Neuroprotective Role of Atractylenolide-I in an In Vitro and In Vivo Model of Parkinson’s Disease



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Material
2.2. BV-2 Cell Culture
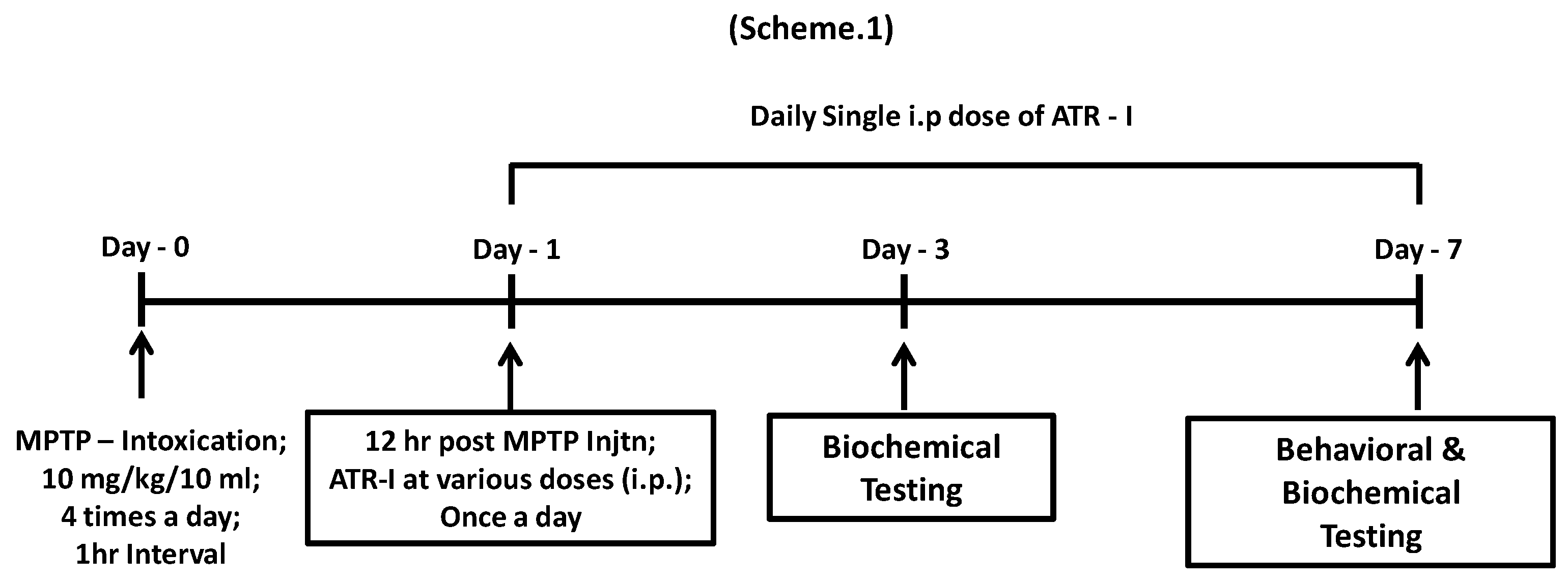
2.3. Animals and MPTP Administration
2.4. Pole Test
2.5. Nitrite Measurement Assay
2.6. Immunohistochemistry
2.7. Isolation of Total RNA and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.8. SDS-PAGE and Western Blot Analysis
2.9. Detection of Intracellular ROS Generation Using a Flow Cytometer
2.10. Immunofluorescence Assay
2.11. Statistical Analyses
3. Results
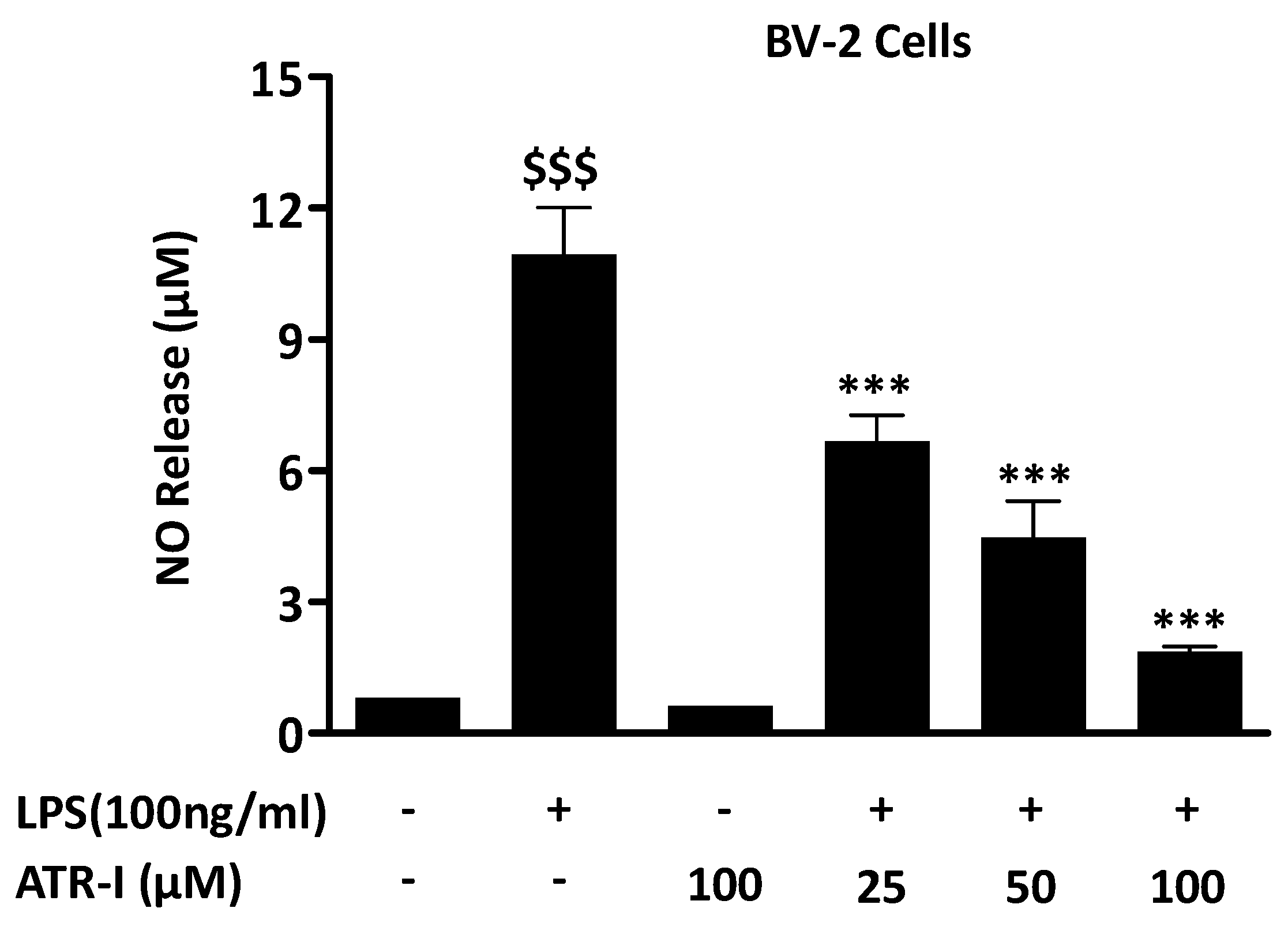
3.1. ATR-I Suppresses LPS-Induced NO Release in BV-2 Cells
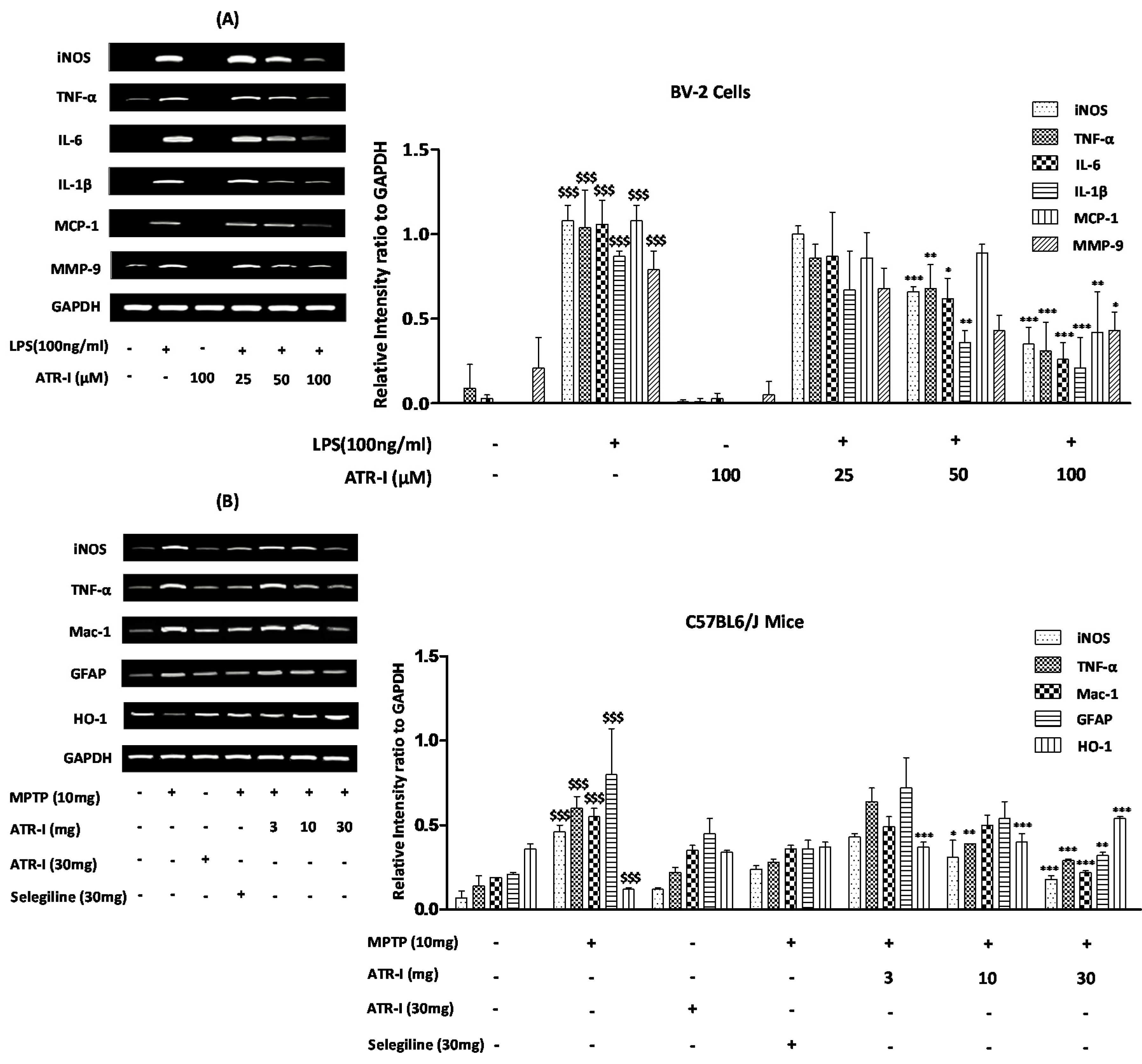
3.2. ATR-I Diminishes iNOS, TNF-α, IL-6, IL-1β, MCP-1, and MMP-9 mRNA-Expression in LPS-Stimulated BV-2 Cells
3.3. ATR-I Diminishes mRNA-Expression of Microglia-Derived Pro-Inflammatory Mediators and Induces HO-1 Expression in MPTP-Induced Model of Neuroinflammation
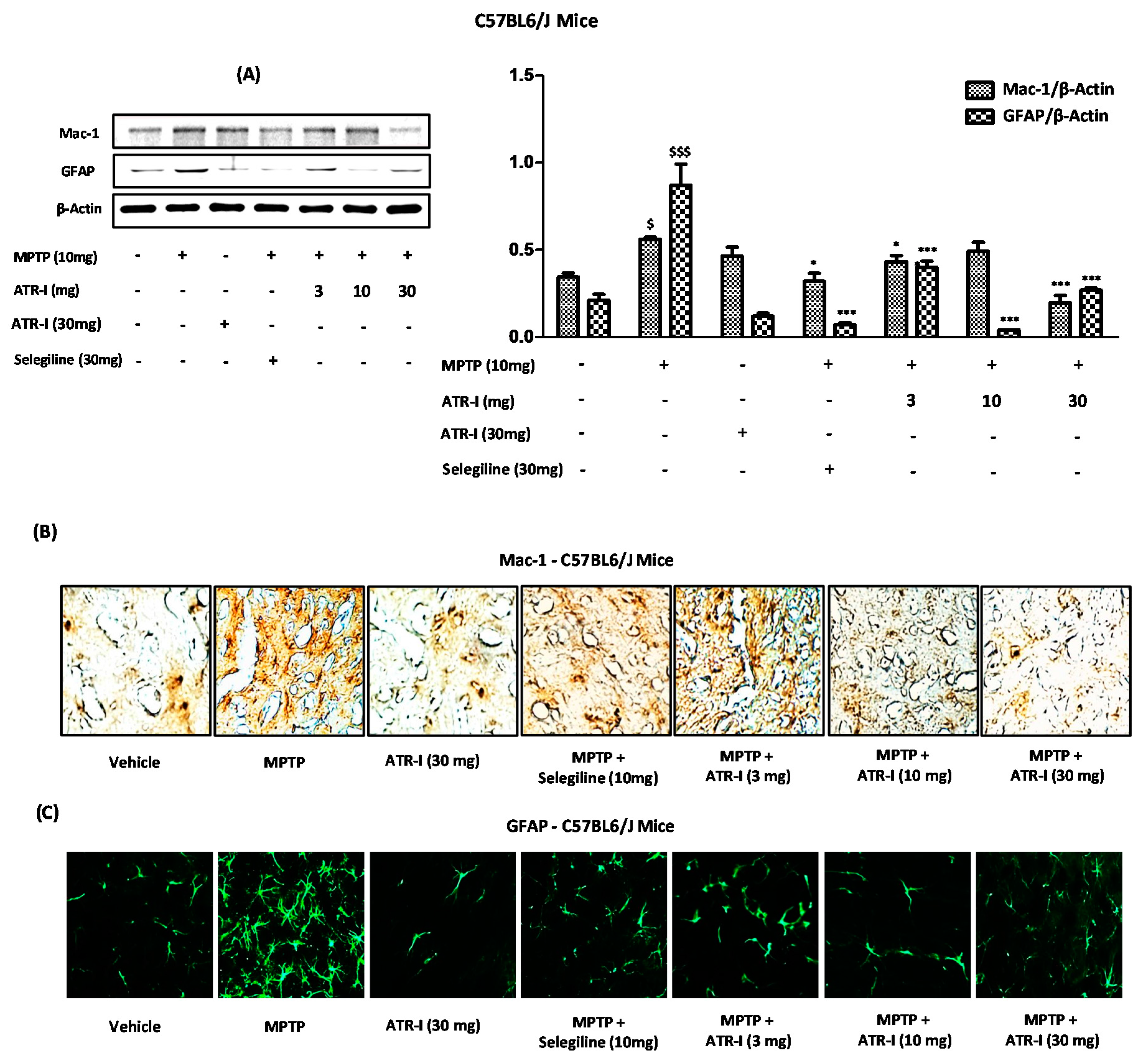
3.4. ATR-I Diminishes the Microglial and Astroglial Markers in Striatum of MPTP-Intoxicated Mice
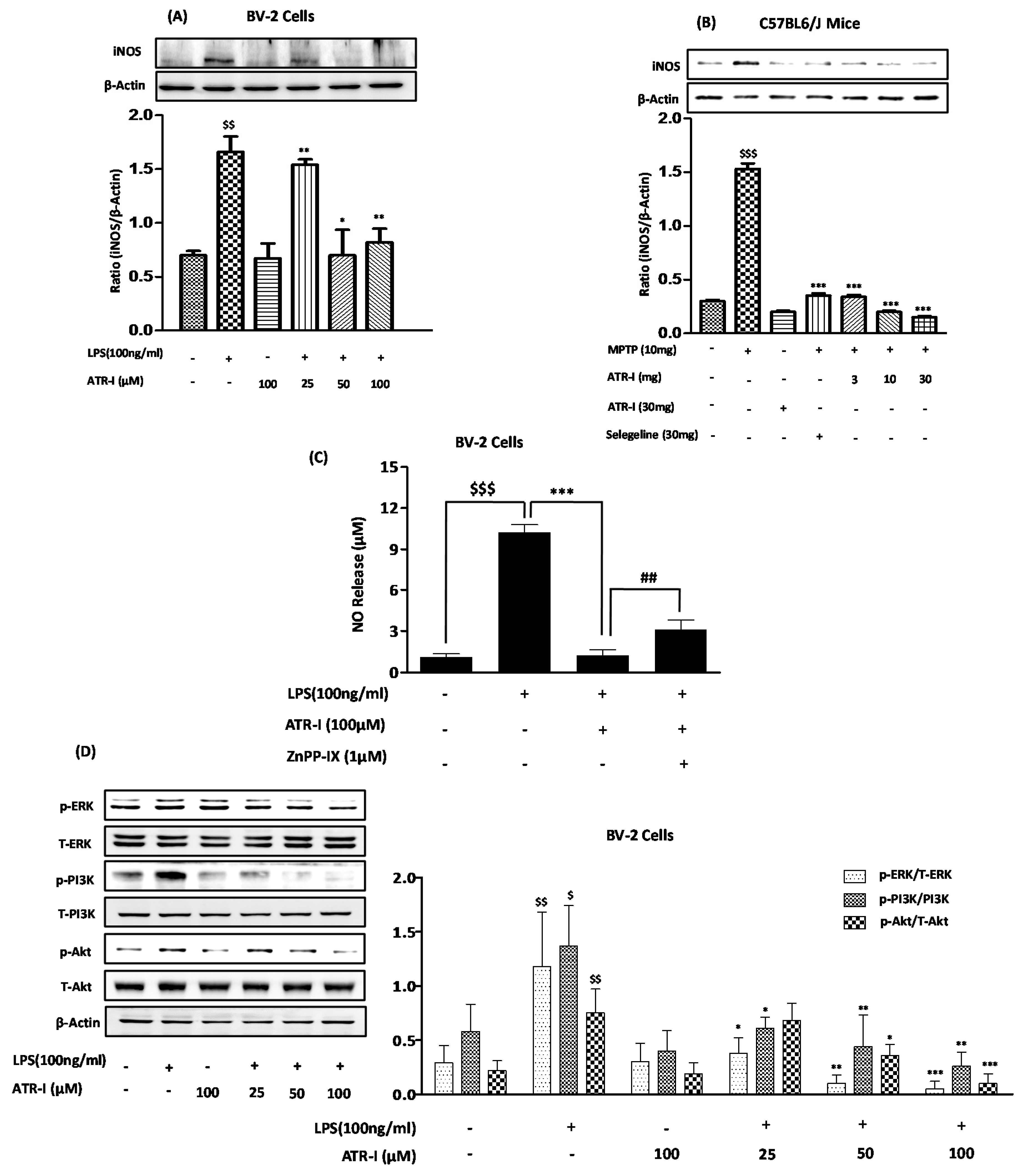
3.5. ATR-I Subsides LPS- and MPTP-Induced Increases of iNOS Expression and Modulates LPS-Induced Phosphorylation of ERK, PI3K, and Akt
3.6. HO-1 Mediates the Inhibitory Effect of ATR-I on NO Release in LPS-Stimulated BV-2 Cells
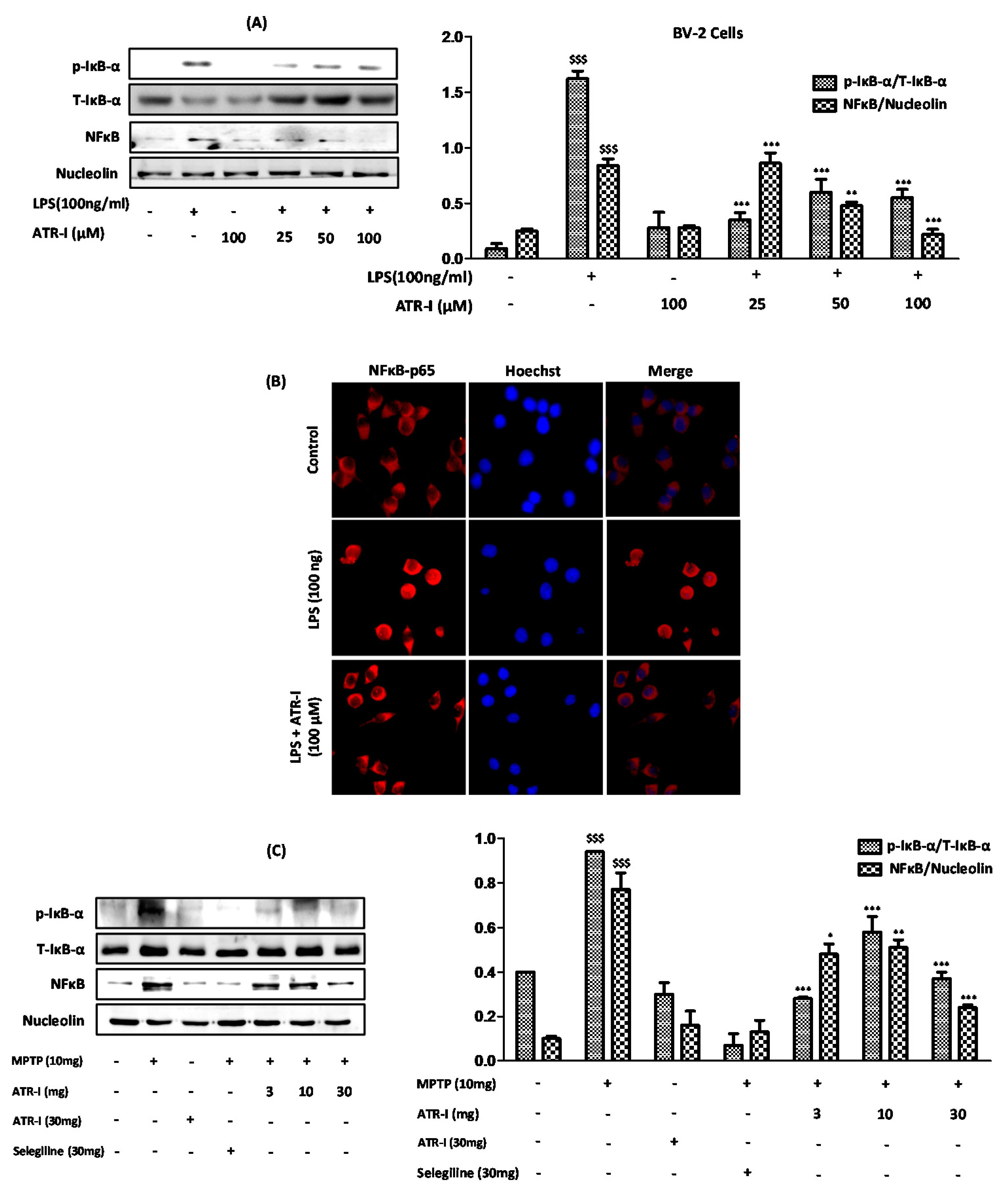
3.7. ATR-I Ameliorates the Degradation of IκB-α and Inhibits Nuclear Translocation of NF-κB in LPS and MPTP Model of Neuroinflammation
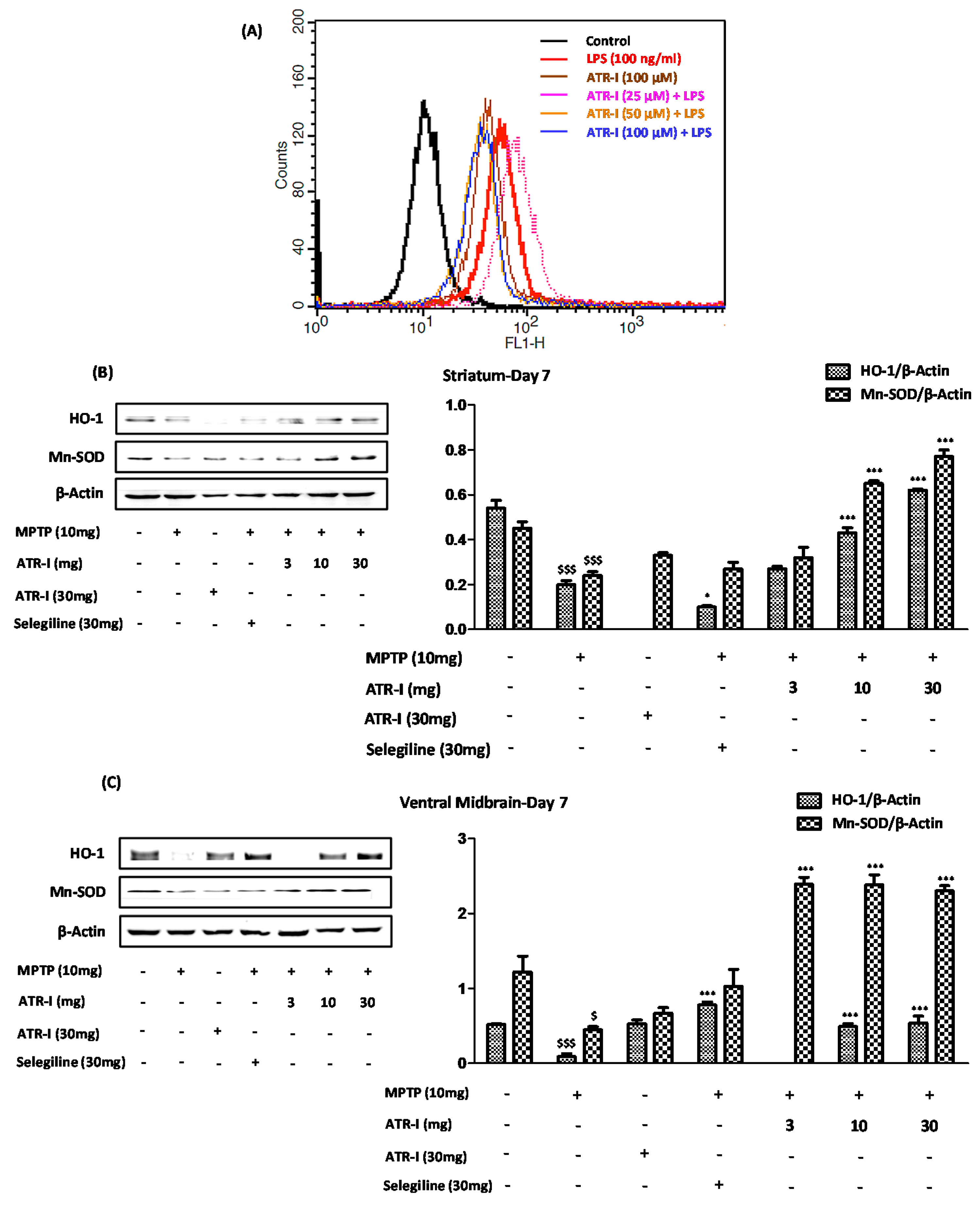
3.8. ATR-I Suppresses the LPS-Induced Production of ROS in BV-2 Cells, While Upregulates the Anti-Oxidant Defense System in the MPTP-Induced Model of Neuroinflammation
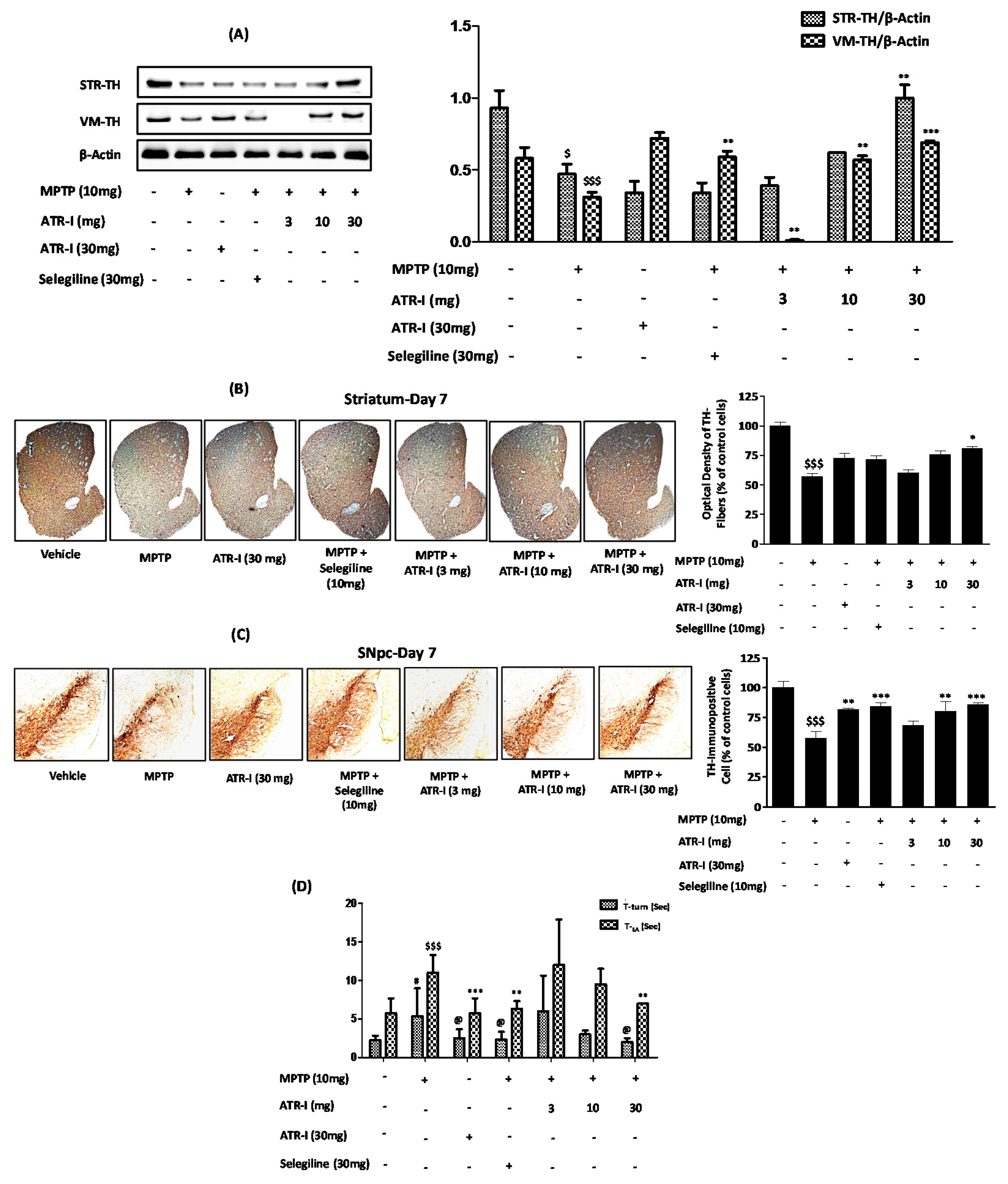
3.9. ATR-I Mitigates MPTP-Induced Dopaminergic Neurodegeneration and Motor Dysfunction in a Mouse Model of Neuroinflammation
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Henchcliffe, C.; Beal, M.F. Mitochondrial biology and oxidative stress in parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 2008, 4, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Vivekanantham, S.; Shah, S.; Dewji, R.; Dewji, A.; Khatri, C.; Ologunde, R. Neuroinflammation in parkinson’s disease: Role in neurodegeneration and tissue repair. Int. J. Neurosci. 2015, 125, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Koike, M.; Kuzumaki, N.; Hayakawa, H.; Nihira, T.; Kobayashi, T.; Ohyama, M.; Sato, S.; et al. Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in park2 ipsc-derived neurons and postmortem brain tissue. Mol. Brain 2012, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Licker, V.; Turck, N.; Kovari, E.; Burkhardt, K.; Cote, M.; Surini-Demiri, M.; Lobrinus, J.A.; Sanchez, J.C.; Burkhard, P.R. Proteomic analysis of human substantia nigra identifies novel candidates involved in parkinson’s disease pathogenesis. Proteomics 2014, 14, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.K.; Ou, Y.C.; Lin, S.Y.; Pan, H.C.; Song, P.J.; Raung, S.L.; Lai, C.Y.; Liao, S.L.; Lu, H.C.; Chen, C.J. Luteolin inhibits cytokine expression in endotoxin/cytokine-stimulated microglia. J. Nutr. Biochem. 2011, 22, 612–624. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; He, H.; Li, P.; Zhu, J.; Xiong, M. Identification and quantification of atractylenolide i and atractylenolide iii in rhizoma atractylodes macrocephala by liquid chromatography-ion trap mass spectrometry. Biomed. Chromatogr. 2013, 27, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, S.; Lin, Y. gastrointestinal inhibitory effects of sesquiterpene lactones from atractylodes macrocephala. Zhong Yao Cai 1999, 22, 636–640. [Google Scholar] [PubMed]
- Wang, C.C.; Lin, S.Y.; Cheng, H.C.; Hou, W.C. Pro-oxidant and cytotoxic activities of atractylenolide i in human promyeloleukemic hl-60 cells. Food Chem. Toxicol. 2006, 44, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.L.; Chen, C.C.; Yeh, C.Y.; Huang, R.L. Reactive oxygen species mediation of baizhu-induced apoptosis in human leukemia cells. J. Ethnopharmacol. 2005, 97, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ye, F.; Qiu, G.Q.; Zhang, M.; Wang, R.; He, Q.Y.; Cai, Y. Effects of lactone i from atractylodes macrocephala koidz on cytokines and proteolysis-inducing factors in cachectic cancer patients. Di Yi Jun Yi Da Xue Xue Bao 2005, 25, 1308–1311. [Google Scholar] [PubMed]
- Li, C.; He, L. Establishment of the model of white blood cell membrane chromatography and screening of antagonizing tlr4 receptor component from atractylodes macrocephala koidz. Sci. China C Life Sci. 2006, 49, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Q.; He, L.C.; Dong, H.Y.; Jin, J.Q. Screening for the anti-inflammatory activity of fractions and compounds from atractylodes macrocephala koidz. J. Ethnopharmacol. 2007, 114, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Kostic, V.; Przedborski, S.; Flaster, E.; Sternic, N. Early development of levodopa-induced dyskinesias and response fluctuations in young-onset parkinson’s disease. Neurology 1991, 41, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Bocchini, V.; Mazzolla, R.; Barluzzi, R.; Blasi, E.; Sick, P.; Kettenmann, H. An immortalized cell line expresses properties of activated microglial cells. J. Neurosci. Res. 1992, 31, 616–621. [Google Scholar] [CrossRef] [PubMed]
- More, S.V.; Park, J.Y.; Kim, B.W.; Kumar, H.; Lim, H.W.; Kang, S.M.; Koppula, S.; Yoon, S.H.; Choi, D.K. Anti-neuroinflammatory activity of a novel cannabinoid derivative by inhibiting the nf-kappab signaling pathway in lipopolysaccharide-induced bv-2 microglial cells. J. Pharmacol. Sci. 2013, 121, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Kohutnicka, M.; Lewandowska, E.; Kurkowska-Jastrzebska, I.; Czlonkowski, A.; Czlonkowska, A. Microglial and astrocytic involvement in a murine model of parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (mptp). Immunopharmacology 1998, 39, 167–180. [Google Scholar] [CrossRef]
- Himeda, T.; Watanabe, Y.; Tounai, H.; Hayakawa, N.; Kato, H.; Araki, T. Time dependent alterations of co-localization of s100beta and gfap in the mptp-treated mice. J. Neural Transm. 2006, 113, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Kabuto, H.; Makino, H.; Ogawa, N. Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J. Neurosci. Methods 1997, 73, 45–48. [Google Scholar] [CrossRef]
- Green, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of nitrate, nitrite, and [15n]nitrate in biological fluids. Anal. Biochem. 1982, 126, 131–138. [Google Scholar] [CrossRef]
- Kim, B.W.; Koppula, S.; Kumar, H.; Park, J.Y.; Kim, I.W.; More, S.V.; Kim, I.S.; Han, S.D.; Kim, S.K.; Yoon, S.H. A-asarone attenuates microglia-mediated neuroinflammation by inhibiting nf kappa b activation and mitigates mptp-induced behavioral deficits in a mouse model of parkinson’s disease. Neuropharmacology 2015, 97, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kim, I.S.; More, S.V.; Kim, B.W.; Bahk, Y.Y.; Choi, D.K. Gastrodin protects apoptotic dopaminergic neurons in a toxin-induced parkinson’s disease model. Evid. Based Complement. Altern. Med. 2013, 2013, 514095. [Google Scholar] [CrossRef] [PubMed]
- Bass, D.A.; Parce, J.W.; Dechatelet, L.R.; Szejda, P.; Seeds, M.C.; Thomas, M. Flow cytometric studies of oxidative product formation by neutrophils: A graded response to membrane stimulation. J. Immunol. 1983, 130, 1910–1917. [Google Scholar] [PubMed]
- Eruslanov, E.; Kusmartsev, S. Identification of ros using oxidized dcfda and flow-cytometry. Methods Mol. Biol. 2010, 594, 57–72. [Google Scholar] [PubMed]
- Wagner, S.; Arce, R.; Murillo, R.; Terfloth, L.; Gasteiger, J.; Merfort, I. Neural networks as valuable tools to differentiate between sesquiterpene lactones’ inhibitory activity on serotonin release and on nf-kappab. J. Med. Chem. 2008, 51, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Willuhn, G. New findings from research on arnica. Pharm. Unserer Zeit 1981, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Commission, C.P. Pharmacopoeia of the People’s Republic of China; China Medical Science and Technology Press: Beijing, China, 2010; Volume 1. [Google Scholar]
- Lin, Y.C.; Kuo, H.C.; Wang, J.S.; Lin, W.W. Regulation of inflammatory response by 3-methyladenine involves the coordinative actions on akt and glycogen synthase kinase 3beta rather than autophagy. J. Immunol. 2012, 189, 4154–4164. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Taguchi, T.; Taguchi, F.; Hikino, H.; Yamahara, J.; Fujimura, H. Antiinflammatory principles of atractylodes rhizomes. Chem. Pharm. Bull. (Tokyo) 1979, 27, 2954–2958. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Lim, H.; Kim, H.P.; Kwon, Y.S. Downregulation of matrix metalloproteinase-13 by the root extract of cyathula officinalis kuan and its constituents in il-1beta-treated chondrocytes. Planta Med. 2011, 77, 1528–1530. [Google Scholar] [CrossRef] [PubMed]
- Sin, K.S.; Kim, H.P.; Lee, W.C.; Pachaly, P. Pharmacological activities of the constituents of atractylodes rhizomes. Arch. Pharm. Res. 1989, 12, 236–238. [Google Scholar] [CrossRef]
- More, S.V.; Kumar, H.; Kim, I.S.; Song, S.Y.; Choi, D.K. Cellular and molecular mediators of neuroinflammation in the pathogenesis of parkinson’s disease. Mediat. Inflamm. 2013, 2013, 952375. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Experimental models of parkinson’s disease. Nat. Rev. Neurosci. 2001, 2, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Ferger, B. Neurochemical findings in the mptp model of parkinson’s disease. J. Neural Transm. 2001, 108, 1263–1282. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.K.; Moon, E.; Kim, S.Y. Chrysin suppresses lps-stimulated proinflammatory responses by blocking nf-kappab and jnk activations in microglia cells. Neurosci. Lett. 2010, 485, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Shahani, N.; Sawa, A. Nitric oxide signaling and nitrosative stress in neurons: Role for s-nitrosylation. Antioxid. Redox Signal. 2011, 14, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Hunot, S.; Dugas, N.; Faucheux, B.; Hartmann, A.; Tardieu, M.; Debré, P.; Agid, Y.; Dugas, B.; Hirsch, E.C. Fcεrii/cd23 is expressed in parkinson’s disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-α in glial cells. J. Neurosci. 1999, 19, 3440–3447. [Google Scholar] [PubMed]
- Thompson, W.L.; Van Eldik, L.J. Inflammatory cytokines stimulate the chemokines ccl2/mcp-1 and ccl7/mcp-3 through nfkb and mapk dependent pathways in rat astrocytes. Brain Res. 2009, 1287, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Jayasooriya, R.G.; Choi, Y.H.; Kim, G.Y. Glutamine-free condition inhibits lipopolysaccharide-induced invasion of bv2 microglial cells by suppressing of matrix metalloproteinase-9 expression. Environ. Toxicol. Pharmacol. 2013, 36, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Q.; He, L.C.; Jin, J.Q. Atractylenolide i and atractylenolide iii inhibit lipopolysaccharide-induced tnf-alpha and no production in macrophages. Phytother. Res. 2007, 21, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Liberatore, G.T.; Jackson-Lewis, V.; Vukosavic, S.; Mandir, A.S.; Vila, M.; McAuliffe, W.G.; Dawson, V.L.; Dawson, T.M.; Przedborski, S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the mptp model of parkinson disease. Nat. Med. 1999, 5, 1403–1409. [Google Scholar] [PubMed]
- Bruck, D.; Wenning, G.K.; Stefanova, N.; Fellner, L. Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiol. Dis. 2016, 85, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, J.; Wu, J.; Zhu, C.; Hui, Y.; Han, Y.; Huang, Z.; Ellsworth, K.; Fan, W. Alpha7 nicotinic acetylcholine receptor-mediated neuroprotection against dopaminergic neuron loss in an mptp mouse model via inhibition of astrocyte activation. J. Neuroinflamm. 2012, 9, 98. [Google Scholar] [CrossRef] [PubMed]
- Hunot, S.; Hirsch, E. Neuroinflammatory processes in parkinson’s disease. Ann. Neurol. 2003, 53, S49–S60. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11 c](r)-pk11195 pet in idiopathic parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Kochanek, P.M.; Berger, R.P.; Bayr, H.; Wagner, A.K.; Jenkins, L.W.; Clark, R.S. Biomarkers of primary and evolving damage in traumatic and ischemic brain injury: Diagnosis, prognosis, probing mechanisms, and therapeutic decision making. Curr. Opin. Crit. Care 2008, 14, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Van Eldik, L.J.; Thompson, W.L.; Ranaivo, H.R.; Behanna, H.A.; Watterson, D.M. Glia proinflammatory cytokine upregulation as a therapeutic target for neurodegenerative diseases: Function-based and target-based discovery approaches. Int. Rev. Neurobiol. 2007, 82, 277–296. [Google Scholar] [PubMed]
- McGeer, P.; Itagaki, S.; Boyes, B.; McGeer, E. Reactive microglia are positive for hla-dr in the substantia nigra of parkinson’s and alzheimer’s disease brains. Neurology 1988, 38, 1285. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.K.; Ischiropoulos, H.; Przedborski, S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of parkinson disease. J. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [PubMed]
- Choi, W.S.; Seo, Y.B.; Shin, P.G.; Kim, W.Y.; Lee, S.Y.; Choi, Y.J.; Kim, G.D. Veratric acid inhibits inos expression through the regulation of pi3k activation and histone acetylation in lps-stimulated raw264.7 cells. Int. J. Mol. Med. 2015, 35, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Feng, J. Signaling pathways associated with inflammatory bowel disease. Recent Pat. Inflamm. Allergy Drug Discov. 2010, 4, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Han, M.H.; Park, C.; Jin, C.-Y.; Kim, G.-Y.; Choi, I.-W.; Kim, N.D.; Nam, T.-J.; Kwon, T.K.; Choi, Y.H. Anti-inflammatory effects of fucoidan through inhibition of nf-κb, mapk and akt activation in lipopolysaccharide-induced bv2 microglia cells. Food Chem. Toxicol. 2011, 49, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.C.; Hou, R.C.; Wang, J.C.; Ping, L.I. Sesamin inhibits lipopolysaccharide-induced cytokine production by suppression of p38 mitogen-activated protein kinase and nuclear factor-kappab. Immunol. Lett. 2005, 97, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.M.; Kim, E.A.; Hahn, H.G.; Nam, K.D.; Yang, S.J.; Choi, S.Y.; Kim, T.U.; Cho, S.W.; Huh, J.W. Protective effect of benzothiazole derivative khg21834 on amyloid beta-induced neurotoxicity in pc12 cells and cortical and mesencephalic neurons. Toxicology 2007, 239, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Moron, J.A.; Zakharova, I.; Ferrer, J.V.; Merrill, G.A.; Hope, B.; Lafer, E.M.; Lin, Z.C.; Wang, J.B.; Javitch, J.A.; Galli, A.; et al. Mitogen-activated protein kinase regulates dopamine transporter surface expression and dopamine transport capacity. J. Neurosci. 2003, 23, 8480–8488. [Google Scholar] [PubMed]
- Zhu, C.B.; Blakely, R.D.; Hewlett, W.A. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology 2006, 31, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Trabucco, S.E.; Zhang, H. Oxidative stress, mitochondrial dysfunction and the mitochondria theory of aging. Interdiscip. Top. Gerontol. 2014, 39, 86–107. [Google Scholar] [PubMed]
- Hwang, O. Role of oxidative stress in parkinson’s disease. Exp. Neurobiol. 2013, 22, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Flohe, L.; Brigelius-Flohe, R.; Saliou, C.; Traber, M.G.; Packer, L. Redox regulation of nf-kappa b activation. Free Radic. Biol. Med. 1997, 22, 1115–1126. [Google Scholar] [CrossRef]
- Schoonbroodt, S.; Piette, J. Oxidative stress interference with the nuclear factor-kappa b activation pathways. Biochem. Pharmacol. 2000, 60, 1075–1083. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chen, C.H.; Ko, W.S.; Cheng, C.Y.; Sue, Y.M.; Chen, T.H. Dipyridamole inhibits lipopolysaccharide-induced cyclooxygenase-2 and monocyte chemoattractant protein-1 via heme oxygenase-1-mediated reactive oxygen species reduction in rat mesangial cells. Eur. J. Pharmacol. 2011, 650, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.N.; Jeon, W.K.; Lee, J.J.; Kim, B.C. Up-regulation of heme oxygenase-1 expression through camkii-erk1/2-nrf2 signaling mediates the anti-inflammatory effect of bisdemethoxycurcumin in lps-stimulated macrophages. Free Radic. Biol. Med. 2010, 49, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Shen, S.C.; Chen, Y.C. Anti-inflammatory effect of heme oxygenase 1: Glycosylation and nitric oxide inhibition in macrophages. J. Cell. Physiol. 2005, 202, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.S.; Pae, H.O.; Choi, B.M.; Chae, S.C.; Lee, H.S.; Ryu, D.G.; Chung, H.T. 3-hydroxyanthranilic acid, one of metabolites of tryptophan via indoleamine 2,3-dioxygenase pathway, suppresses inducible nitric oxide synthase expression by enhancing heme oxygenase-1 expression. Biochem. Biophys. Res. Commun. 2004, 320, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Miriyala, S.; Holley, A.K.; St Clair, D.K. Mitochondrial superoxide dismutase—Signals of distinction. Anticancer Agents Med. Chem. 2011, 11, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R. Superoxide dismutase in redox biology: The roles of superoxide and hydrogen peroxide. Anticancer Agents Med. Chem. 2011, 11, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Klivenyi, P.; St Clair, D.; Wermer, M.; Yen, H.C.; Oberley, T.; Yang, L.; Flint Beal, M. Manganese superoxide dismutase overexpression attenuates mptp toxicity. Neurobiol. Dis. 1998, 5, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Datta, P.K.; Koukouritaki, S.B.; Hopp, K.A.; Lianos, E.A. Heme oxygenase-1 induction attenuates inducible nitric oxide synthase expression and proteinuria in glomerulonephritis. J. Am. Soc. Nephrol. 1999, 10, 2540–2550. [Google Scholar] [PubMed]
- Mosley, K.; Wembridge, D.E.; Cattell, V.; Cook, H.T. Heme oxygenase is induced in nephrotoxic nephritis and hemin, a stimulator of heme oxygenase synthesis, ameliorates disease. Kidney Int. 1998, 53, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Dal-Cim, T.; Molz, S.; Egea, J.; Parada, E.; Romero, A.; Budni, J.; Martin de Saavedra, M.D.; del Barrio, L.; Tasca, C.I.; Lopez, M.G. Guanosine protects human neuroblastoma sh-sy5y cells against mitochondrial oxidative stress by inducing heme oxigenase-1 via pi3k/akt/gsk-3beta pathway. Neurochem. Int. 2012, 61, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Cha, Y.N.; Surh, Y.J. Peroxynitrite induces ho-1 expression via pi3k/akt-dependent activation of nf-e2-related factor 2 in pc12 cells. Free Radic. Biol. Med. 2006, 41, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Samoylenko, A.; Dimova, E.Y.; Horbach, T.; Teplyuk, N.; Immenschuh, S.; Kietzmann, T. Opposite expression of the antioxidant heme oxygenase-1 in primary cells and tumor cells: Regulation by interaction of usf-2 and fra-1. Antioxid. Redox Signal. 2008, 10, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.H.; Liou, H.H.; Hour, M.J.; Liou, H.C.; Fu, W.M. Protection of dopaminergic neurons by 5-lipoxygenase inhibitor. Neuropharmacology 2013, 73, 380–387. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
More, S.; Choi, D.-K. Neuroprotective Role of Atractylenolide-I in an In Vitro and In Vivo Model of Parkinson’s Disease. Nutrients 2017, 9, 451. https://doi.org/10.3390/nu9050451
More S, Choi D-K. Neuroprotective Role of Atractylenolide-I in an In Vitro and In Vivo Model of Parkinson’s Disease. Nutrients. 2017; 9(5):451. https://doi.org/10.3390/nu9050451
Chicago/Turabian StyleMore, Sandeep, and Dong-Kug Choi. 2017. "Neuroprotective Role of Atractylenolide-I in an In Vitro and In Vivo Model of Parkinson’s Disease" Nutrients 9, no. 5: 451. https://doi.org/10.3390/nu9050451
APA StyleMore, S., & Choi, D. -K. (2017). Neuroprotective Role of Atractylenolide-I in an In Vitro and In Vivo Model of Parkinson’s Disease. Nutrients, 9(5), 451. https://doi.org/10.3390/nu9050451