1. Introduction
Staphylococcus aureus is a Gram-positive bacterium that resides in the anterior nares of approximately one-third of the population. Diseases caused by
S. aureus range in severity from minor skin and soft tissue infections, to life-threatening infections such as endocarditis, necrotizing fasciitis, and sepsis [
1]. This incredible diversity in diseases is largely due to the multitude of virulence factors that
S. aureus produces, such as exoenzymes that assist in the degradation of host molecules, adhesins that aid in attachment to surfaces, and toxins that lyse host cells [
2,
3,
4,
5]. Two of the best characterized toxins for their role in infection are α-toxin (encoded by the
hla gene) and the phenol-soluble modulins (PSMs) [
6,
7].
Hla is a receptor-mediated pore-forming toxin, which binds the sheddase ADAM10 on the surface of host cells and disrupts their cellular membranes. Hla has been implicated as the primary toxin responsible for the lysis of rabbit erythrocytes, which have high amounts of ADAM10 coating their surface [
6,
8,
9]. Interestingly, human red blood cells have very little ADAM10 on their surface and consequently, it takes high levels of accumulated Hla to lyse human erythrocytes. Lysis of human erythrocytes is more efficiently accomplished by the alpha phenol-soluble modulins (αPSMs) [
10].
S. aureus encodes four αPSMs, each approximately 20–25 amino acids in size, on a single polycistronic transcript, called the αPSM transcript. A fifth αPSM (Hld or the delta-toxin) is encoded within the regulatory RNA molecule RNAIII. Studies on the regulation of both Hla and the αPSMs have been largely centered around the
agr system. AgrA has been shown to bind directly to the promoter region of the αPSM transcript where it activates transcription, while
hla translation is regulated by the
agr effector RNAIII [
11]. Interestingly, it has also been demonstrated that the αPSMs can regulate Hla production in murine skin and lung models of infection [
12], although the exact mechanism is unclear. Investigating the regulation of these toxins will improve our understanding of their relative contribution during human infection and may help in the development of “anti-virulence” approaches to combat infections caused by
S. aureus.
Peptidyl-prolyl
cis/trans isomerases (PPIases) are a family of enzymes that catalyze the
cis-to-
trans isomerization of proline peptide bonds. Proline is a unique amino acid in that it can exist in both the
cis and
trans isomerization state in vivo. Correct protein folding is often not possible when a proline peptide bond is in the incorrect configuration and therefore the isomerization rate of proline peptide bonds can be the rate-limiting step in protein folding [
13]. PPIase enzymes accelerate this isomerization and therefore assist in the regulation of proteins via a post-translational mechanism. Numerous studies have identified bacterial PPIases that contribute to virulence [
14,
15,
16,
17,
18]. In addition to acting as foldases proteins, PPIases often have moonlighting roles and functions not limited to prolyl isomerization in the cell [
19,
20,
21].
S. aureus encodes three PPIase enzymes—trigger factor (Tig), PrsA, and PpiB. Recent work in our lab (and others) has demonstrated that both PrsA and PpiB influence the activity of secreted virulence factors [
21,
22,
23].
Initial work in our lab showed that a Δ
prsA mutant has reduced phospholipase C (PI-PLC) activity and decreased protease activity, and that a Δ
ppiB mutant has reduced hemolysis and nuclease activity [
24]. A follow-up study demonstrated that PpiB contributes to virulence in a murine abscess model of infection, independently of its PPIase activity [
21]. Recently, work by Lin et al. on the methicillin-sensitive
S. aureus strain HG001, demonstrated that a Δ
prsA mutant had better survival than wild-type (WT) in a murine systemic model of infection [
22]. They also concluded that there was altered abundance of 67 exoproteins and 163 cell wall-associated proteins in a Δ
prsA mutant, including the virulence factors surface protein A (SpA), immunodominant staphylococcal antigen B (IsaB) and the αPSMs.
Due to the contribution of these two PPIases to the regulation of multiple virulence determinants, we hypothesized that PrsA and PpiB would contribute to virulence in a murine systemic model of infection using a community acquired methicillin-resistant S. aureus (CA-MRSA) USA300 strain. In this study, we demonstrate that a USA300 ΔppiB mutant is attenuated in both an abscess and systemic model of infection but there is no significant attenuation with a ΔprsA mutant. The same virulence trend was observed during intracellular survival assays using macrophage and human nasal-epithelial cells. To understand the molecular mechanism underlying the attenuation of virulence in the ΔppiB mutant, we examined toxin production by measuring the hemolytic activity of culture supernatants against human and rabbit erythrocytes. We identify a significant reduction in the activity and secretion of the αPSMs in a ΔppiB mutant as well as a reduction in the activity of Hla in both the ΔprsA and ΔppiB mutants. Immunoprecipitation analysis of PpiB and PrsA suggests a role for PpiB in the Sec secretion pathway and that PrsA is involved in cell-wall processing. Together, these data suggest a role for two PPIase proteins in the regulation of S. aureus hemolytic toxins via two distinct mechanisms.
3. Discussion
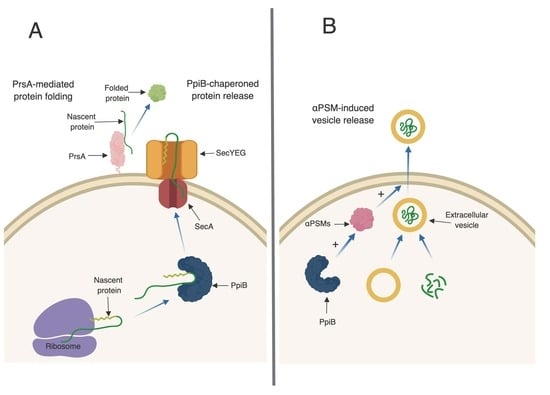
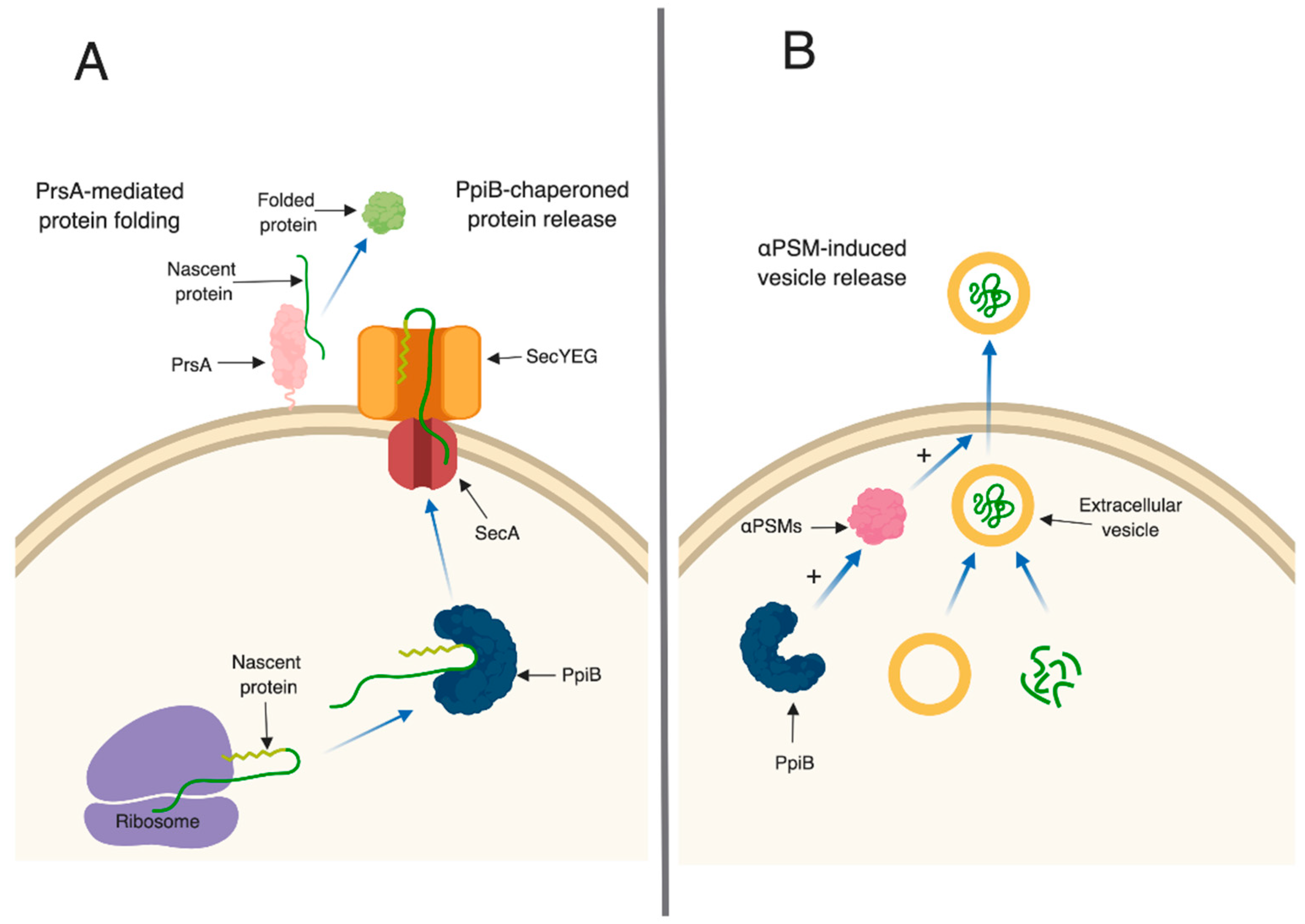
In bacteria, PPIase enzymes are traditionally studied for their roles in protein (re)folding. In Gram-positive bacteria, parvulin-type PPIases (including PrsA) are often anchored on the external leaflet of the cell membrane, and are thought to fold secreted proteins after they have been translocated out of the cell.
S. aureus PrsA appears to function in a similar manner. A
prsA mutant strain has decreased proteolytic and phospholipase activity [
24], increased sensitivity to β-lactam antibiotics [
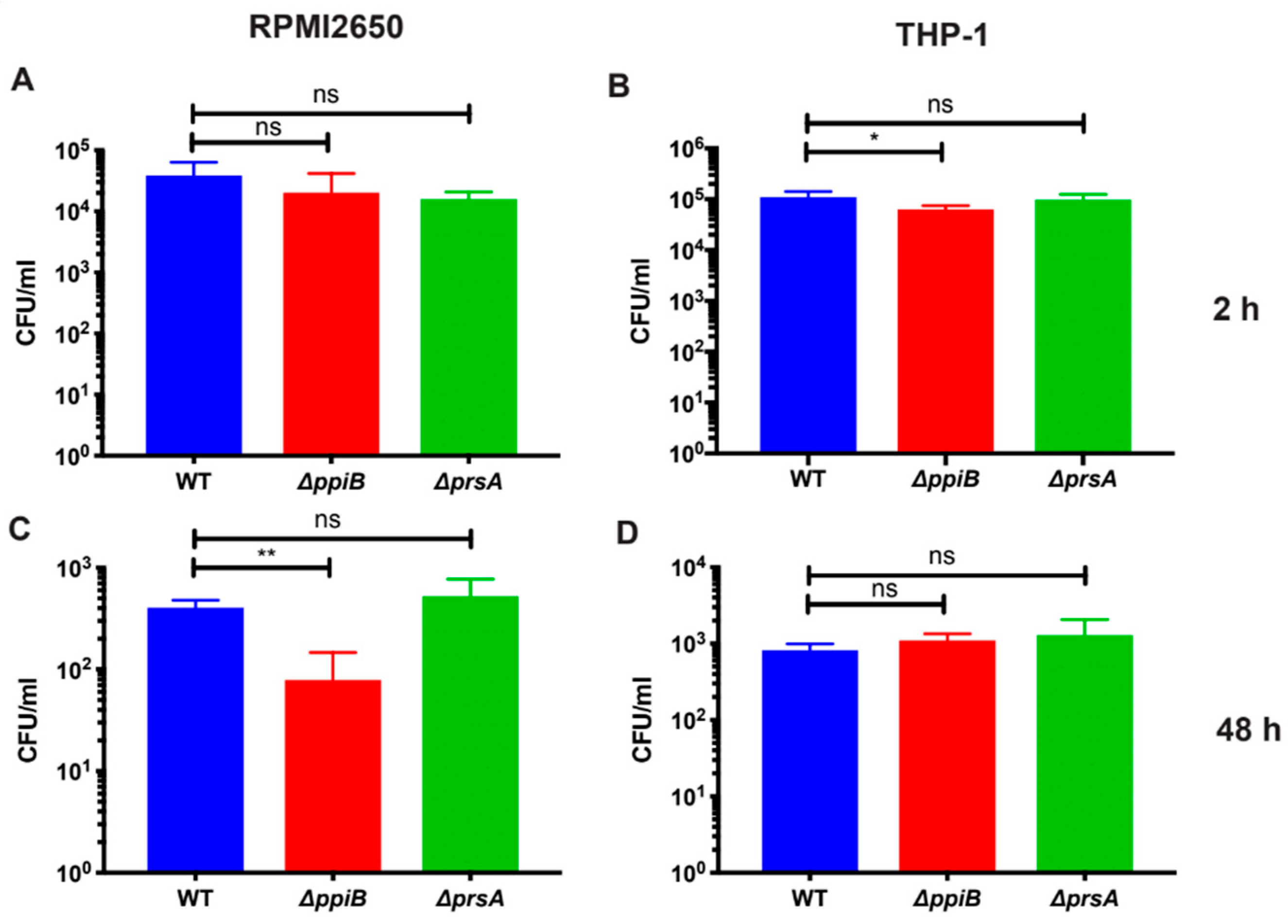
23] and decreased hemolytic activity (this study). We hypothesize that all of these defects arise as a result of defective protein folding in the absence of PrsA. This idea is supported by a number of observations in our proteomic and immunoprecipitation data analysis. First, the number of secreted proteins displaying altered levels in
prsA mutant culture supernatants was relatively low (16 proteins,
Table 2,
Table S2). This suggests that PrsA targets are present in these supernatants, however, they may exhibit reduced activity due to incorrect folding (
Figure 6A). Second, the majority of proteins found to interact with PrsA by immunoprecipitation are localized to the cell envelope and play important roles in cell wall synthesis and stability. Of particular interest was the identification of Pbp2a in the immunoprecipitation assay. This result supports the findings of Jousselin et al. and validates the approach used in this study to identify proteins that interact with PrsA [
23]. Interestingly, EsaA, a component of the recently identified
S. aureus type 7 secretion system, was identified in the immunoprecipitation assay as interacting with PrsA and was also found at lower levels in the culture supernatant. This may indicate a potential role for PrsA in type 7 secretion.
An unexpected finding in this study was that PrsA did not contribute to virulence in either a murine abscess or sepsis model of infection. This result conflicts with the results obtained by Lin et al., who demonstrate attenuation of virulence in a HG001
prsA mutant [
22]. One possible explanation for the discrepancy in results observed is the strain background used in each study. HG001, the strain used by Lin et al., is a methicillin-sensitive strain derived from NCTC8325 (with the
rsbU gene repaired) [
22]. The strain used in this study, TCH1516, is a methicillin-resistant USA300 strain. USA300 strains typically produce high levels of toxins and previously it was shown that a USA300 strain is more hemolytic than HG001 [
39]. We speculate that, in USA300 strains, the high level of toxin production and overlapping activities of
S. aureus toxins may negate the requirement for PrsA activity when it comes to causing disease in a mouse model of infection. In strains with relatively lower levels of toxin production (such as HG001), the loss of PrsA activity may have a more dramatic effect and result in attenuation. To test this hypothesis, we are currently broadening our investigations and exploring the role of PrsA in a variety of
S. aureus strains.
While the role of PrsA in
S. aureus appears to be similar to that of homologues in other Gram-positive bacteria (such as
Listeria monocytogenes), the mechanism of action of PpiB remains elusive. PpiB clearly plays an important role in infection, as a
ppiB mutant is attenuated in abscess and sepsis models of infection, and displays reduced intracellular survival. The activity and abundance of several virulence factors (including nuclease, alpha toxin, and the PSMs) is reduced in
ppiB culture supernatants, which explains the attenuation of virulence, however the mechanism through which PpiB functions is not clear. We previously demonstrated that PpiB (i) is found in the cytoplasm, (ii) possesses PPIase activity, and (iii) its PPIase activity was dispensable during infection [
21,
24]. Taken together, this implies that PpiB functions inside the cell, in a PPIase-independent manner, to influence virulence factor production. Our first indication of a potential biological role for PpiB comes from the secreted proteomic and immunoprecipitation data presented herein. SecA, a component of the general secretion pathway in
S. aureus, was found to interact with PpiB and was also found at significantly reduced levels in
ppiB mutant culture supernatants. In
E. coli, during general secretion, a chaperone protein called SecB binds to newly synthesized proteins and delivers them to SecA for translocation. No SecB homologue has been identified in
S. aureus, therefore, based on our data, we hypothesize that one potential role for PpiB could be to functionally compensate for SecB and chaperone proteins prior to secretion (
Figure 6A). Thus, the loss of PpiB could result in general secretion defects and/or the secretion of misfolded proteins. This might explain why so many proteins had altered abundance in the culture supernatants of
ppiB mutant strains compared to
prsA mutants (86 proteins altered in
ppiB mutant, 16 in
prsA mutant). Another role of chaperone proteins is to prevent the aggregation of misfolded proteins under stress [
40]. If PpiB is functioning as a chaperone, it could additionally function to protect the αPSM peptides within the
S. aureus cell. If this is the case, it could explain why there are less active αPSMs in culture supernatants of a Δ
ppiB mutant.
S. aureus, like many other cells, has the ability to produce and secrete extracellular vesicles (EVs) [
27]. Recently, the formation of EVs in
S. aureus has been shown to be enhanced by the αPSMs [
27]. EVs are loaded with proteins, including many proteins typically thought of as being cytoplasmic. The presence of EVs in culture supernatants may explain why cytoplasmic proteins are commonly identified during proteomic studies of secreted fractions, which we also observed in our proteomic data set. Interestingly, of the 86 proteins that were altered in abundance in
ppiB mutant supernatants, a large proportion of them are considered cytoplasmic. These proteins were present in WT supernatants, but decreased in
ppiB mutant supernatants. It is possible that PpiB plays a role in EV biogenesis and that in the mutant there is reduced EV production, which accounts for the variation in cytoplasmic proteins. Since a
ppiB mutant produces less αPSMs which are necessary for EV release, it is also possible that the same number of EVs are being generated in a Δ
ppiB mutant but they are unable to be released from the cell due to the decrease in αPSMs (
Figure 6B). We are currently investigating EV production in a
ppiB mutant.
In conclusion, while the role of PpiB in pathogenesis is clear, its molecular mechanism of action remains uncertain. The results generated in this study further our understanding of S. aureus virulence factors regulated by PpiB and suggest that PpiB may be functioning in a secretion-related manner. Further studies are being conducted to investigate this novel protein to better understand how it regulates virulence factor production and virulence.
4. Materials and Methods
4.1. Strains and Strain Construction
All bacterial strains and plasmids are listed in
Table 5 and oligonucleotides in
Table 6. The Δ
ppiB and Δ
prsA mutant strains were constructed via allelic exchange as previously described [
21,
24,
41]. For overexpresser strains used in the immunoprecipitation, plasmids pRKC0131, pRKC0126 and pMK4 (empty vector) were transduced into RKC0323 (Δ
ppiB) and RKC0085 (Δ
prsA) respectively. A Δ
αpsm strain was constructed by allelic exchange using plasmid pJB38 [
42]. DNA sequences flanking the αPSM transcript were amplified using primer pairs 490/644 and 645/493 and a third sequence containing the erythromycin cassette from the
bursa aurealis transposon was amplified using primers 638 and 639 [
42]. These three fragments were cloned to generate plasmid pRKC0674. This plasmid was recombined onto the
S. aureus chromosome and excised to make the deletion strain according to previously published protocol [
41].
4.2. Bacterial Growth Conditions
S. aureus cultures were grown in tryptic soy broth (TSB) shaking at 37 °C. Where appropriate, the antibiotic chloramphenicol was used at the concentration of 5 μg mL−1. For analysis of culture supernatants, S. aureus cultures were synchronized as follows. Replicate overnight cultures were grown in 5 mL of TSB for 15 h. The next day, cultures were diluted 1:100 in 10 mL of pre-warmed TSB and grown for 3 h to mid-exponential phase. The 3-h cultures were then diluted into 25 mL of TSB in 250 flasks and normalized to an optical density at 600 nm (OD600) of 0.05. Resulting flasks were grown overnight for 15 h.
4.3. Murine Abscess Model of Infection
A subcutaneous abscess infection was performed as described by us previously [
21]. Cultures were grown for 2.5 h in TSB to an OD
600 of 0.75. Resulting bacterial cells were pelleted via centrifugation and resuspended in sterile phosphate-buffered saline (PBS) to prepare an inocula of 10
6 CFU/50 μL. Each inoculum was then confirmed via serial diluting and plating. Six-week-old female BALB/c mice were anesthetized by isoflourane inhalation before being shaved at the right flank and treated with Nair to remove fur. Mice were then injected with 50 μL of their respective strains and the infection was allowed to proceed for 7 days. Following 7 days of infection, mice were euthanized with CO
2 and abscesses were excised and homogenized. Homogenates were serial diluted and plated onto TSB to count recovered CFU/ abscess.
4.4. Murine Systemic Model of Infection
A systemic model of dissemination was chosen to mimic septic infection and cultures were prepared as previously described by Spaan et al. [
49]. Cultures were grown for approximately 2.5 h in TSB to an OD
600 of 0.75. Resulting bacterial cells were pelleted via centrifugation and resuspended in sterile phosphate-buffered saline (PBS) to prepare an inocula of 10
7 CFU/100 μL. Six-week-old BALB/c mice were injected retro-orbitally with 100 μL of bacterial cultures and the infection was allowed to proceed for 3 days. Following 3 days of infection, mice were euthanized with CO
2 and the brain, lungs, heart, liver, kidneys and spleen were harvested. Each organ was weighed and homogenized before serial dilution and plating was conducted to quantify recovered bacteria/ organ.
4.5. Macrophage Infection and Cell Differentiation
THP-1 infection assays were performed as outlined in Carroll et al. [
50]. Macrophages were seeded at a density of 2 × 10
5 macrophages/well in a volume of 500 µL of RPMI 1640 with 10% FBS and 1% penicillin/streptomycin. A multiplicity of infection (MOI) of 10 was used to infect each well (2 × 10
6 bacteria/well). Strains were prepared by taking 250 µL of overnight culture grown at 37 °C and inoculating it into a 250 mL flask containing 25 mL TSB. The bacteria were then grown in a shaking 37 °C incubator until their optical density (OD
600) reached 0.4–0.6. The volume of bacteria needed to perform the infection was then transferred into a 1.5 mL centrifuge tube and pelleted for 20 min at 3000 rpm. The supernatant was removed and discarded. The pellet was then washed with 500 µL of phosphate buffered saline (PBS) and pelleted for 20 min at 3000 rpm. The pellet was resuspended in 20% human serum and 80% PBS. This solution was then incubated in a 37 °C water bath for 30 min to opsonize the bacteria. Opsonized bacteria were resuspended into the correct volume of 1640 RPMI medium with 10% FBS.
The macrophages were washed twice with 37 °C prewarmed PBS. Each well received 500 µL of the opsonized bacteria resuspended in 1640 RPMI with 10% FBS. Plates were centrifuged at 1000 rpm for 10 min at room temperature and placed into a 37 °C incubator supplemented with 5% CO2 for 60 min. After 60 min, the medium was aspirated, and the wells were washed once with prewarmed PBS as described above. To each well, 500 µL of RPMI 1640 with 10% FBS and 30 µg/mL of gentamicin was added. Cells were incubated in a 37 °C incubator supplemented with 5% CO2 for 60 min. The end of this 60 min incubation marked hour 0 of intracellular. After this incubation, the medium was aspirated and replaced with 500 µL of RPMI 1640 with 10% FBS and 5 µg/mL of gentamicin for the rest of the infection.
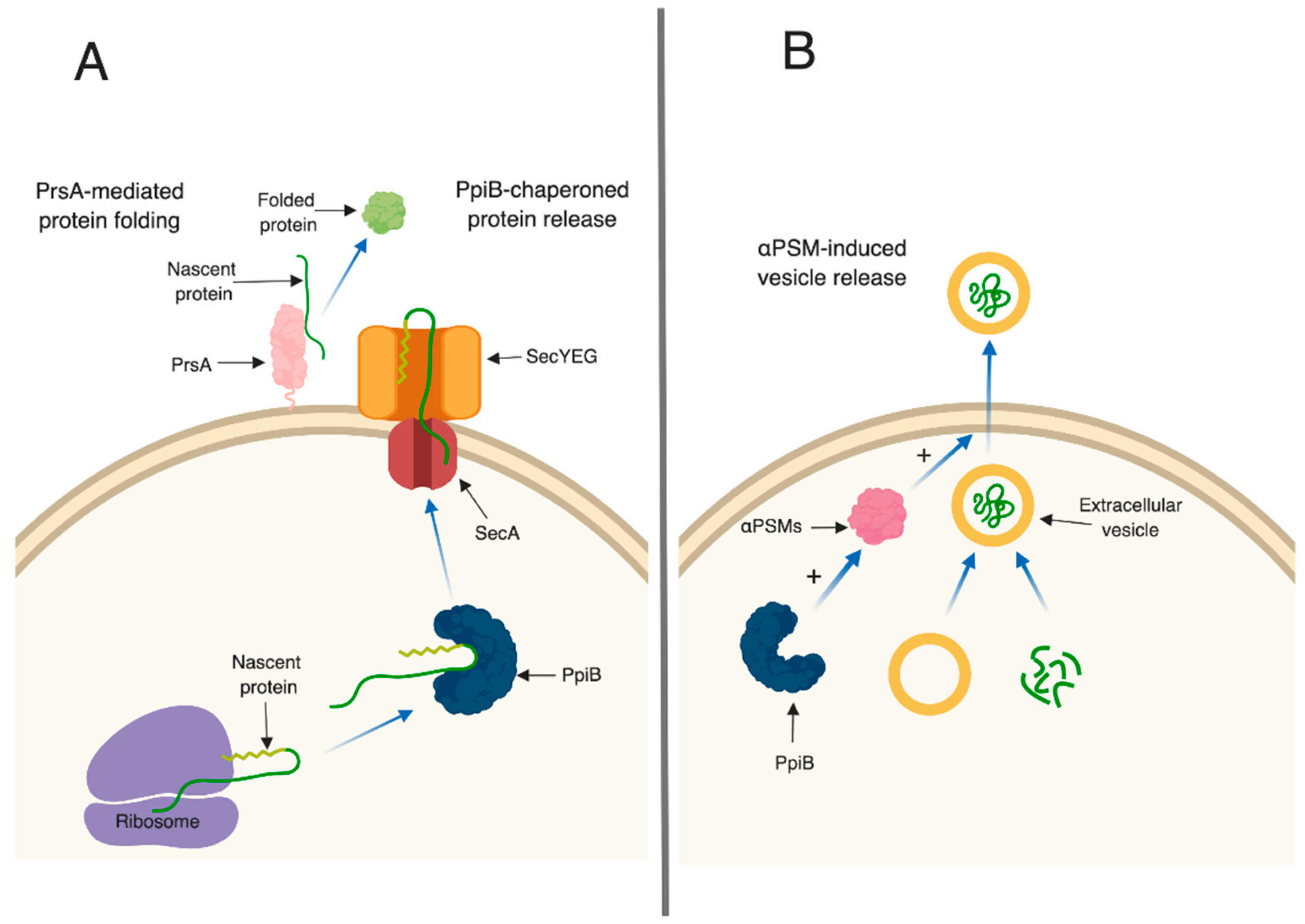
Intracellular bacteria were harvested from the macrophages at 2 h and 48 h, representing initial and late stages of infection. Medium was aspirated, and the cells were washed twice with prewarmed PBS as described above. A 500 µL aliquot of 0.5% Triton X-100 in PBS was added to each well to lyse the macrophages and release the intracellular bacteria. The lysates for each time point were serially diluted in PBS in sterile 96-well plates. A volume of 100 µL of each of the diluted lysate was plated on TSA plates. Plates were incubated in a 37 °C incubator and colonies were counted to determine the number of recovered intracellular bacteria and calculated to determine CFU/mL.
4.6. Nasal Epithelial Cell Infection
Strains were prepared and washed in the same way as described above except the bacteria were not opsonized. After washing the bacterial pellet with PBS, the bacteria were resuspended into the correct volume of EMEM with 10% FBS. RPMI 2650 cells were seeded at the same density and volume as described above except using EMEM with 10% FBS and 1% penicillin/streptomycin. Cells were washed twice with prewarmed PBS and infected with bacteria at an MOI of 10 (2 x 106 bacteria/well).
4.7. Exoproteome Analysis
Bacterial strains were grown in triplicate in 5 mL overnight cultures shaking at 37 °C. The next day, cultures were diluted 1:100 in 10 mL of TSB and incubated for 3 h shaking at 37 °C. After 3 h, the OD600 of each culture was measured and each sample was normalized to an OD600 of 0.05 in 100 mL of TSB. After 15 h of growth in the flask, samples were split into 2 × 50 mL tubes and centrifuged at 3000 rpm for 15 min. Cell-free supernatants were harvested and filter sterilized through a 0.45 μm filter disk to ensure all bacterial cells were removed from the sample. An amount of 10 mL of TCA was added to the culture supernatants and incubated overnight at 4 °C. The following day, samples were centrifuged at 11,000 RPM for 10 min and the supernatant was removed. Resulting pellets containing concentrated proteins were washed with ice-cold acetone and sent to the University of Nebraska Lincoln Proteomics Facility for mass spectrometry analysis.
TCA precipitated samples were dissolved in a solution of 7M urea, 2M thiourea, 5 mM DTT, 0.1 M Tris, pH 8 and 1× PhosStop by gentle shaking for 1.5 h at 24 °C. After reduction, the samples were alkylated for 30 min using a 3-fold molar excess of iodoacetamide. The protein concentration was determined using the CB-X protein assay kit. An amount of 100 μg of protein in 10 μL of the urea solution was diluted and digested with 2 μg trypsin (1:50 enzyme:substrate ratio) for 16 h overnight, then an additional 1 μg of trypsin was added and digestion continued for a further 4 h. An amount of 200 ng of each of the 9 samples was run by nanoLC-MS/MS using a 2 h gradient on a 0.075 mm × 250 mm C18 Waters CSH column feeding into a Q-Exactive HF mass spectrometer.
Raw datafiles were loaded directly in to Progenesis QI for proteomics version 2.0 and alignment of the chromatograms showed a ≥95% match. Features (peaks) were extracted across all runs and areas under the peaks calculated. Runs were normalized using the “normalize to all proteins” setting. Data were exported from Progenesis and analyzed using the search engine Mascot (Matrix Science, London, UK; version 2.6.1, 2016. Mascot was set up to search 2 databases: the common contaminants database cRAP_20150130 database (117 entries) and a combined database containing 2 uniprot reference proteomes for S. aureus NCTC8325 & C0673_20170607 (5705 entries) assuming the digestion enzyme trypsin. Mascot was searched with a fragment ion mass tolerance of 0.060 Da and a parent ion tolerance of 10.0 PPM. Oxidation of methionine; deamidated of asparagine and glutamine; and carbamidomethyl of cysteine were specified in Mascot as variable modifications.
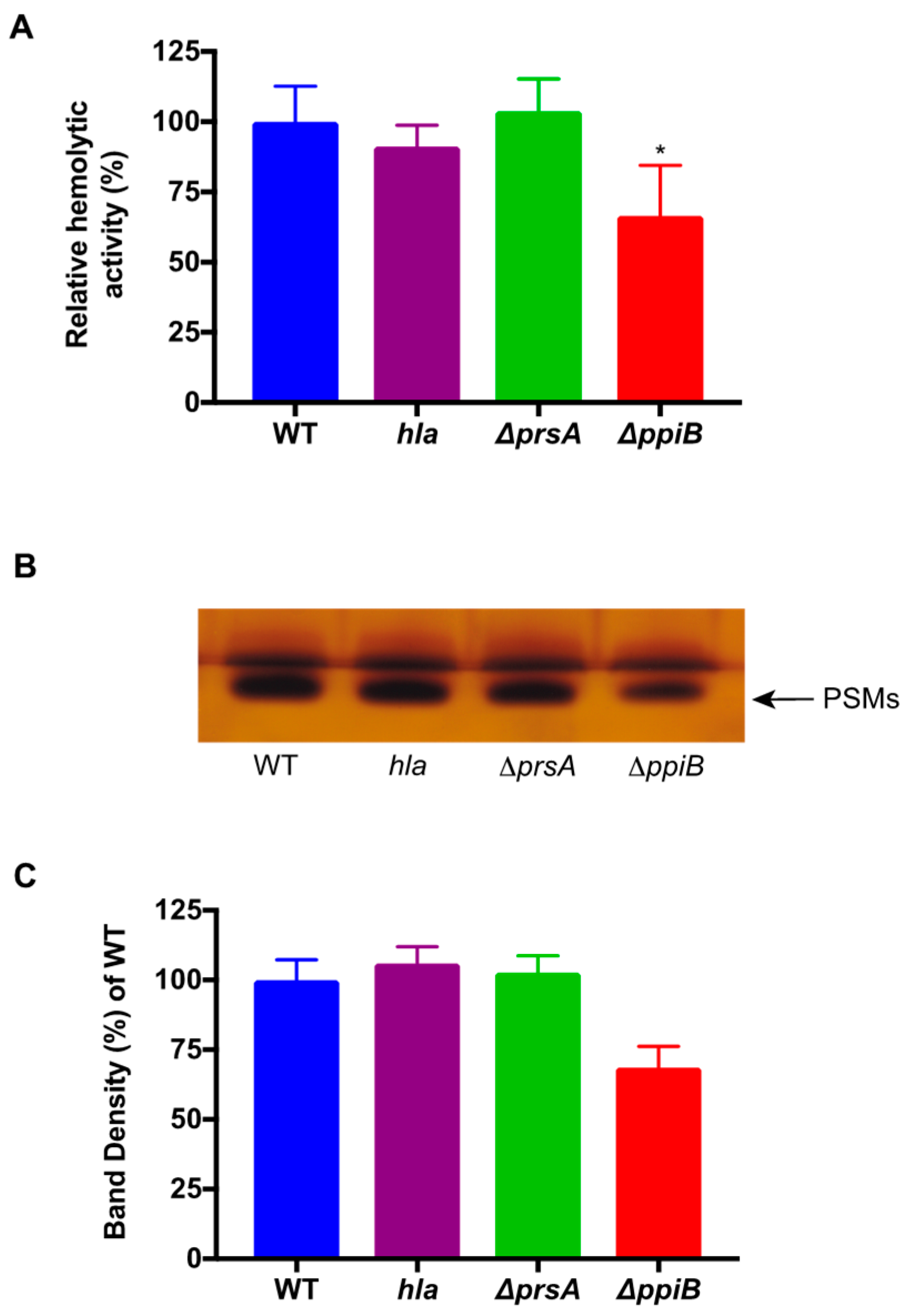
4.8. Butanol Extraction of PSMs and Densitometry Analysis
PSM peptides were isolated as previously described [
29]. An amount of 5 mL of synchronized, cell-free culture supernatants was incubated shaking at 37 °C with 3 mL of
n-butanol for 2 h. Cultures were then centrifuged and 1 mL of the organic layer was removed for vacuum centrifugation for 12 h at 5000 rpm. Samples containing the PSMs were then resuspended in water and utilized for human erythrocyte hemolysis assays. The same set of samples was run on SDS-PAGE gels in duplicate and silver stained. Densitometry analysis of the duplicate extract samples was performed using Image J software. Samples were averaged and normalized to the percent density of WT.
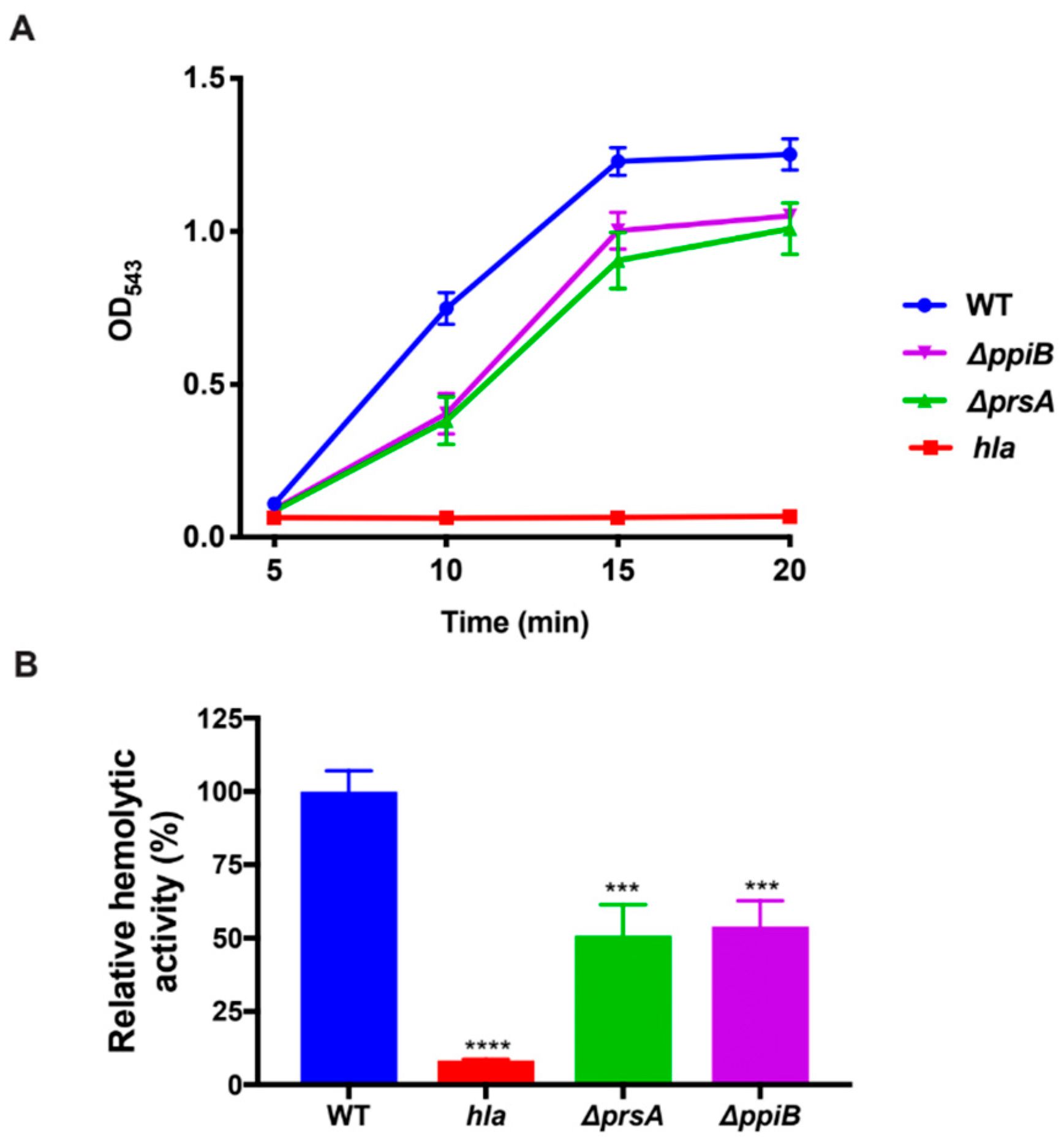
4.9. Human-Erythrocyte Hemolysis Assay
Synchronized bacterial cultures were grown in quadruplicate as described in bacterial growth conditions for 15 h. Samples were centrifuged and cell-free culture supernatants were harvested and diluted 1:2 in reaction buffer containing 40 mM CaCl2, and 1.7% NaCl. An amount of 25 µL of whole-human blood was added to samples and they were incubated at 37 °C while rotating. After 10 min, samples were centrifuged at 5500× g and the resulting supernatant was transferred to a 96-well plate. The degree of erythrocyte lysis was determined by reading sample absorbance at an OD543.
4.10. Rabbit Erythrocyte Hemolysis Assay
Bacterial strains were grown in quadruplicate in a 25 mL flask of TSB as described above. Cultures were then centrifuged to obtain the cell-free supernatant. Supernatants were diluted 1:10 before being further diluted 1:2 in reaction buffer containing 40 mM CaCl2, and 1.7% NaCl and incubated in a 37 °C water bath with 125 µL of rabbit blood. An amount of 200 µL of samples was removed every 5 min after incubation and centrifuged at 5500× g. An amount of 100 µl of supernatant was transferred to a 96-well plate and erythrocyte lysis was determined by reading the absorbance of the samples at OD543.
4.11. Protein Immunoprecipitation Assay
Duplicate bacterial cultures were inoculated into 100 mL of TSB with appropriate antibiotics in 250 mL flasks for 3 h to mid-exponential phase. Samples were centrifuged at 3000 rpm for 20 min and pellets were resuspended in sterile PBS. In total, 37% formaldehyde was added to cultures to a final concentration of 1% and a 20 min shaking incubation was performed at room temperature. The reaction was quenched with the addition of glycine to a final concentration of 200 mM. Cells were centrifuged and the remaining pellets were washed with sterile PBS and then re-suspended in sterile water. Cells were then lysed with lysostaphin at a concentration of 10 mg/mL and incubated at 37 °C for 30 min. Following this, cells were treated with DNase I and incubated at 37 °C for 30 min. Resulting lysates were then sonicated at 20% amplitude, 2 × 15 s. Samples were then centrifuged at 13,000 rpm for 1 min and supernatants were used for immunoprecipitation.
For immunoprecipitation, anti-HA or anti-His magnetic beads were added to respective samples and incubated at 4 °C for 1 h. Beads were collected with a magnetic rack and washed 10 times with 1× PBS. Final washed beads were resuspended in 20 µL of water and sent to the University of Nebraska Lincoln Proteomics Facility for analysis.
The 2 magnetic bead samples were resuspended in ammonium bicarbonate containing 5 mM DTT and reduced at 37 °C for 1 h. Samples were then alkylated (10 mM IAM for 20 min at 22 °C in the dark). The IAM was quenched with a molar equivalent of DTT. Trypsin was added and digestion carried out overnight at 37 °C. The digest was dried down and dissolved in 2.5% acetonitrile, 0.1% formic acid. An amount of 5 uL of one digest was run by nanoLC-MS/MS using a 2 h gradient on a 0.075 mm × 250 mm C18 Waters CSH column feeding into a Q-Exactive HF mass spectrometer.
All MS/MS samples were analyzed using Mascot (Matrix Science, London, UK; version 2.6.1, 2016). Mascot was set up to search the cRAP_20150130.fasta (117 entries); uniprot_S_aureus_USA300_20170823 database (2607 entries) assuming the digestion enzyme trypsin. Mascot was searched with a fragment ion mass tolerance of 0.060 Da and a parent ion tolerance of 10.0 PPM. Deamidated of asparagine and glutamine, oxidation of methionine and carbamidomethyl of cysteine were specified in Mascot as variable modifications.
Scaffold (Proteome Software Inc., Portland, OR, USA, version Scaffold_4.8.4, 2017) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 80.0% probability by the Peptide Prophet algorithm [
51] with Scaffold delta-mass correction. Protein identifications were accepted if they could be established at greater than 99.0% probability and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm [
52]. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters.
4.12. Reverse Transcriptase-Quantitative PCR (RT-qPCR)
RT-qPCR was performed as described previously [
47]. Briefly, bacterial pellets were collected 6 h after subculture and total RNA was isolated. Complimentary DNA (cDNA) was synthesized from 1 µg of total RNA using iScript reverse transcriptase (Bio-Rad) according to the manufacturer’s directions. The cDNA was diluted 10 times and used in SYBR Green reactions in technical duplicates to analyze the expression of
αPSM, and
hla. Transcription of the housekeeping gene
gyrB was used as the endogenous control in each strain. Relative expression was determined by first comparing the amount of each individual gene transcript to
gyrB within the same strain, followed by expression of these values in comparison to each respective gene in WT strain TCH1516.
4.13. Ethics Statement
Whole human-blood was isolated from donors in agreement with the Ohio University Institutional Review Board (Identification code; 17-X-79 date of approval: 22 February 2019). Rabbit blood was purchased from Hemostat-laboratories. Six-week-old female BALB/c mice were purchased from Envigo and held at the Ohio University Office of Laboratory Animal Resources. Animal work was performed under approval of the Institutional Animal Care and Use Committee (Identification code; 18-H-029 date of approval: 04 September 2018) at Ohio University and performed by trained lab personnel.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}