
Unexplored Roles of Erythrocytes in Atherothrombotic Stroke

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Red Blood Cells Participate in Innate Immunity
3. Red Blood Cells in Atherosclerosis
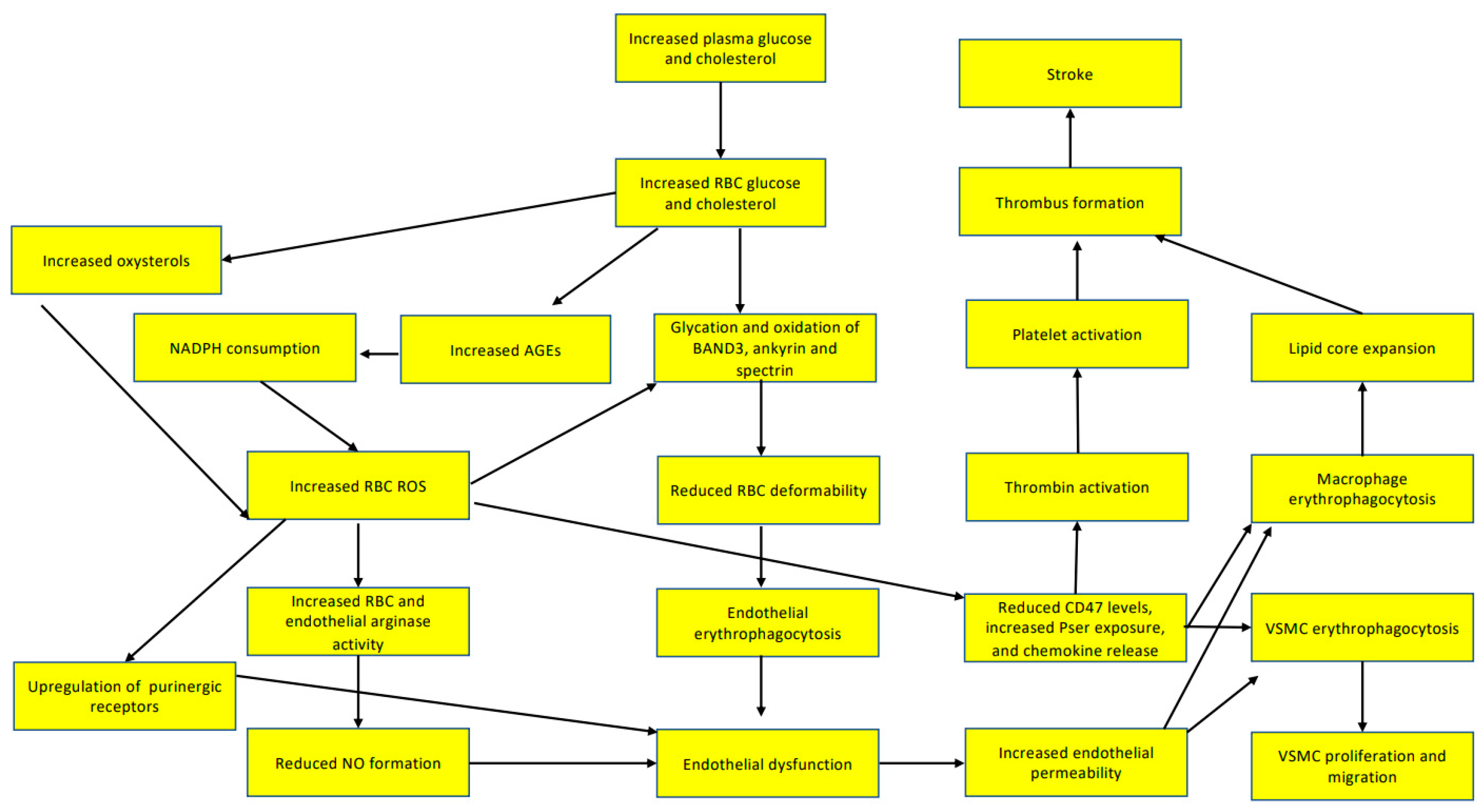
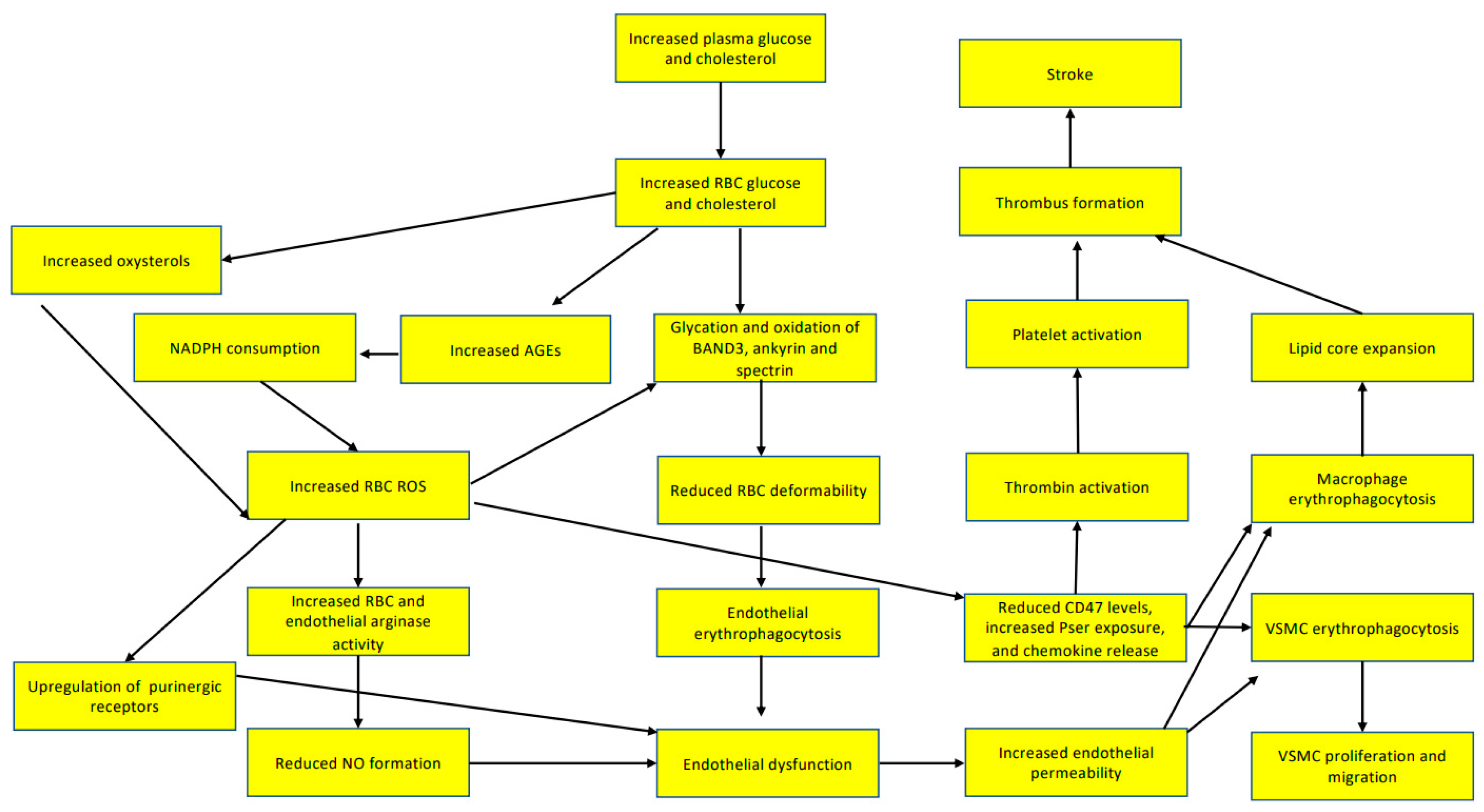
3.1. Red Blood Cells Participate in Endothelial Dysfunction
3.2. Red Blood Cells Participate in Lipid Core Expansion of Atherosclerotic Plaques
3.3. Red Blood Cells Participate in the Inflammatory Activation of Macrophages in Atherosclerotic Plaques
3.4. Red Blood Cells Participate in Vascular Smooth Muscle Cell Function
3.5. Red Blood Cells Participate in T-Cell Activation
4. Red Blood Cells Participate in Thrombus Formation
4.1. Red Blood Cells Determine Blood Viscosity
4.2. Red Blood Cells Participate in Neutrophil Extracellular Traps of Atherothrombotic Lesions
4.3. Red Blood Cells Participate in Platelet Activation
5. Red Blood Cells Participate in Thrombus Stabilization
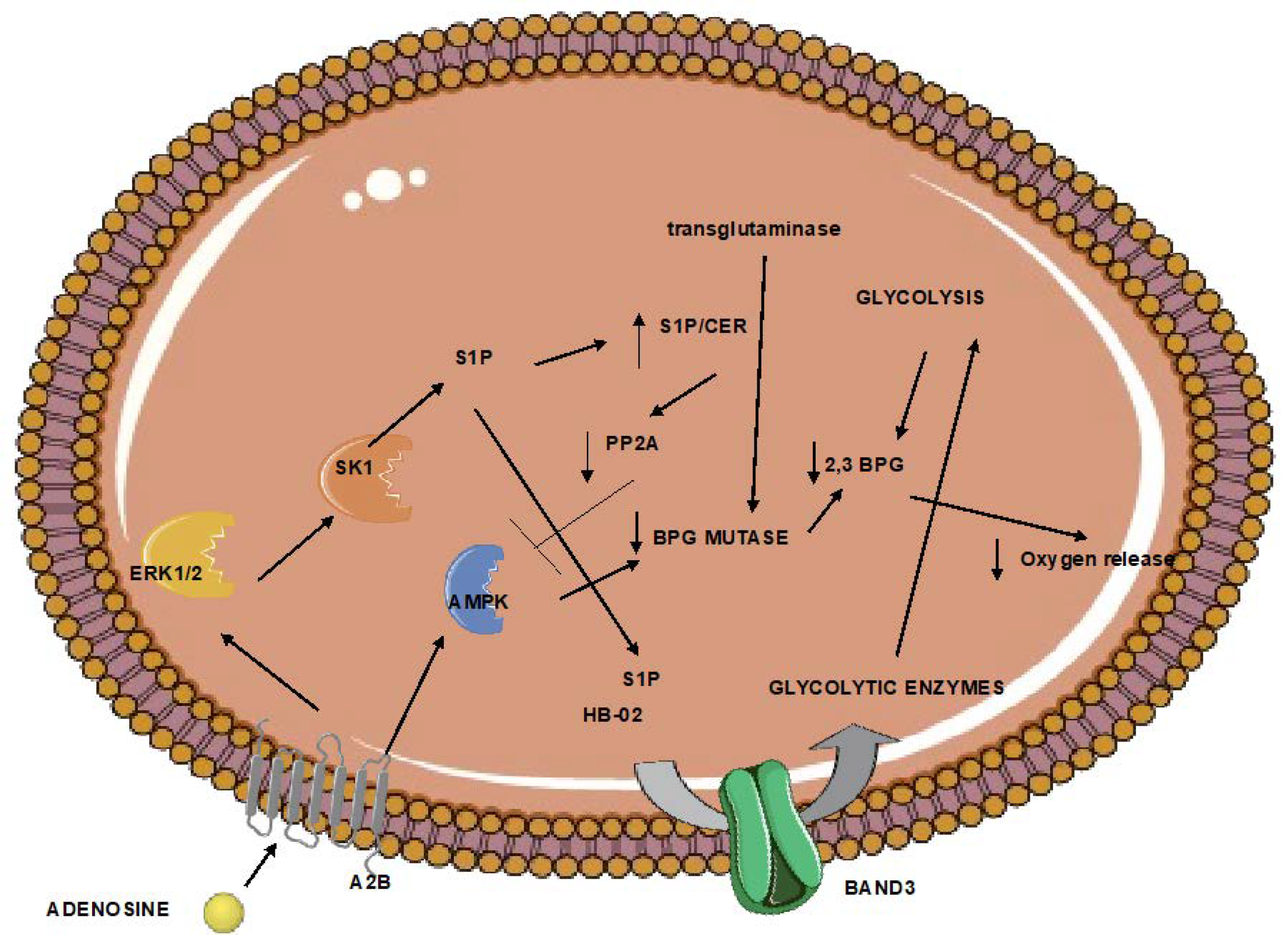
6. Red Blood Cells Participate in Hypoxia after Atherothrombotic Stroke
7. Red blood Cells Respond to Damage-Associated Molecular Patterns Released after Ischemic Stroke
7.1. Cell-Free Mitochondrial DNA and CpG DNA
7.2. ATP
7.3. Extracellular Histones
7.4. Lipopolysaccharides (LPS)
7.5. Amyloids
8. Red Blood Cells Could Connect Non-Alcoholic Fatty Liver Disease with the Risk of Atherothrombotic Strokes
9. Future Perspectives
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2,3-BPG | 2,3 biphosphoglycerate |
| ADP | adenosine diphosphate |
| AMPK | adenosine monophosphate-dependent kinase |
| ATP | adenosine triphosphate |
| ADMA | asymmetric dimethylarginine |
| BPG mutase | Biphosphoglycerate mutase |
| CD47 | cluster of differentiation 47 |
| CER | ceramide |
| DALYs | disability-adjusted life-years |
| DAMP | damage-associated molecular patterns |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| FASR | FAS cell surface death receptor |
| FASL | FAS ligand |
| I-CAM | intercellular adhesion molecule 1 |
| IL-8 | interleukin 8 |
| IL-17 | interleukin 17 |
| IFN-γ | interferon-γ |
| KA | kynurenic acid |
| LPS | lipopolysaccharide |
| MGF-E8 | milk fat globule-EGF factor 8 protein |
| MCP1 | monocyte chemoattractant protein 1 |
| mtDNA | mitochondrial DNA |
| NET | neutrophil extracellular traps |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NF-Κβ | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | nitric oxide |
| PAMP | pathogen-associated molecular patterns |
| PKCζ | protein kinase C ζ |
| ROS | reactive oxygen species |
| S1P | sphingosine 1-phosphate |
| SK1 | Sphingosine kinase 1 |
| Th17 | T helper cells 17 |
| TLR2 | toll-like receptor 2 |
| TLR9 | Toll-like receptor 9 |
| V-CAM | vascular cell adhesion protein |
| VSMC | vascular smooth muscle cell |
References
- Saini, V.; Guada, L.; Yavagal, D.R. Global Epidemiology of Stroke and Access to Acute Ischemic Stroke Interventions. Neurology 2021, 97 (Suppl. S2), S6–S16. [Google Scholar] [CrossRef] [PubMed]
- Rosales, S.; Alet, M.; Lereis, V.P.; Ameriso, S.F. Abstract P561: Fall in the Proportion of Atherothrombotic Stroke in the Last Decade. Success of Current Medical Treatment? Circulation 2020, 141 (Suppl. S1). [Google Scholar] [CrossRef]
- Asada, Y.; Yamashita, A.; Sato, Y.; Hatakeyama, K. Pathophysiology of Atherothrombosis: Mechanisms of Thrombus Formation on Disrupted Atherosclerotic Plaques. Pathol. Int. 2020, 70, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, P.R.; Fuster, V. New Aspects in the Pathogenesis of Diabetic Atherothrombosis. J. Am. Coll. Cardiol. 2004, 44, 2293–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lentz, S.R. Mechanisms of Homocysteine-Induced Atherothrombosis. J. Thromb. Haemost. 2005, 3, 1646–1654. [Google Scholar] [CrossRef]
- Rocha, V.Z.; Santos, R.D. Cholesterol and Inflammation: The Lesser the Better in Atherothrombosis. Eur. J. Prev. Cardiol. 2018, 25, 944–947. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity 2017, 47, 406–420. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, C.; Panopoulou, M.; Anagnostopoulos, K.; Tentes, I. Immune and Metabolic Interactions of Human Erythrocytes: A Molecular Perspective. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 843–853. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Anagnostopoulos, K.; Tentes, I. Erythrocytes contribute to the immunometabolic cross-talk. Immunometabolism 2021, 3, e210015. [Google Scholar]
- Papadopoulos, C.; Tentes, I.; Anagnostopoulos, K. Lipotoxicity Disrupts Erythrocyte Function: A Perspective. Cardiovasc. Hematol. Disord. Drug Targets 2021, 21, 91–94. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Tentes, I.; Anagnostopoulos, K. Molecular Interactions between Erythrocytes and the Endocrine System. Maedica 2021, 16, 489. [Google Scholar] [CrossRef]
- Papadopoulos, C. Erythrocyte Glucotoxicity Results in Vascular Inflammation. Endocr. Metab. Immune Disord. Drug Targets 2022, 22, 901–903. [Google Scholar] [CrossRef]
- Papadopoulos, C. Immunosuppressive Function of Intratumor Red Blood Cells: An Immune-Metabolic Perspective. Curr. Cancer Rev. 2022, 18, 224–226. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Wautier, J.L.; Wautier, M.P.; Schmidt, A.M.; Anderson, G.M.; Hori, O.; Zoukourian, C.; Capron, L.; Chappey, O.; Yan, S.D.; Brett, J.; et al. Advanced Glycation End Products (AGEs) on the Surface of Diabetic Erythrocytes Bind to the Vessel Wall via a Specific Receptor Inducing Oxidant Stress in the Vasculature: A Link between Surface-Associated AGEs and Diabetic Complications. Proc. Natl. Acad. Sci. USA 1994, 91, 7742–7746. [Google Scholar] [CrossRef] [Green Version]
- Catan, A.; Turpin, C.; Diotel, N.; Patche, J.; Guerin-Dubourg, A.; Debussche, X.; Bourdon, E.; Ah-You, N.; le Moullec, N.; Besnard, M.; et al. Aging and Glycation Promote Erythrocyte Phagocytosis by Human Endothelial Cells: Potential Impact in Atherothrombosis under Diabetic Conditions. Atherosclerosis 2019, 291, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Mahdi, A.; Tratsiakovich, Y.; Zahorán, S.; Kövamees, O.; Nordin, F.; Uribe Gonzalez, A.E.; Alvarsson, M.; Östenson, C.G.; Andersson, D.C.; et al. Erythrocytes from Patients with Type 2 Diabetes Induce Endothelial Dysfunction via Arginase I. J. Am. Coll. Cardiol. 2018, 72, 769–780. [Google Scholar] [CrossRef]
- Mahdi, A.; Tratsiakovich, Y.; Tengbom, J.; Jiao, T.; Garib, L.; Alvarsson, M.; Yang, J.; Pernow, J.; Zhou, Z. Erythrocytes Induce Endothelial Injury in Type 2 Diabetes Through Alteration of Vascular Purinergic Signaling. Front. Pharmacol. 2020, 11, 603226. [Google Scholar] [CrossRef]
- Minetti, M.; Agati, L.; Malorni, W. The microenvironment can shift erythrocytes from a friendly to a harmful behavior: Pathogenetic implications for vascular diseases. Cardiovasc. Res. 2007, 75, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Mahdi, A.; Tengbom, J.; Alvarsson, M.; Wernly, B.; Zhou, Z.; Pernow, J. Red Blood Cell Peroxynitrite Causes Endothelial Dysfunction in Type 2 Diabetes Mellitus via Arginase. Cells 2020, 9, 1712. [Google Scholar] [CrossRef]
- Zhou, Z.; Collado, A.; Sun, C.; Tratsiakovich, Y.; Mahdi, A.; Winter, H.; Chernogubova, E.; Seime, T.; Narayanan, S.; Jiao, T.; et al. Downregulation of Erythrocyte MiR-210 Induces Endothelial Dysfunction in Type 2 Diabetes. Diabetes 2022, 71, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, Y.; Fujii, S.; Kaneko, T. Human Erythrocyte Sorbitol Metabolism and the Role of Sorbitol Dehydrogenase. Diabetologia 1988, 31, 766–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suissa, L.; Guigonis, J.M.; Graslin, F.; Doche, E.; Osman, O.; Chau, Y.; Sedat, J.; Lindenthal, S.; Pourcher, T. Metabolome of Cerebral Thrombi Reveals an Association between High Glycemia at Stroke Onset and Good Clinical Outcome. Metabolites 2020, 10, 483. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, A.; Wodaje, T.; Kövamees, O.; Tengbom, J.; Zhao, A.; Jiao, T.; Henricsson, M.; Yang, J.; Zhou, Z.; Nieminen, A.I.; et al. The Red Blood Cell as a Mediator of Endothelial Dysfunction in Patients with Familial Hypercholesterolemia and Dyslipidemia. J. Intern. Med. 2022, 293, 228–245. [Google Scholar] [CrossRef] [PubMed]
- Unruh, D.; Srinivasan, R.; Benson, T.; Haigh, S.; Coyle, D.; Batra, N.; Keil, R.; Sturm, R.; Blanco, V.; Palascak, M.; et al. Red Blood Cell Dysfunction Induced by High-Fat Diet. Circulation 2015, 132, 1898–1908. [Google Scholar] [CrossRef] [Green Version]
- Cilla, A.; López-García, G.; Collado-Díaz, V.; Amparo Blanch-Ruiz, M.; Garcia-Llatas, G.; Barberá, R.; Martinez-Cuesta, M.A.; Real, J.T.; Álvarez, Á.; Martínez-Hervás, S. Hypercholesterolemic Patients Have Higher Eryptosis and Erythrocyte Adhesion to Human Endothelium Independently of Statin Therapy. Int. J. Clin. Pract. 2021, 75, e14771. [Google Scholar] [CrossRef]
- Tesoriere, L.; Attanzio, A.; Allegra, M.; Cilla, A.; Gentile, C.; Livrea, M.A. Oxysterol Mixture in Hypercholesterolemia-Relevant Proportion Causes Oxidative Stress-Dependent Eryptosis. Cell. Physiol. Biochem. 2014, 34, 1075–1089. [Google Scholar] [CrossRef] [Green Version]
- Attanzio, A.; Frazzitta, A.; Cilla, A.; Livrea, M.A.; Tesoriere, L.; Allegra, M. 7-Keto-Cholesterol and Cholestan-3β, 5α, 6β-Triol Induce Eryptosis through Distinct Pathways Leading to NADPH Oxidase and Nitric Oxide Synthase Activation. Cell. Physiol. Biochem. 2019, 53, 933–947. [Google Scholar] [CrossRef] [Green Version]
- Ballas, S.K.; Mohandas, N.; Marton, L.J.; Shohet, S.B. Stabilization of Erythrocyte Membranes by Polyamines. Proc. Natl. Acad. Sci. USA 1983, 80, 1942–1946. [Google Scholar] [CrossRef] [Green Version]
- Fens, M.H.A.M.; Storm, G.; Pelgrim, R.C.M.; Ultee, A.; Byrne, A.T.; Gaillard, C.A.; van Solinge, W.W.; Schiffelers, R.M. Erythrophagocytosis by Angiogenic Endothelial Cells Is Enhanced by Loss of Erythrocyte Deformability. Exp. Hematol. 2010, 38, 282–291. [Google Scholar] [CrossRef]
- Sun, J.; Vyas, P.; Mann, S.; Paganini-Hill, A.; Nunes, A.C.F.; Lau, W.L.; Cribbs, D.H.; Fisher, M.J.; Sumbria, R.K. Insights Into the Mechanisms of Brain Endothelial Erythrophagocytosis. Front. Cell Dev. Biol. 2021, 9, 1795. [Google Scholar] [CrossRef]
- Chang, R.; Castillo, J.; Zambon, A.C.; Krasieva, T.B.; Fisher, M.J.; Sumbria, R.K. Brain Endothelial Erythrophagocytosis and Hemoglobin Transmigration across Brain Endothelium: Implications for Pathogenesis of Cerebral Microbleeds. Front. Cell. Neurosci. 2018, 12, 279. [Google Scholar] [CrossRef] [Green Version]
- López, V.; Uribe, E.; Moraga, F.A. Activation of Arginase II by Asymmetric Dimethylarginine and Homocysteine in Hypertensive Rats Induced by Hypoxia: A New Model of Nitric Oxide Synthesis Regulation in Hypertensive Processes? Hypertens. Res. 2021, 44, 263–275. [Google Scholar] [CrossRef]
- Stühlinger, M.C.; Tsao, P.S.; Her, J.H.; Kimoto, M.; Balint, R.F.; Cooke, J.P. Homocysteine Impairs the Nitric Oxide Synthase Pathway. Circulation 2001, 104, 2569–2575. [Google Scholar] [CrossRef]
- Nemkov, T.; Sun, K.; Reisz, J.A.; Song, A.; Yoshida, T.; Dunham, A.; Wither, M.J.; Francis, R.O.; Roach, R.C.; Dzieciatkowska, M.; et al. Hypoxia Modulates the Purine Salvage Pathway and Decreases Red Blood Cell and Supernatant Levels of Hypoxanthine during Refrigerated Storage. Haematologica 2018, 103, 361. [Google Scholar] [CrossRef] [Green Version]
- Xie, R.; Jia, D.; Gao, C.; Zhou, J.; Sui, H.; Wei, X.; Zhang, T.; Han, Y.; Shi, J.; Bai, Y. Homocysteine Induces Procoagulant Activity of Red Blood Cells via Phosphatidylserine Exposure and Microparticles Generation. Amino Acids 2014, 46, 1997–2004. [Google Scholar] [CrossRef]
- Tziakas, D.N.; Chalikias, G.K.; Stakos, D.; Boudoulas, H. The Role of Red Blood Cells in the Progression and Instability of Atherosclerotic Plaque. Int. J. Cardiol. 2010, 142, 2–7. [Google Scholar] [CrossRef]
- Pavlaki, M.; Kourkouli, A.; Chalikias, G.; Kikas, P.; Tziakas, D. Cholesterol content of erythrocyte membranes and elusive target. Thromb. Res. 2020, 185, 32. [Google Scholar] [CrossRef] [Green Version]
- Tziakas, D.N.; Chalikias, G.K.; Boudoulas, H. Significance of the cholesterol content of erythrocyte membranes in atherosclerosis. Clin. Lipidol. 2010, 5, 449–452. [Google Scholar] [CrossRef]
- Tziakas, D.; Chalikias, G.; Kapelouzou, A.; Tentes, I.; Schäfer, K.; Karayannakos, P.; Kostakis, A.; Boudoulas, H.; Konstantinides, S. Erythrocyte Membrane Cholesterol and Lipid Core Growth in a Rabbit Model of Atherosclerosis: Modulatory Effects of Rosuvastatin. Int. J. Cardiol. 2013, 170, 173–181. [Google Scholar] [CrossRef]
- Giannoglou, G.D.; Koskinas, K.C.; Tziakas, D.N.; Ziakas, A.G.; Antoniadis, A.P.; Tentes, I.K.; Parcharidis, G.E. Total Cholesterol Content of Erythrocyte Membranes and Coronary Atherosclerosis: An Intravascular Ultrasound Pilot Study. Angiology 2009, 60, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Tziakas, D.N.; Chalikias, G.K.; Stakos, D.; Tentes, I.K.; Papazoglou, D.; Thomaidi, A.; Grapsa, A.; Gioka, G.; Kaski, J.C.; Boudoulas, H. Independent and Additive Predictive Value of Total Cholesterol Content of Erythrocyte Membranes with Regard to Coronary Artery Disease Clinical Presentation. Int. J. Cardiol. 2011, 150, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Tziakas, D.N.; Chalikias, G.K.; Stakos, D.; Tentes, I.K.; Chatzikyriakou, S.v.; Mitrousi, K.; Kortsaris, A.X.; Boudoulas, H.; Kaski, J.C. Cholesterol Composition of Erythrocyte Membranes and Its Association with Clinical Presentation of Coronary Artery Disease. Coron. Artery Dis. 2008, 19, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Tziakas, D.N.; Chalikias, G.K.; Tentes, I.K.; Stakos, D.; Chatzikyriakou, S.v.; Mitrousi, K.; Kortsaris, A.X.; Kaski, J.C.; Boudoulas, H. Interleukin-8 Is Increased in the Membrane of Circulating Erythrocytes in Patients with Acute Coronary Syndrome. Eur. Heart J. 2008, 29, 2713–2722. [Google Scholar] [CrossRef] [Green Version]
- Delbosc, S.; Bayles, R.G.; Laschet, J.; Ollivier, V.; Ho-Tin-Noé, B.; Touat, Z.; Deschildre, C.; Morvan, M.; Louedec, L.; Gouya, L.; et al. Erythrocyte Efferocytosis by the Arterial Wall Promotes Oxidation in Early-Stage Atheroma in Humans. Front. Cardiovasc. Med. 2017, 4, 43. [Google Scholar] [CrossRef] [Green Version]
- Pan, D.; Wu, W.; Zuo, G.; Xie, X.; Li, H.; Ren, X.; Kong, C.; Zhou, W.; Zhang, Z.; Waterfall, M.; et al. Sphingosine 1-Phosphate Receptor 2 Promotes Erythrocyte Clearance by Vascular Smooth Muscle Cells in Intraplaque Hemorrhage through MFG-E8 Production. Cell Signal. 2022, 98, 110419. [Google Scholar] [CrossRef]
- Gerner, M.C.; Bileck, A.; Janker, L.; Ziegler, L.S.; Öhlinger, T.; Raeven, P.; Müllner, E.W.; Salzer, U.; Gerner, C.; Schmetterer, K.G.; et al. Packed Red Blood Cells Inhibit T-Cell Activation via ROS-Dependent Signaling Pathways. J. Biol. Chem. 2021, 296, 100487. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Anagnostopoulos, K.; Tentes, I. An Immunometabolic Pathway for Hyperglycemia?—Commentary on: Packed Red Blood Cells Inhibit T-Cell Activation via ROS-Dependent Signaling Pathways. Immunometabolism 2021, 3, e210027. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Morabito, R.; Remigante, A.; Spinelli, S.; Vitale, G.; Trichilo, V.; Loddo, S.; Marino, A. high glucose concentrations affect band 3 protein in human erythrocytes. Antioxidants 2020, 9, 365. [Google Scholar] [CrossRef]
- Schwartz, R.S.; Madsen, J.W.; Rybicki, A.C.; Nagel, R.L. Oxidation of spectrin and deformability defects in diabetic erythrocytes. Diabetes 1991, 40, 701–708. [Google Scholar] [CrossRef]
- Shin, S.; Ku, Y.; Park, M.S.; Suh, S.J. Deformability of red blood cells: A determinant of blood viscosity. J. Mech. Sci. Technol. 2005, 19, 216–223. [Google Scholar] [CrossRef]
- Mostafa, M.N.; Osama, M. The Implications of Neutrophil Extracellular Traps in the pathophysiology of Atherosclerosis and Atherothrombosis. Exp. Biol. Med. 2020, 245, 1376–1384. [Google Scholar] [CrossRef]
- Chilingaryan, Z.; Deshmukh, T.; Leung, H.H.L.; Perdomo, J.; Emerson, P.; Kurup, R.; Chong, B.H.; Chong, J.J.H. Erythrocyte Interaction with Neutrophil Extracellular Traps in Coronary Artery Thrombosis Following Myocardial Infarction. Pathology 2022, 54, 87–94. [Google Scholar] [CrossRef]
- Chung, S.M.; Bae, O.N.; Lim, K.M.; Noh, J.Y.; Lee, M.Y.; Jung, Y.S.; Chung, J.H. Lysophosphatidic Acid Induces Thrombogenic Activity Through Phosphatidylserine Exposure and Procoagulant Microvesicle Generation in Human Erythrocytes. Arter. Thromb. Vasc. Biol. 2007, 27, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Vallés, J.; Teresa Santos, M.; Aznar, J.; Martínez, M.; Moscardó, A.; Piñón, M.; Johan Broekman, M.; Marcus, A.J. Platelet-Erythrocyte Interactions Enhance AIIbβ3 Integrin Receptor Activation and P-Selectin Expression during Platelet Recruitment: Down-Regulation by Aspirin Ex Vivo. Blood 2002, 99, 3978–3984. [Google Scholar] [CrossRef] [Green Version]
- Klatt, C.; Krüger, I.; Zey, S.; Krott, K.J.; Spelleken, M.; Gowert, N.S.; Oberhuber, A.; Pfaff, L.; Lückstädt, W.; Jurk, K.; et al. Platelet-RBC Interaction Mediated by FasL/FasR Induces Procoagulant Activity Important for Thrombosis. J. Clin. Investig. 2018, 128, 3906–3925. [Google Scholar] [CrossRef]
- Carroll, J.S.; Ku, C.J.; Karunarathne, W.; Spence, D.M. Red Blood Cell Stimulation of Platelet Nitric Oxide Production Indicated by Quantitative Monitoring of the Communication between Cells in the Bloodstream. Anal. Chem. 2007, 79, 5133–5138. [Google Scholar] [CrossRef]
- Bell, D.N.; Spain, S.; Goldsmith, H.L. The Effect of Red Blood Cells on the ADP-Induced Aggregation of Human Platelets in Flow through Tubes. Thromb. Haemost. 1990, 63, 112–121. [Google Scholar] [CrossRef]
- Aleman, M.M.; Byrnes, J.R.; Wang, J.G.; Tran, R.; Lam, W.A.; di Paola, J.; Mackman, N.; Degen, J.L.; Flick, M.J.; Wolberg, A.S. Factor XIII Activity Mediates Red Blood Cell Retention in Venous Thrombi. J. Clin. Investig. 2014, 124, 3590–3600. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, F.A.; Connell, S.; Miltenberger-Miltenyi, G.; Pereira, S.V.; Tavares, A.; Ariëns, R.A.S.; Santos, N.C. Atomic Force Microscopy-Based Molecular Recognition of a Fibrinogen Receptor on Human Erythrocytes. ACS Nano 2010, 4, 4609–4620. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, S.; de Almeida, V.V.; Calado, A.; Rosário, H.S.; Saldanha, C. Integrin-Associated Protein (CD47) Is a Putative Mediator for Soluble Fibrinogen Interaction with Human Red Blood Cells Membrane. Biochim. Biophys. Acta 2012, 1818, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maekawa, K.; Shibata, M.; Nakajima, H.; Mizutani, A.; Kitano, Y.; Seguchi, M.; Yamasaki, M.; Kobayashi, K.; Sano, T.; Mori, G.; et al. Erythrocyte-Rich Thrombus Is Associated with Reduced Number of Maneuvers and Procedure Time in Patients with Acute Ischemic Stroke Undergoing Mechanical Thrombectomy. Cereb. Dis. Extra 2018, 8, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Asaro, R.J.; Cabrales, P. Red Blood Cells: Tethering, Vesiculation, and Disease in Micro-Vascular Flow. Diagnostics 2021, 11, 971. [Google Scholar] [CrossRef]
- Xu, P.; Chen, C.; Zhang, Y.; Dzieciatkowska, M.; Brown, B.C.; Zhang, W.; Xie, T.; Abdulmalik, O.; Song, A.; Tong, C.; et al. Erythrocyte Transglutaminase-2 Combats Hypoxia and Chronic Kidney Disease by Promoting Oxygen Delivery and Carnitine Homeostasis. Cell Metab. 2022, 34, 299–316.e6. [Google Scholar] [CrossRef]
- Qiang, Q.; Manalo, J.M.; Sun, H.; Zhang, Y.; Song, A.; Wen, A.Q.; Edward Wen, Y.; Chen, C.; Liu, H.; Cui, Y.; et al. Erythrocyte Adenosine A2B Receptor Prevents Cognitive and Auditory Dysfunction by Promoting Hypoxic and Metabolic Reprogramming. PLoS Biol. 2021, 19, e3001239. [Google Scholar] [CrossRef]
- Sun, K.; Zhang, Y.; Bogdanov, M.v.; Wu, H.; Song, A.; Li, J.; Dowhan, W.; Idowu, M.; Juneja, H.S.; Molina, J.G.; et al. Elevated Adenosine Signaling via Adenosine A2B Receptor Induces Normal and Sickle Erythrocyte Sphingosine Kinase 1 Activity. Blood 2015, 125, 1643–1652. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Zhang, Y.; D’Alessandro, A.; Nemkov, T.; Song, A.; Wu, H.; Liu, H.; Adebiyi, M.; Huang, A.; Wen, Y.E.; et al. Sphingosine-1-Phosphate Promotes Erythrocyte Glycolysis and Oxygen Release for Adaptation to High-Altitude Hypoxia. Nat. Commun. 2016, 7, 12086. [Google Scholar] [CrossRef]
- Kimura, H.; Hamasaki, N.; Yamamoto, M.; Tomonaga, M. Circulation of Red Blood Cells Having High Levels of 2,3-Bisphosphoglycerate Protects Rat Brain from Ischemic Metabolic Changes during Hemodilution. Stroke 1995, 26, 1431–1437. [Google Scholar] [CrossRef]
- Domingo-Ortí, I.; Lamas-Domingo, R.; Ciudin, A.; Hernández, C.; Herance, J.R.; Palomino-Schätzlein, M.; Pineda-Lucena, A. Metabolic Footprint of Aging and Obesity in Red Blood Cells. Aging 2021, 13, 4850. [Google Scholar] [CrossRef]
- Stanzione, R.; Forte, M.; Cotugno, M.; Bianchi, F.; Marchitti, S.; Rubattu, S. Role of DAMPs and of Leukocytes Infiltration in Ischemic Stroke: Insights from Animal Models and Translation to the Human Disease. Cell. Mol. Neurobiol. 2022, 42, 545–556. [Google Scholar] [CrossRef]
- Hakoupian, M.; Ferino, E.; Jickling, G.C.; Amini, H.; Stamova, B.; Ander, B.P.; Alomar, N.; Sharp, F.R.; Zhan, X. Bacterial lipopolysaccharide is associated with stroke. Sci. Rep. 2021, 11, 6570. [Google Scholar] [CrossRef]
- Pfefferlé, M.; Ingoglia, G.; Schaer, C.A.; Yalamanoglu, A.; Buzzi, R.; Dubach, I.L.; Tan, G.; López-Cano, E.Y.; Schulthess, N.; Hansen, K.; et al. Hemolysis transforms liver macrophages into antiinflammatory erythrophagocytes. J. Clin. Investig. 2020, 130, 5576–5590. [Google Scholar] [CrossRef]
- Ni, W.; Mao, S.; Xi, G.; Keep, R.F.; Hua, Y. Role of Erythrocyte CD47 in Intracerebral Hematoma Clearance. Stroke 2016, 47, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Hotz, M.J.; Qing, D.; Shashaty, M.G.S.; Zhang, P.; Faust, H.; Sondheimer, N.; Rivella, S.; Worthen, G.S.; Mangalmurti, N.S. Red Blood Cells Homeostatically Bind Mitochondrial DNA through TLR9 to Maintain Quiescence and to Prevent Lung Injury. Am. J. Respir. Crit. Care Med. 2018, 197, 470–480. [Google Scholar] [CrossRef]
- Lam, L.K.M.; Murphy, S.; Kokkinaki, D.; Venosa, A.; Sherrill-Mix, S.; Casu, C.; Rivella, S.; Weiner, A.; Park, J.; Shin, S.; et al. DNA binding to TLR9 expressed by red blood cells promotes innate immune activation and anemia. Sci. Transl. Med. 2021, 13, eabj1008. [Google Scholar] [CrossRef]
- Parker, J.C.; Snow, R.L. Influence of external ATP on permeability and metabolism of dog red blood cells. Am. J. Physiol. 1972, 223, 888–893. [Google Scholar] [CrossRef] [Green Version]
- Sluyter, R.; Shemon, A.N.; Barden, J.; Wiley, J.S. Extracellular ATP increases cation fluxes in human erythrocytes by activation of the P2X7 receptor. J. Biol. Chem. 2004, 279, 44749–44755. [Google Scholar] [CrossRef] [Green Version]
- Sluyter, R.; Shemon, A.N.; Hughes, W.E.; Stevenson, R.O.; Georgiou, J.G.; Eslick, G.D.; Taylor, R.M.; Wiley, J.S. Canine erythrocytes express the P2X7 receptor: Greatly increased function compared with human erythrocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, 2090–2098. [Google Scholar] [CrossRef] [Green Version]
- Faulks, M.; Kuit, T.A.; Sophocleous, R.A.; Curtis, B.L.; Curtis, S.J.; Jurak, L.M.; Sluyter, R. P2X7 receptor activation causes phosphatidylserine exposure in canine erythrocytes. World J. Hematol. 2016, 5, 88–93. [Google Scholar] [CrossRef]
- Sophocleous, R.A.; Mullany, P.R.F.; Winter, K.M.; Marks, D.C.; Sluyter, R. Propensity of red blood cells to undergo P2X7 receptor-mediated phosphatidylserine exposure does not alter during in vivo or ex vivo aging. Transfusion 2015, 55, 1946–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semeraro, F.; Ammollo, C.T.; Esmon, N.L.; Esmon, C.T. Histones induce phosphatidylserine exposure and a procoagulant phenotype in human red blood cells. J. Thromb. Haemost. 2014, 12, 1697–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordbacheh, F.; O’Meara, C.H.; Coupland, L.A.; Lelliott, P.M.; Parish, C.R. Extracellular histones induce erythrocyte fragility and anemia. Blood 2017, 130, 2884–2888. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.W.; Lau, P.M.; Tsang, H.L.; Ho, H.P.; Kwan, Y.W.; Kong, S.K. Extracellular histones induced eryptotic death in human erythrocytes. Cell. Physiol. Biochem. 2019, 53, 229–241. [Google Scholar]
- Noubouossie, D.; Sokol, J.; Piegore, M.G.; Ilich, A.; Henderson, M.W.; Mooberry, M.; Monroe, D.; Key, S.N. Histones Induce the Release of Extracellular Hemoglobin and Red Blood Cell-Derived Microvesicles with Procoagulant Activity. Blood 2018, 132 (Suppl. S1), 2514. [Google Scholar] [CrossRef]
- Gwoździński, K.; Pienia̧zek, A.; Kaca, W. Lipopolysaccharide from Proteus mirabilis O29 induces changes in red blood cell membrane lipids and proteins. Int. J. Biochem. Cell Biol. 2003, 35, 333–338. [Google Scholar] [CrossRef]
- Gwozdzinski, K.; Pieniazek, A.; Sudak, B.; Kaca, W. Alterations in human red blood cell membrane properties induced by the lipopolysaccharide from Proteus mirabilis S1959. Chem. Biol. Interact. 2003, 146, 73–80. [Google Scholar] [CrossRef]
- Brauckmann, S.; Effenberger-Neidnicht, K.; De Groot, H.; Nagel, M.; Mayer, C.; Peters, J.; Hartmann, M. Lipopolysaccharide-induced hemolysis: Evidence for direct membrane interactions. Sci. Rep. 2016, 6, 35508. [Google Scholar] [CrossRef] [Green Version]
- Nicolay, J.P.; Gatz, S.; Liebig, G.; Gulbins, E.; Lang, F. Amyloid induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2007, 19, 175–184. [Google Scholar] [CrossRef]
- Tikhonova, L.A.; Kaminskii, Y.G.; Kosenko, E.A. Effects of amyloid-β peptide Aβ25-35 on glycolytic and antioxidant enzymes in erythrocytes of different ages. Biol. Bull. 2014, 41, 312–317. [Google Scholar] [CrossRef]
- Clementi, M.E.; Giardina, B.; Colucci, D.; Galtieri, A.; Misiti, F. Amyloid-beta peptide affects the oxygen dependence of erythrocyte metabolism: A role for caspase 3. Int. J. Biochem. Cell Biol. 2007, 39, 727–735. [Google Scholar] [CrossRef]
- Misiti, F.; Orsini, F.; Clementi, M.E.; Masala, D.; Tellone, E.; Galtieri, A.; Giardina, B. Amyloid peptide inhibits ATP release from human erythrocytes. Biochem. Cell Biol. 2008, 86, 501–508. [Google Scholar] [CrossRef]
- Carelli-Alinovi, C.; Giardina, B.; Misiti, F. Amyloid beta peptide (1-42)-mediated antioxidant imbalance is associated with activation of protein kinase C in red blood cells. Cell Biochem. Funct. 2015, 33, 196–201. [Google Scholar] [CrossRef]
- Carelli-Alinovi, C.; Dinarelli, S.; Sampaolese, B.; Misiti, F.; Girasole, M. Morphological changes induced in erythrocyte by amyloid beta peptide and glucose depletion: A combined atomic force microscopy and biochemical study. Biochim. Biophys. Acta-Biomembr. 2019, 1861, 236–244. [Google Scholar] [CrossRef]
- Xu, J.; Dai, L.; Zhang, Y.; Wang, A.; Li, H.; Wang, Y.; Meng, X.; Wu, S.; Wang, Y. Severity of Nonalcoholic Fatty Liver Disease and Risk of Future Ischemic Stroke Events. Stroke 2021, 52, 103–110. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Spourita, E.; Mimidis, K.; Kolios, G.; Tentes, L.; Anagnostopoulos, K. Nonalcoholic Fatty Liver Disease Patients Exhibit Reduced CD47 and Increased Sphingosine, Cholesterol, and Monocyte Chemoattractant Protein-1 Levels in the Erythrocyte Membranes. Metab. Syndr. Relat. Disord. 2022, 20, 377–383. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Mimidis, K.; Tentes, I.; Tente, T.; Anagnostopoulos, K. Validation and application of a protocol for the extraction and quantitative analysis of sphingomyelin in erythrocyte membranes of patients with non-alcoholic fatty liver disease. JPC-J. Planar Chromatogr. 2021, 34, 411–418. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Mimidis, K.; Papazoglou, D.; Kolios, G.; Tentes, I.; Anagnostopoulos, K. Red Blood Cell-Conditioned Media from Non-Alcoholic Fatty Liver Disease Patients Contain Increased MCP1 and Induce TNF-α Release. Rep. Biochem. Mol. Biol. 2022, 11, 54–62. [Google Scholar] [CrossRef]
- Otogawa, K.; Kinoshita, K.; Fujii, H.; Sakabe, M.; Shiga, R.; Nakatani, K.; Ikeda, K.; Nakajima, Y.; Ikura, Y.; Ueda, M.; et al. Erythrophagocytosis by liver macrophages (Kupffer cells) promotes oxidative stress, inflammation, and fibrosis in a rabbit model of steatohepatitis: Implications for the pathogenesis of human nonalcoholic steatohepatitis. Am. J. Pathol. 2007, 170, 967–980. [Google Scholar] [CrossRef] [Green Version]
- Jing, C.; Bian, L.; Wang, M.; Keep, R.F.; Xi, G.; Hua, Y. Enhancement of Hematoma Clearance with CD47 Blocking Antibody in Experimental Intracerebral Hemorrhage. Stroke 2019, 50, 1539–1547. [Google Scholar] [CrossRef]
- Chang, C.F.; Goods, B.A.; Askenase, M.H.; Hammond, M.D.; Renfroe, S.C.; Steinschneider, A.F.; Landreneau, M.J.; Ai, Y.; Beatty, H.E.; da Costa, L.H.A.; et al. Erythrocyte Efferocytosis Modulates Macrophages towards Recovery after Intracerebral Hemorrhage. J. Clin. Investig. 2018, 128, 607–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Damp/Pamp | Receptor | Effect |
|---|---|---|
| mtDNA | TLR9 |
|
| CpG DNA | TLR9 |
|
| ATP | P2X7 |
|
| Extracellular histones | TLR2? |
|
| LPS | - |
|
| Amyloids | - |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadopoulos, C.; Anagnostopoulos, K.; Tsiptsios, D.; Karatzetzou, S.; Liaptsi, E.; Lazaridou, I.Z.; Kokkotis, C.; Makri, E.; Ioannidou, M.; Aggelousis, N.; et al. Unexplored Roles of Erythrocytes in Atherothrombotic Stroke. Neurol. Int. 2023, 15, 124-139. https://doi.org/10.3390/neurolint15010011
Papadopoulos C, Anagnostopoulos K, Tsiptsios D, Karatzetzou S, Liaptsi E, Lazaridou IZ, Kokkotis C, Makri E, Ioannidou M, Aggelousis N, et al. Unexplored Roles of Erythrocytes in Atherothrombotic Stroke. Neurology International. 2023; 15(1):124-139. https://doi.org/10.3390/neurolint15010011
Chicago/Turabian StylePapadopoulos, Charalampos, Konstantinos Anagnostopoulos, Dimitrios Tsiptsios, Stella Karatzetzou, Eirini Liaptsi, Irene Zacharo Lazaridou, Christos Kokkotis, Evangelia Makri, Maria Ioannidou, Nikolaos Aggelousis, and et al. 2023. "Unexplored Roles of Erythrocytes in Atherothrombotic Stroke" Neurology International 15, no. 1: 124-139. https://doi.org/10.3390/neurolint15010011
APA StylePapadopoulos, C., Anagnostopoulos, K., Tsiptsios, D., Karatzetzou, S., Liaptsi, E., Lazaridou, I. Z., Kokkotis, C., Makri, E., Ioannidou, M., Aggelousis, N., & Vadikolias, K. (2023). Unexplored Roles of Erythrocytes in Atherothrombotic Stroke. Neurology International, 15(1), 124-139. https://doi.org/10.3390/neurolint15010011