. CD40 Agonists Alter the Pancreatic Cancer Microenvironment by Shifting the Macrophage Phenotype toward M1 and Suppress Human Pancreatic Cancer in Organotypic Slice Cultures. Gut Liver 2022, 16, 645–659. [Google Scholar] [CrossRef]
- Wattenberg, M.M.; Herrera, V.M.; Giannone, M.A.; Gladney, W.L.; Carpenter, E.L.; Beatty, G.L. Systemic inflammation is a determinant of outcomes of CD40 agonist–based therapy in pancreatic cancer patients. JCI Insight 2021, 6, e145389. [Google Scholar] [CrossRef]
- Beck, J.D.; Diken, M.; Suchan, M.; Streuber, M.; Diken, E.; Kolb, L.; Allnoch, L.; Vascotto, F.; Peters, D.; Beißert, T.; et al. Long-lasting mRNA-encoded interleukin-2 restores CD8+ T cell neoantigen immunity in MHC class I-deficient cancers. Cancer Cell 2024, 42, 568–582. [Google Scholar] [CrossRef]
- The moving target of cancer cell plasticity. Nat. Cancer 2022, 3, 1013–1014. [CrossRef]
- Wang, L.; Sun, Y. Efflux mechanism and pathway of verapamil pumping by human P-glycoprotein. Arch. Biochem. Biophys. 2020, 696, 108675. [Google Scholar] [CrossRef]
- Hoeben, A.; Joosten, E.A.J.; van den Beuken-van Everdingen, M.H.J. Personalized Medicine: Recent Progress in Cancer Therapy. Cancers 2021, 13, 242. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Bender, R.J.; Halverson, D.; Rahib, L.; Hendifar, A.E.; Mikhail, S.; Chung, V.; Picozzi, V.J.; Sohal, D.; Blais, E.M.; et al. Molecular Profiling of Patients with Pancreatic Cancer: Initial Results from the Know Your Tumor Initiative. Clin. Cancer Res. 2018, 24, 5018–5027. [Google Scholar] [CrossRef]
- Sahin, U.; Muik, A.; Vogler, I.; Derhovanessian, E.; Kranz, L.M.; Vormehr, M.; Quandt, J.; Bidmon, N.; Ulges, A.; Baum, A.; et al. BNT162b2 vaccine induces neutralizing antibodies and poly-specific T cells in humans. Nature 2021, 595, 572–577. [Google Scholar] [CrossRef]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef]
- Jocham, D.; Richter, A.; Hoffmann, L.; Iwig, K.; Fahlenkamp, D.; Zakrzewski, G.; Schmitt, E.; Dannenberg, T.; Lehmacher, W.; von Wietersheim, J.; et al. Adjuvant autologous renal tumour cell vaccine and risk of tumour progression in patients with renal-cell carcinoma after radical nephrectomy: Phase III, randomised controlled trial. Lancet Lond. Engl. 2004, 363, 594–599. [Google Scholar] [CrossRef]
- Vaccentis Pipeline (o. J.). Available online: https://vaccentis.com/pipeline/ (accessed on 1 August 2024).
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- BioNTech Press Release (7 April 2024) (o. J.). Available online: https://investors.biontech.de/news-releases/news-release-details/three-year-phase-1-follow-data-mrna-based-individualized/ (accessed on 1 August 2024).
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Wang, H.; Li, J.; He, J.; Liu, Y.; Feng, W.; Zhou, H.; Zhou, M.; Wei, H.; Lu, Y.; Peng, W.; et al. Methyl-CpG-binding protein 2 drives the Furin/TGF-β1/Smad axis to promote epithelial-mesenchymal transition in pancreatic cancer cells. Oncogenesis 2020, 9, 76. [Google Scholar] [CrossRef]
- Yang, Y.; Ye, W.-L.; Zhang, R.-N.; He, X.-S.; Wang, J.-R.; Liu, Y.-X.; Wang, Y.; Yang, X.-M.; Zhang, Y.-J.; Gan, W.-J. The Role of TGF-β Signaling Pathways in Cancer and Its Potential as a Therapeutic Target. Evid. Based Complement. Altern. Med. 2021, 2021, 6675208. [Google Scholar] [CrossRef]
- Feldmann, G.; Dhara, S.; Fendrich, V.; Bedja, D.; Beaty, R.; Mullendore, M.; Karikari, C.; Alvarez, H.; Iacobuzio-Donahue, C.; Jimeno, A.; et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: A new paradigm for combination therapy in solid cancers. Cancer Res. 2007, 67, 2187–2196. [Google Scholar] [CrossRef]
- Xie, H.; Paradise, B.D.; Ma, W.W.; Fernandez-Zapico, M.E. Recent Advances in the Clinical Targeting of Hedgehog/GLI Signaling in Cancer. Cells 2019, 8, 394. [Google Scholar] [CrossRef]
- Liu, Y.; Feng, M.; Chen, H.; Yang, G.; Qiu, J.; Zhao, F.; Cao, Z.; Luo, W.; Xiao, J.; You, L.; et al. Mechanistic target of rapamycin in the tumor microenvironment and its potential as a therapeutic target for pancreatic cancer. Cancer Lett. 2020, 485, 1–13. [Google Scholar] [CrossRef]
- Liu, Y.; Azizian, N.G.; Sullivan, D.K.; Li, Y. mTOR inhibition attenuates chemosensitivity through the induction of chemotherapy resistant persisters. Nat. Commun. 2022, 13, 7047. [Google Scholar] [CrossRef]
- Takeshita, F.; Patrawala, L.; Osaki, M.; Takahashi, R.; Yamamoto, Y.; Kosaka, N.; Kawamata, M.; Kelnar, K.; Bader, A.G.; Brown, D.; et al. Systemic delivery of synthetic microRNA-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 181–187. [Google Scholar] [CrossRef]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef]
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef]
- Kay, M.A. State-of-the-art gene-based therapies: The road ahead. Nat. Rev. Genet. 2011, 12, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Ertl, H.C.J. Immunogenicity and toxicity of AAV gene therapy. Front. Immunol. 2022, 13, 975803. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Samulski, R.J.; Berns, K.I.; Tan, M.; Muzyczka, N. Cloning of adeno-associated virus into pBR322: Rescue of intact virus from the recombinant plasmid in human cells. Proc. Natl. Acad. Sci. USA 1982, 79, 2077–2081. [Google Scholar] [CrossRef] [PubMed]
- Ragni, M.V. Hemophilia as a blueprint for gene therapy. Science 2021, 374, 40–41. [Google Scholar] [CrossRef]
- Hamann, M.V.; Beschorner, N.; Vu, X.-K.; Hauber, I.; Lange, U.C.; Traenkle, B.; Kaiser, P.D.; Foth, D.; Schneider, C.; Büning, H.; et al. Improved targeting of human CD4+ T cells by nanobody-modified AAV2 gene therapy vectors. PLoS ONE 2021, 16, e0261269. [Google Scholar] [CrossRef]
- Dudek, A.M.; Zabaleta, N.; Zinn, E.; Pillay, S.; Zengel, J.; Porter, C.; Franceschini, J.S.; Estelien, R.; Carette, J.E.; Zhou, G.L.; et al. GPR108 Is a Highly Conserved AAV Entry Factor. Mol. Ther. 2020, 28, 367–381. [Google Scholar] [CrossRef]
- Asokan, A.; Shen, S. Redirecting AAV vectors to extrahepatic tissues. Mol. Ther. J. Am. Soc. Gene Ther. 2023, 31, 3371–3375. [Google Scholar] [CrossRef]
- Fong, S.; Yates, B.; Sihn, C.-R.; Mattis, A.N.; Mitchell, N.; Liu, S.; Russell, C.B.; Kim, B.; Lawal, A.; Rangarajan, S.; et al. Interindividual variability in transgene mRNA and protein production following adeno-associated virus gene therapy for hemophilia A. Nat. Med. 2022, 28, 789–797. [Google Scholar] [CrossRef]
- Atchison, R.W.; Casto, B.C.; Hammon, W.M. Adenovirus-Associated Defective Virus Particles. Science 1965, 149, 754–756. [Google Scholar] [CrossRef]
- Gonzalez-Sandoval, A.; Pekrun, K.; Tsuji, S.; Zhang, F.; Hung, K.L.; Chang, H.Y.; Kay, M.A. The AAV capsid can influence the epigenetic marking of rAAV delivered episomal genomes in a species dependent manner. Nat. Commun. 2023, 14, 2448. [Google Scholar] [CrossRef]
- Büning, H. AAV Entry: Filling in the Blanks. Mol. Ther. 2020, 28, 346–347. [Google Scholar] [CrossRef] [PubMed]
- Summerford, C.; Johnson, J.S.; Samulski, R.J. AAVR: A Multi-Serotype Receptor for AAV. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Sihn, C.-R.; Handyside, B.; Liu, S.; Zhang, L.; Murphy, R.; Yates, B.; Xie, L.; Torres, R.; Russell, C.B.; O’Neill, C.A.; et al. Molecular analysis of AAV5-hFVIII-SQ vector-genome-processing kinetics in transduced mouse and nonhuman primate livers. Mol. Ther. Methods Clin. Dev. 2022, 24, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Malech, H.L.; Garabedian, E.K.; Hsieh, M.M. Evolution of Gene Therapy, Historical Perspective. Hematol. Oncol. Clin. North Am. 2022, 36, 627–645. [Google Scholar] [CrossRef]
- Wade, N. Patient Dies during a Trial of Therapy Using Genes. New York Times 1999, 29, A24. [Google Scholar]
- Conese, M. Engineered Virus to Treat Lipoprotein Lipase Deficiency. Adv. Genet. Eng. 2013, 2, e104. [Google Scholar] [CrossRef]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef]
- Ay, C.; Frenzel, L.; Pinachyan, K.; Le Quellec, S. Gene therapy for haemophilia A and B, from basic principles to clinical implementation: An illustrated review. Haemophilia 2024, 30, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Muhuri, M.; Levy, D.I.; Schulz, M.; McCarty, D.; Gao, G. Durability of transgene expression after rAAV gene therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2022, 30, 1364–1380. [Google Scholar] [CrossRef] [PubMed]
- Handyside, B.; Ismail, A.M.; Zhang, L.; Yates, B.; Xie, L.; Sihn, C.-R.; Murphy, R.; Bouwman, T.; Kim, C.K.; De Angelis, R.; et al. Vector genome loss and epigenetic modifications mediate decline in transgene expression of AAV5 vectors produced in mammalian and insect cells. Mol. Ther. 2022, 30, 3570–3586. [Google Scholar] [CrossRef]
- Sheridan, C. For hemophilia and thalassemia, a new era of “one-and-done” gene therapies has arrived. Nat. Biotechnol. 2022, 40, 1531–1533. [Google Scholar] [CrossRef]
- High, K.A. The gene therapy journey for hemophilia: Are we there yet? Blood 2012, 120, 4482–4487. [Google Scholar] [CrossRef] [PubMed]
- Cecchin, R.; Troyer, Z.; Witwer, K.; Morris, K.V. Extracellular vesicles: The next generation in gene therapy delivery. Mol. Ther. J. Am. Soc. Gene Ther. 2023, 31, 1225–1230. [Google Scholar] [CrossRef]
- Ay, C.; Reinisch, A. Gene therapy: Principles, challenges and use in clinical practice. Wien. Klin. Wochenschr. 2024; ahead of print. [Google Scholar] [CrossRef]
- Anguela, X.M.; High, K.A. Hemophilia B and gene therapy: A new chapter with etranacogene dezaparvovec. Blood Adv. 2024, 8, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Samelson-Jones, B.J.; George, L.A. Adeno-Associated Virus Gene Therapy for Hemophilia. Annu. Rev. Med. 2023, 74, 231–247. [Google Scholar] [CrossRef] [PubMed]
- Ten Ham, R.M.T.; Walker, S.M.; Soares, M.O.; Frederix, G.W.J.; Leebeek, F.W.G.; Fischer, K.; Coppens, M.; Palmer, S.J. Modeling Benefits, Costs, and Affordability of a Novel Gene Therapy in Hemophilia A. HemaSphere 2022, 6, e679. [Google Scholar] [CrossRef]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated virus serotypes: Vector toolkit for human gene therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef]
- Coppens, M.; Pipe, S.W.; Miesbach, W.; Astermark, J.; Recht, M.; van der Valk, P.; Ewenstein, B.; Pinachyan, K.; Galante, N.; Le Quellec, S.; et al. Etranacogene dezaparvovec gene therapy for haemophilia B (HOPE-B): 24-month post-hoc efficacy and safety data from a single-arm, multicentre, phase 3 trial. Lancet Haematol. 2024, 11, e265–e275. [Google Scholar] [CrossRef]
- Mahlangu, J.; Kaczmarek, R.; von Drygalski, A.; Shapiro, S.; Chou, S.-C.; Ozelo, M.C.; Kenet, G.; Peyvandi, F.; Wang, M.; Madan, B.; et al. Two-Year Outcomes of Valoctocogene Roxaparvovec Therapy for Hemophilia A. N. Engl. J. Med. 2023, 388, 694–705. [Google Scholar] [CrossRef]
- Wang, J.-H.; Gessler, D.J.; Zhan, W.; Gallagher, T.L.; Gao, G. Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduct Target Ther. 2024, 9, 78. [Google Scholar] [CrossRef]
- Li, J.; Røise, J.J.; He, M.; Das, R.; Murthy, N. Non-viral strategies for delivering genome editing enzymes. Adv. Drug Deliv. Rev. 2021, 168, 99–117. [Google Scholar] [CrossRef]
- Khalil, A.M. The genome editing revolution: Review. J. Genet. Eng. Biotechnol. 2020, 18, 68. [Google Scholar] [CrossRef]
- Sayed, N.; Allawadhi, P.; Khurana, A.; Singh, V.; Navik, U.; Pasumarthi, S.K.; Khurana, I.; Banothu, A.K.; Weiskirchen, R.; Bharani, K.K. Gene therapy: Comprehensive overview and therapeutic applications. Life Sci. 2022, 294, 120375. [Google Scholar] [CrossRef]
- Frangoul, H.; Locatelli, F.; Sharma, A.; Bhatia, M.; Mapara, M.; Molinari, L.; Wall, D.; Liem, R.I.; Telfer, P.; Shah, A.J.; et al. Exagamglogene Autotemcel for Severe Sickle Cell Disease. N. Engl. J. Med. 2024, 390, 1649–1662. [Google Scholar] [CrossRef]
- Alliance for Regenerative Medicine (o. J.). Cell and Gene Therapy Sector Data (Zugriff vom 13.07.2024). Available online: https://alliancerm.org/wp-content/uploads/2024/05/Trials_Final_Q1_2024.pdf (accessed on 15 July 2024).
- Madigan, V.; Zhang, F.; Dahlman, J.E. Drug delivery systems for CRISPR-based genome editors. Nat. Rev. Drug Discov. 2023, 22, 875–894. [Google Scholar] [CrossRef]

| Hallmarks of Pancreatic Cancer | Challenges for Drug Delivery | References | |
|---|---|---|---|
| Tumor Microenvironment | Abnormal Vasculature: Poor Perfusion & Low Oxygen | Poor access to target cells | [76,79,109] |
| Abnormal Vasculature: IFP | Pressure gradient | [79,109] | |
| Abnormal Vasculature: Acidic pH | Degradation | [98] | |
| Fibrotic Stroma | Physical barrier | [79,109] | |
| Immunologic Barrier | Immunosuppressive milieu, PD-L1 expression | [76,79,80,81,83] | |
| Genetic Diversity and Clonal Evolution | Tumor Heterogeneity | Multitude of targets Differences between patients | [93,94,97,98,99] |
| Clonal Evolution | Changing targets and escape mutations | [97,98] | |
| Chemotherapy Resistance | Efflux Pumps, mTOR inhibition | [98,99,100,109,140] | |
| Metastatic Potential | EMT | Changing targets due to phenotypic alterations | [109] |
| Circulating Tumor Cells | Moving targets & protection by a shield of platelets | [102] | |
Distant Organs
poor access to bone marrow | Physical barriers (BBB) Uneven poor drug distribution No access to target cells | [110,113] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basar, E.; Mead, H.; Shum, B.; Rauter, I.; Ay, C.; Skaletz-Rorowski, A.; Brockmeyer, N.H. Biological Barriers for Drug Delivery and Development of Innovative Therapeutic Approaches in HIV, Pancreatic Cancer, and Hemophilia A/B. Pharmaceutics 2024, 16, 1207. https://doi.org/10.3390/pharmaceutics16091207
Basar E, Mead H, Shum B, Rauter I, Ay C, Skaletz-Rorowski A, Brockmeyer NH. Biological Barriers for Drug Delivery and Development of Innovative Therapeutic Approaches in HIV, Pancreatic Cancer, and Hemophilia A/B. Pharmaceutics. 2024; 16(9):1207. https://doi.org/10.3390/pharmaceutics16091207
Chicago/Turabian StyleBasar, Emre, Henry Mead, Bennett Shum, Ingrid Rauter, Cihan Ay, Adriane Skaletz-Rorowski, and Norbert H. Brockmeyer. 2024. "Biological Barriers for Drug Delivery and Development of Innovative Therapeutic Approaches in HIV, Pancreatic Cancer, and Hemophilia A/B" Pharmaceutics 16, no. 9: 1207. https://doi.org/10.3390/pharmaceutics16091207