Expression Patterns of Coagulation Factor XIII Subunit A on Leukemic Lymphoblasts Correlate with Clinical Outcome and Genetic Subtypes in Childhood B-cell Progenitor Acute Lymphoblastic Leukemia

 , , , , , , , ,
, , , , , , , ,  , and
, and  add
Show full author list
add
Show full author list
Abstract
:1. Introduction
Presence and Role of FXIII-A in Different Cell Types
2. Results
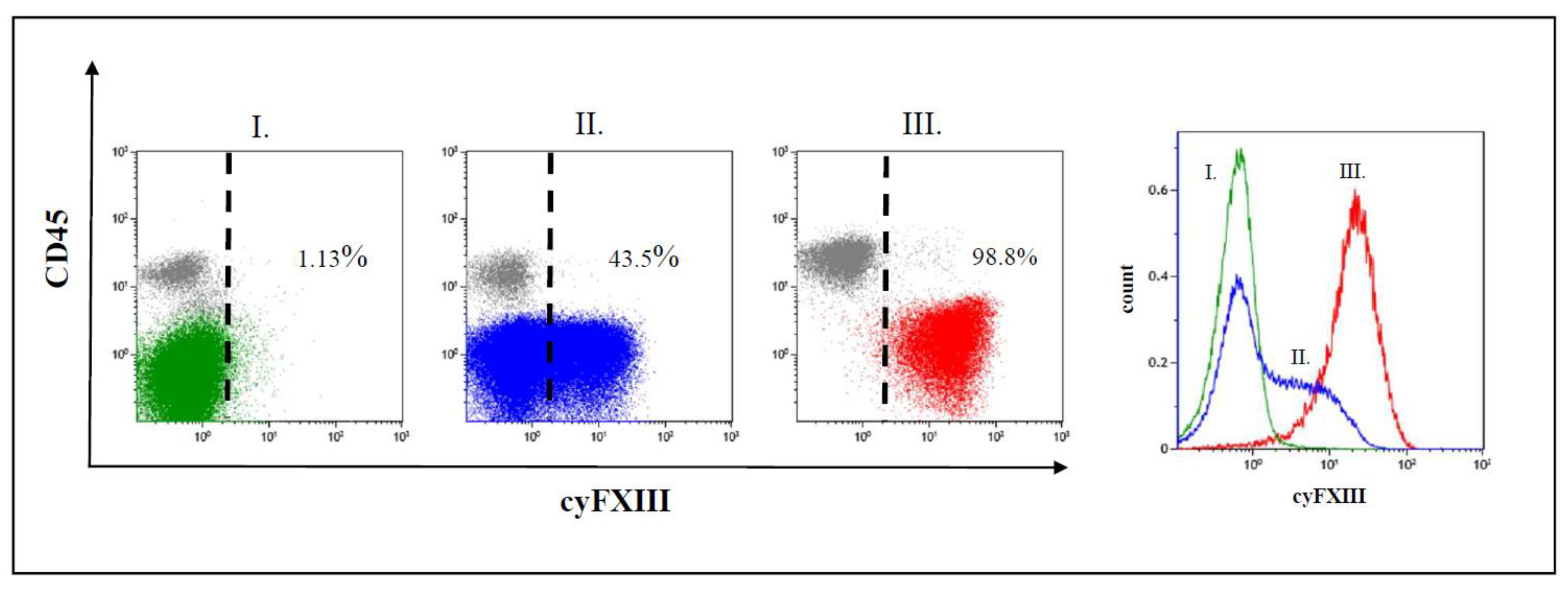
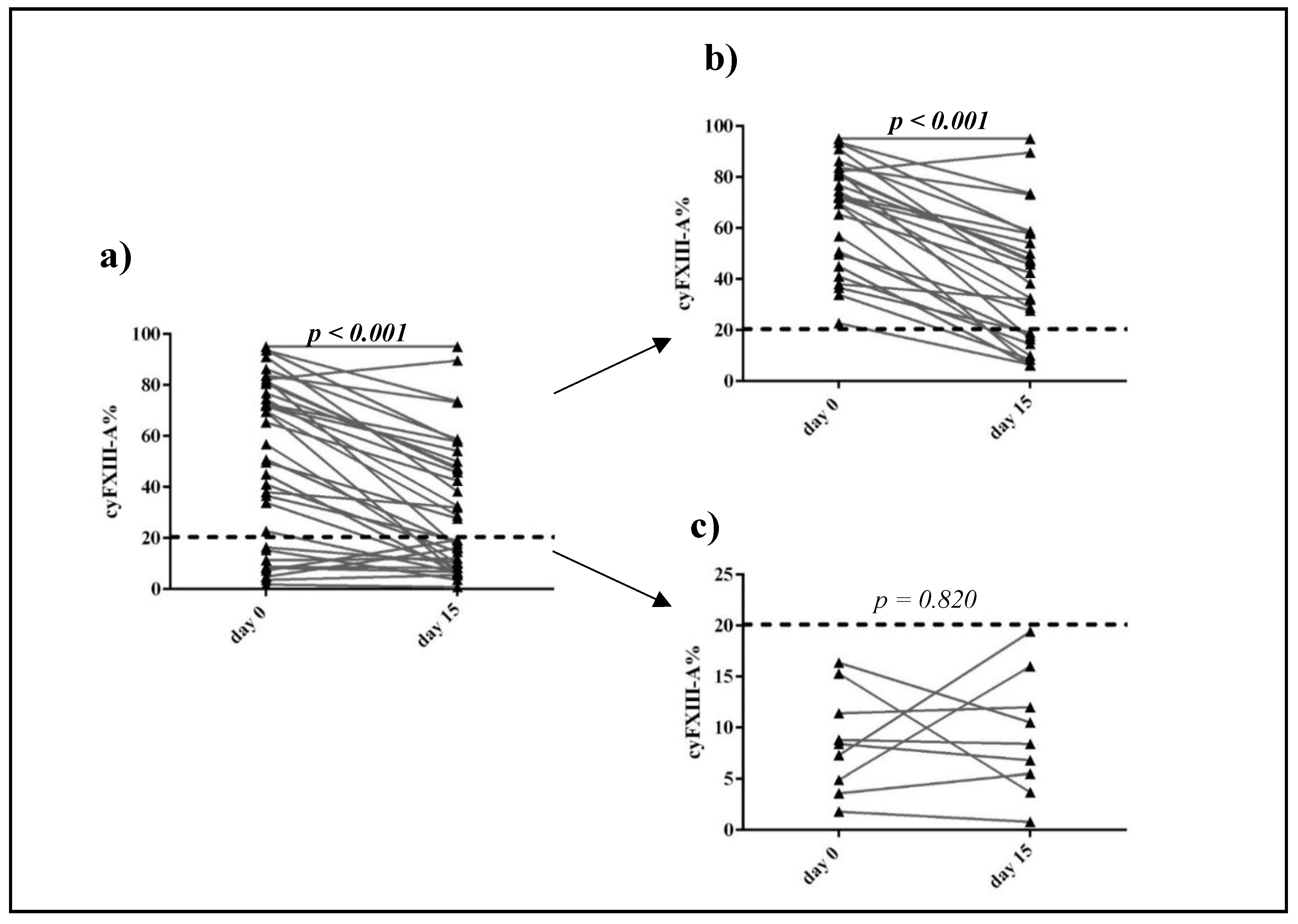
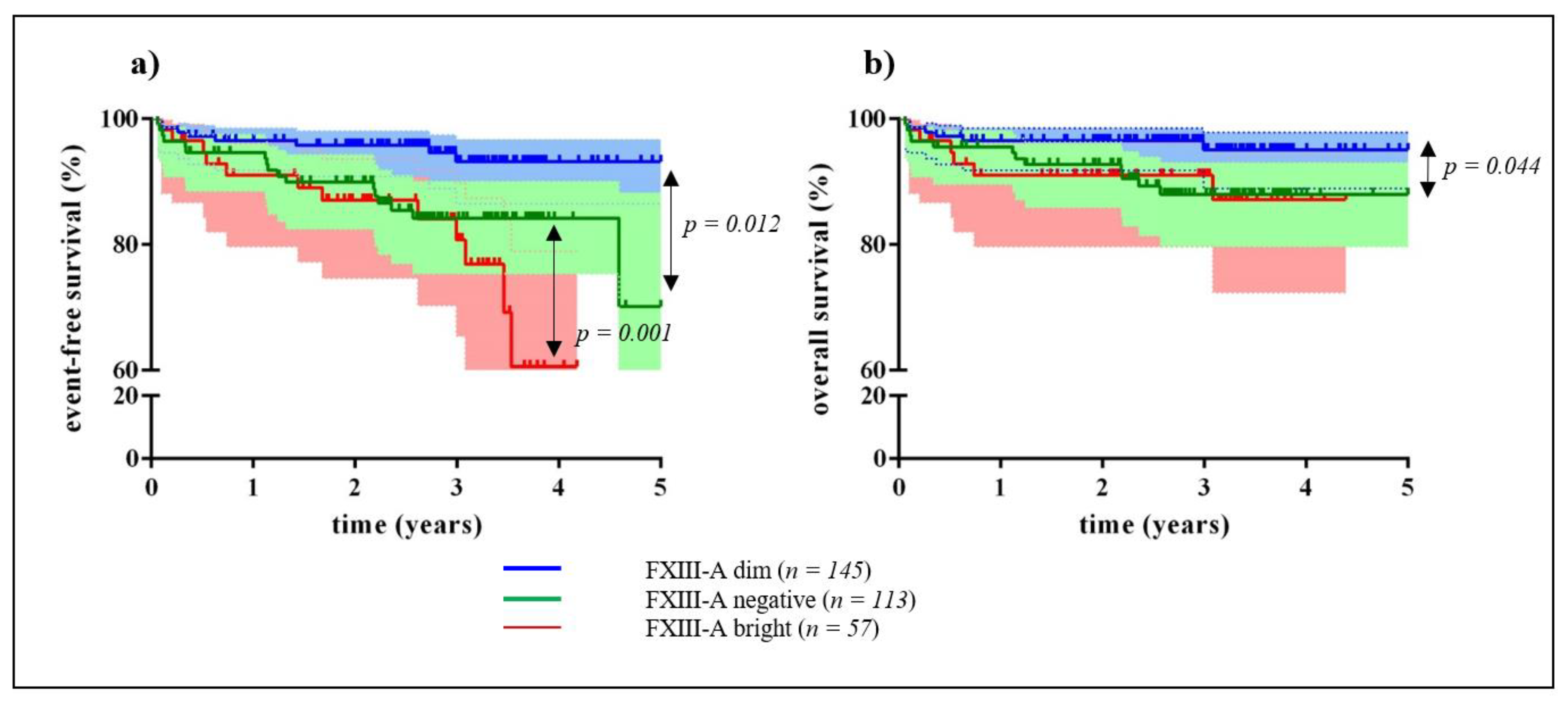
2.1. Clinical Significance of Different Expression Patterns of FXIII-A by B-Cell Progenitor (BCP) Lymphoblasts
2.2. Correlation of FXIII-A Expression Patterns with Conventional Clinical, Minimal Residual Disease (MRD), and Genetic Risk Factors
3. Discussion
4. Materials and Methods
4.1. Study Cohort
4.2. Immunophenotype Analysis
4.3. Genetic Investigations
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FXIII-A | coagulation factor XIII subunit A |
| BCP | B-cell progenitor |
| ALL | acute lymphoblastic leukemia |
| BFM ALL-IC 2009 | Berlin-Frankfurt-Münster inter-Continental 2009 clinical trial |
| iBFM-SG | international Berlin-Frankfurt-Münster Study Group |
| EFS | event-free survival |
| OS | overall survival |
| COG | Children’s Oncology Group |
| WBC | white blood cell count |
| ic | Intracellular |
| FC | flow cytometry |
| MRD | minimal residual disease |
| BM | bone marrow |
| MLPA | multiplex ligation dependent probe amplification |
| CNA | copy number alterations |
| AIEOP-BFM 2009 | International Collaborative Treatment Protocol For Children And Adolescents With Acute Lymphoblastic Leukemia clinical trial |
| cyFXIII-A | cytoplasmic coagulation factor XIII subunit A |
| PB | peripheral blood |
| HR | high-risk |
| IR | intermediate risk |
| SR | standard risk |
| FLR | flow low-risk |
| FMR | flow medium-risk |
| FHR | flow high-risk |
| BFM-HR | BFM-high risk |
| BFM-SR | BFM standrard risk |
| BFM-IR | BFM intermediate risk |
| CCG | Children’s cancer group |
| Mtx | Methotrexate |
| FITC | fluorescent isotihocyanate |
| FISH | fluorescence in situ hybridization |
| CI | cinfidence intervals |
| HR | Hazard-ratio |
| CCC | clear cell carcinomas |
| APL | acute promyelocytic leukemia |
| PGR | prednisone good response |
| PPR | prednisone poor response |
| OR | Odds ratio |
| NA | not available |
| N | Number |
References
- Hunger, S.P. Expanding clinical trial networks in pediatric acute lymphoblastic leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 169–170. [Google Scholar] [CrossRef] [PubMed]
- Acute Lymphoblastic Leukaemia Committee (All)—International Bfm Study Group. Available online: https://bfminternational.wordpress.com/structure/committees/acute-lymphoblastic-leukaemia-committee-all (accessed on 18 June 2020).
- Howard, S.C.; Davidson, A.; Luna-Fineman, S.; Israels, T.; Chantada, G.; Lam, C.G.; Hunger, S.P.; Bailey, S.; Ribeiro, R.C.; Arora, R.S.; et al. A framework to develop adapted treatment regimens to manage pediatric cancer in low- and middle-income countries: The pediatric oncology in developing countries (podc) committee of the international pediatric oncology society (siop). Pediatr. Blood Cancer 2017, 64, e26879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stary, J.; Zimmermann, M.; Campbell, M.; Castillo, L.; Dibar, E.; Donska, S.; Gonzalez, A.; Izraeli, S.; Janic, D.; Jazbec, J.; et al. Intensive chemotherapy for childhood acute lymphoblastic leukemia: Results of the randomized intercontinental trial all ic-bfm 2002. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 174–184. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the world health organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute lymphoblastic leukaemia. Lancet 2013, 381, 1943–1955. [Google Scholar] [CrossRef] [Green Version]
- Moorman, A.V. New and emerging prognostic and predictive genetic biomarkers in b-cell precursor acute lymphoblastic leukemia. Haematologica 2016, 101, 407–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobucci, I.; Mullighan, C.G. Genetic basis of acute lymphoblastic leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef]
- Mirkowska, P.; Hofmann, A.; Sedek, L.; Slamova, L.; Mejstrikova, E.; Szczepanski, T.; Schmitz, M.; Cario, G.; Stanulla, M.; Schrappe, M.; et al. Leukemia surfaceome analysis reveals new disease-associated features. Blood 2013, 121, e149–e159. [Google Scholar] [CrossRef] [Green Version]
- Sedek, L.; Theunissen, P.; Sobral da Costa, E.; van der Sluijs-Gelling, A.; Mejstrikova, E.; Gaipa, G.; Sonsala, A.; Twardoch, M.; Oliveira, E.; Novakova, M.; et al. Differential expression of cd73, cd86 and cd304 in normal vs. Leukemic b-cell precursors and their utility as stable minimal residual disease markers in childhood b-cell precursor acute lymphoblastic leukemia. J. Immunol. Methods 2019, 475, 112429. [Google Scholar] [CrossRef]
- Navarrete, M.; Rossi, E.; Brivio, E.; Carrillo, J.M.; Bonilla, M.; Vasquez, R.; Pena, A.; Fu, L.; Martinez, R.; Espinoza, C.M.; et al. Treatment of childhood acute lymphoblastic leukemia in central america: A lower-middle income countries experience. Pediatr. Blood Cancer 2014, 61, 803–809. [Google Scholar] [CrossRef]
- Theunissen, P.; Mejstrikova, E.; Sedek, L.; van der Sluijs-Gelling, A.J.; Gaipa, G.; Bartels, M.; Sobral da Costa, E.; Kotrova, M.; Novakova, M.; Sonneveld, E.; et al. Standardized flow cytometry for highly sensitive mrd measurements in b-cell acute lymphoblastic leukemia. Blood 2017, 129, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Mejstrikova, E.; Fronkova, E.; Kalina, T.; Omelka, M.; Batinic, D.; Dubravcic, K.; Pospisilova, K.; Vaskova, M.; Luria, D.; Cheng, S.H.; et al. Detection of residual b precursor lymphoblastic leukemia by uniform gating flow cytometry. Pediatr. Blood Cancer 2010, 54, 62–70. [Google Scholar] [CrossRef]
- Radu, L.E.; Colita, A.; Pasca, S.; Tomuleasa, C.; Popa, C.; Serban, C.; Gheorghe, A.; Serbanica, A.; Jercan, C.; Marcu, A.; et al. Day 15 and day 33 minimal residual disease assessment for acute lymphoblastic leukemia patients treated according to the bfm all ic 2009 protocol: Single-center experience of 133 cases. Front. Oncol. 2020, 10, 923. [Google Scholar] [CrossRef] [PubMed]
- St. Anna Children’s Cancer Research Institute Science Report 2009–2010. Available online: https://science.ccri.at/fileadmin/content/3Research/6Scientific_Reports/CCRI_ScienceReport_2009–2010.pdf (accessed on 18 June 2020).
- Muszbek, L.; Bereczky, Z.; Bagoly, Z.; Komaromi, I.; Katona, E. Factor xiii: A coagulation factor with multiple plasmatic and cellular functions. Physiol. Rev. 2011, 91, 931–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickneite, G.; Herwald, H.; Korte, W.; Allanore, Y.; Denton, C.P.; Matucci Cerinic, M. Coagulation factor xiii: A multifunctional transglutaminase with clinical potential in a range of conditions. Thromb. Haemost. 2015, 113, 686–697. [Google Scholar]
- Paragh, L.; Torocsik, D. Factor xiii subunit a in the skin: Applications in diagnosis and treatment. BioMed Res. Int. 2017, 2017, 3571861. [Google Scholar] [CrossRef] [Green Version]
- Kiss, F.; Simon, A.; Csathy, L.; Hevessy, Z.; Katona, E.; Kiss, C.; Kappelmayer, J. A coagulation factor becomes useful in the study of acute leukemias: Studies with blood coagulation factor xiii. Cytom. Part A J. Int. Soc. Anal. Cytol. 2008, 73, 194–201. [Google Scholar] [CrossRef]
- Penneys, N.S.; Smith, K.J.; Nemeth, A.J. Factor xiiia in the hamartomas of tuberous sclerosis. J. Dermatol. Sci. 1991, 2, 50–54. [Google Scholar] [CrossRef]
- Bains, A.; Parham, D.M. Langerhans cell histiocytosis preceding the development of juvenile xanthogranuloma: A case and review of recent developments. Pediatr. Dev. Pathol. 2011, 14, 480–484. [Google Scholar] [CrossRef]
- Tran, T.A.; Hayner-Buchan, A.; Jones, D.M.; McRorie, D.; Carlson, J.A. Cutaneous balloon cell dermatofibroma (fibrous histiocytoma). Am. J. Dermatopathol. 2007, 29, 197–200. [Google Scholar] [CrossRef]
- Nemeth, A.J.; Penneys, N.S. Factor xiiia is expressed by fibroblasts in fibrovascular tumors. J. Cutan. Pathol. 1989, 16, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Ogera, P.; Benardete, E.A.; Nicastri, A.D.; Rao, C. Cellular solitary fibrous tumor (hemangiopericytoma) with anaplasia at cerebellopontine angle—A case report. Pathol. Res. Pract. 2012, 208, 493–496. [Google Scholar] [CrossRef]
- Cuff, J.; Salari, K.; Clarke, N.; Esheba, G.E.; Forster, A.D.; Huang, S.; West, R.B.; Higgins, J.P.; Longacre, T.A.; Pollack, J.R. Integrative bioinformatics links hnf1b with clear cell carcinoma and tumor-associated thrombosis. PLoS ONE 2013, 8, e74562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooper, L.M.; Huang, S.C.; Antonescu, C.R.; Westra, W.H.; Bishop, J.A. Biphenotypic sinonasal sarcoma: An expanded immunoprofile including consistent nuclear beta-catenin positivity and absence of sox10 expression. Hum. Pathol. 2016, 55, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Simon, A.; Bagoly, Z.; Hevessy, Z.; Csathy, L.; Katona, E.; Vereb, G.; Ujfalusi, A.; Szerafin, L.; Muszbek, L.; Kappelmayer, J. Expression of coagulation factor xiii subunit a in acute promyelocytic leukemia. Cytom. Part B Clin. Cytom. 2012, 82, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Kiss, F.; Hevessy, Z.; Veszpremi, A.; Katona, E.; Kiss, C.; Vereb, G.; Muszbek, L.; Kappelmayer, J.N. Leukemic lymphoblasts, a novel expression site of coagulation factor xiii subunit a. Thromb. Haemost. 2006, 96, 176–182. [Google Scholar]
- Ueki, S.; Takagi, J.; Saito, Y. Dual functions of transglutaminase in novel cell adhesion. J. Cell Sci. 1996, 109, 2727–2735. [Google Scholar]
- Bonish, B.K.; Foreman, K.E.; Gutierrez-Steil, C.; Nickoloff, B.J. Phenotype and proliferation characteristics of cultured spindle-shaped cells obtained from normal human skin and lesions of dermatofibroma, kaposi‘s sarcoma, and dermatofibrosarcoma protuberans: A comparison with fibroblast and endothelial cells of the dermis. J. Dermatol. Sci. 1997, 16, 52–58. [Google Scholar]
- Karai, B.; Hevessy, Z.; Szantho, E.; Csathy, L.; Ujfalusi, A.; Gyurina, K.; Szegedi, I.; Kappelmayer, J.; Kiss, C. Expression of coagulation factor xiii subunit a correlates with outcome in childhood acute lymphoblastic leukemia. Pathol. Oncol. Res. POR 2018, 24, 345–352. [Google Scholar] [CrossRef]
- Gyurina, K.; Karai, B.; Ujfalusi, A.; Hevessy, Z.; Barna, G.; Jakso, P.; Palfi-Meszaros, G.; Poliska, S.; Scholtz, B.; Kappelmayer, J.; et al. Coagulation fxiii-a protein expression defines three novel sub-populations in pediatric b-cell progenitor acute lymphoblastic leukemia characterized by distinct gene expression signatures. Front. Oncol. 2019, 9, 1063. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Xu, J.; Liu, Q.; Li, J.; Xi, Y. Expression and significance of cd47, pd1 and pdl1 in t-cell acute lymphoblastic lymphoma/leukemia. Pathol. Res. Pract. 2019, 215, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Sasca, D.; Szybinski, J.; Schuler, A.; Shah, V.; Heidelberger, J.; Haehnel, P.S.; Dolnik, A.; Kriege, O.; Fehr, E.M.; Gebhardt, W.H.; et al. Ncam1 (cd56) promotes leukemogenesis and confers drug resistance in aml. Blood 2019, 133, 2305–2319. [Google Scholar] [CrossRef]
- Dworzak, M.N.; Buldini, B.; Gaipa, G.; Ratei, R.; Hrusak, O.; Luria, D.; Rosenthal, E.; Bourquin, J.P.; Sartor, M.; Schumich, A.; et al. Aieop-bfm consensus guidelines 2016 for flow cytometric immunophenotyping of pediatric acute lymphoblastic leukemia. Cytom. Part B Clin. Cytom. 2018, 94, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, W.N.; Wickham, R.; Coombs, N. An introduction to survival statistics: Kaplan-meier analysis. J. Adv. Pract. Oncol. 2016, 7, 91–100. [Google Scholar] [PubMed] [Green Version]
- Raval, J.S.; Berg, A.N.; Djokic, M.; Roth, C.G.; Rollins-Raval, M.A. Factor xiii subunit a immunohistochemical expression is associated with inferior outcomes in acute promyelocytic leukemia. Appl. Immunohistochem. Mol. Morphol. AIMM 2018, 26, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.; Wu, D.; Crossley, B.; Dai, Y.; Williamson, D.; Gawad, C.; Borowitz, M.J.; Devidas, M.; Maloney, K.W.; Larsen, E.; et al. Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric b-all. Blood 2018, 131, 1350–1359. [Google Scholar] [CrossRef]
- Eckert, C.; Henze, G.; Seeger, K.; Hagedorn, N.; Mann, G.; Panzer-Grumayer, R.; Peters, C.; Klingebiel, T.; Borkhardt, A.; Schrappe, M.; et al. Use of allogeneic hematopoietic stem-cell transplantation based on minimal residual disease response improves outcomes for children with relapsed acute lymphoblastic leukemia in the intermediate-risk group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 2736–2742. [Google Scholar] [CrossRef]
- Krentz, S.; Hof, J.; Mendioroz, A.; Vaggopoulou, R.; Dorge, P.; Lottaz, C.; Engelmann, J.C.; Groeneveld, T.W.; Korner, G.; Seeger, K.; et al. Prognostic value of genetic alterations in children with first bone marrow relapse of childhood b-cell precursor acute lymphoblastic leukemia. Leukemia 2013, 27, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Henze, G.; Langermann, H.J.; Bramswig, J.; Breu, H.; Gadner, H.; Schellong, G.; Welte, K.; Riehm, H. The bfm 76/79 acute lymphoblastic leukemia therapy study (author’s transl). Klin. Padiatr. 1981, 193, 145–154. [Google Scholar] [CrossRef]
- Nachman, J.B.; Sather, H.N.; Sensel, M.G.; Trigg, M.E.; Cherlow, J.M.; Lukens, J.N.; Wolff, L.; Uckun, F.M.; Gaynon, P.S. Augmented post-induction therapy for children with high-risk acute lymphoblastic leukemia and a slow response to initial therapy. N. Engl. J. Med. 1998, 338, 1663–1671. [Google Scholar] [CrossRef] [Green Version]
- Reiter, A.; Schrappe, M.; Ludwig, W.D.; Hiddemann, W.; Sauter, S.; Henze, G.; Zimmermann, M.; Lampert, F.; Havers, W.; Niethammer, D.; et al. Chemotherapy in 998 unselected childhood acute lymphoblastic leukemia patients. Results and conclusions of the multicenter trial all-bfm 86. Blood 1994, 84, 3122–3133. [Google Scholar] [PubMed]
- Bene, M.C.; Castoldi, G.; Knapp, W.; Ludwig, W.D.; Matutes, E.; Orfao, A.; van’t Veer, M.B. Proposals for the immunological classification of acute leukemias. European group for the immunological characterization of leukemias (egil). Leukemia 1995, 9, 1783–1786. [Google Scholar] [PubMed]
- Katona, E.E.; Ajzner, E.; Toth, K.; Karpati, L.; Muszbek, L. Enzyme-linked immunosorbent assay for the determination of blood coagulation factor xiii a-subunit in plasma and in cell lysates. J. Immunol. Methods 2001, 258, 127–135. [Google Scholar] [CrossRef]
- Moorman, A.V.; Enshaei, A.; Schwab, C.; Wade, R.; Chilton, L.; Elliott, A.; Richardson, S.; Hancock, J.; Kinsey, S.E.; Mitchell, C.D.; et al. A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood 2014, 124, 1434–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clappier, E.; Grardel, N.; Bakkus, M.; Rapion, J.; De Moerloose, B.; Kastner, P.; Caye, A.; Vivent, J.; Costa, V.; Ferster, A.; et al. Ikzf1 deletion is an independent prognostic marker in childhood b-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: Results of the eortc children‘s leukemia group study 58951. Leukemia 2015, 29, 2154–2161. [Google Scholar] [CrossRef]
- Messina, M.; Chiaretti, S.; Fedullo, A.L.; Piciocchi, A.; Puzzolo, M.C.; Lauretti, A.; Gianfelici, V.; Apicella, V.; Fazi, P.; Te Kronnie, G.; et al. Clinical significance of recurrent copy number aberrations in b-lineage acute lymphoblastic leukaemia without recurrent fusion genes across age cohorts. Br. J. Haematol. 2017, 178, 583–587. [Google Scholar] [CrossRef]
- Gupta, S.K.; Bakhshi, S.; Kumar, L.; Kamal, V.K.; Kumar, R. Gene copy number alteration profile and its clinical correlation in b-cell acute lymphoblastic leukemia. Leuk. Lymphoma 2017, 58, 333–342. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Event-Free Survival | Overall Survival | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p-Value | HR | 95% CI | p-Value | |
| Univariate Analysis | ||||||
| FXIII-A expression pattern | ||||||
| FXIII-A negative | 2.91 | 1.17–7.21 | 0.021 | 2.63 | 0.88–7.84 | 0.084 |
| FXIII-A dim | 0.34 | 0.14–0.85 | 0.38 | 0.13–1.14 | ||
| Age | ||||||
| 1–5 years | 0.39 | 0.17–0.92 | 0.032 | 0.24 | 0.08–0.71 | 0.010 |
| ≥6 years | 2.56 | 1.10–6.05 | 4.20 | 1.41–12.56 | ||
| WBC | ||||||
| <20,000 | 0.69 | 0.29–1.66 | 0.407 | 0.60 | 0.21–1.71 | 0.334 |
| ≥20,000 | 1.45 | 0.60–3.51 | 1.69 | 0.58–4.86 | ||
| Sex | ||||||
| Male | 1.53 | 0.62–3.79 | 0.360 | 1.87 | 0.59–6.00 | 0.291 |
| Female | 0.65 | 0.26–1.62 | 0.54 | 0.17–1.71 | ||
| prednisone response | ||||||
| PGR | 0.27 | 0.09–0.73 | 0.010 | 0.15 | 0.05–0.45 | 0.001 |
| PPR | 3.75 | 1.37–10.24 | 6.71 | 2.25–20.03 | ||
| FC risk categories | ||||||
| FLR/FMR | 1.43 | 0.40–5.13 | 0.581 | 1.18 | 0.25–5.54 | 0.838 |
| FLR/FHR | 3.37 | 0.78–14.58 | 0.104 | 3.40 | 0.62–18.56 | 0.158 |
| ALLIC BFM 2009 classification | ||||||
| BFM-HR/BFM-SR | 0.12 | 0.01–0.95 | 0.044 | 0.00 | ||
| BFM-HR/BFM-IR | 0.41 | 0.16–1.05 | 0.063 | 0.32 | 0.11–0.92 | 0.035 |
| Genetic risk categories | ||||||
| good/intermediate | 7.12 | 2.07–24.73 | 0.002 | 13.8 | 1.77–107.9 | 0.012 |
| good/high | 23.7 | 4.72–119.3 | <0.001 | 63.1 | 6.50–613.3 | <0.001 |
| B-other | ||||||
| Recurrent genetic abnormalities | 0.26 | 0.10–0.68 | 0.006 | 0.27 | 0.08–0.86 | 0.027 |
| B-other | 3.82 | 1.48–9.86 | 3.71 | 1.16–11.85 | ||
| Multivariate Analysis | ||||||
| FXIII negative | 2.29 | 0.85–6.20 | 0.102 | 1.70 | 0.51–5.71 | 0.390 |
| Genetic risk categories | ||||||
| good/intermediate | 5.78 | 1.63–20.52 | 0.007 | 10.0 | 1.24–81.4 | 0.031 |
| good/high | 17.4 | 2.88–104.8 | 0.002 | 30.5 | 2.63–353.4 | 0.006 |
| Variables | Event-Free Survival | Overall Survival | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p-Value | HR | 95% CI | p-Value | |
| Univariate Analysis | ||||||
| FXIII-A expression pattern | ||||||
| FXIII-A dim | 0.24 | 0.09–0.64 | 0.004 | 0.32 | 0.09–1.04 | 0.057 |
| FXIII-A bright | 4.10 | 1.56–10.78 | 3.17 | 0.97–10.38 | ||
| Age | ||||||
| 1–5 years | 0.52 | 0.19–1.37 | 0.186 | 0.32 | 0.10–1.05 | 0.060 |
| ≥6 years | 1.92 | 0.73–5.06 | 3.13 | 0.96–10.28 | ||
| WBC | ||||||
| <20,000 | 0.67 | 0.26–1.76 | 0.417 | 0.56 | 0.17–1.84 | 0.341 |
| ≥20,000 | 1.49 | 0.57–3.92 | 1.78 | 0.54–5.83 | ||
| Sex | ||||||
| Male | 1.43 | 0.53–3.86 | 0.483 | 2.08 | 0.55–7.86 | 0.278 |
| Female | 0.70 | 0.26–1.90 | 0.48 | 0.18–1.81 | ||
| prednisone response | ||||||
| PGR | 0.34 | 0.08–1.47 | 0.146 | 0.43 | 0.06–3.41 | 0.427 |
| PPR | 1.43 | 0.53–3.86 | 2.08 | 0.55–7.86 | ||
| FC risk categories | ||||||
| FLR/FMR | 0.90 | 0.27–2.83 | 0.857 | 1.33 | 0.24–5.54 | 0.863 |
| FLR/FHR | 1.25 | 0.23–6.81 | 0.801 | 2,59 | 0.36–18.43 | 0.342 |
| ALLIC BFM 2009 classification | ||||||
| BFM-HR/BFM-SR | 0.39 | 0.098–1.59 | 0.192 | 0.17 | 0.02–1.49 | 0.110 |
| BFM-HR/BFM-IR | 0.27 | 0.09–0.78 | 0.015 | 0.21 | 0.06–0.73 | 0.014 |
| Genetic risk categories | ||||||
| good/intermediate | 2.08 | 0.75–5.74 | 0.051 | 3.68 | 0.92–14.73 | 0.065 |
| good/high | 4.86 | 0.99–23.76 | 0.157 | 9.65 | 1.59–58.42 | 0.014 |
| B-other | ||||||
| Recurrent genetic abnormalities | 0.55 | 0.21–1.44 | 0.225 | 0.41 | 0.12–1.33 | 0.137 |
| B-other | 1.80 | 0.70–4.78 | 2.46 | 0.75–8.07 | ||
| Multivariate Analysis | ||||||
| FXIII-A bright | 3.92 | 1.41–10.84 | 0.009 | 3.90 | 0.99–15.31 | 0.051 |
| Genetic risk categories | ||||||
| good/intermediate | 2.69 | 0.84–8.55 | 0.095 | 6.07 | 1.17–31.65 | 0.032 |
| good/high | 3.56 | 0.70–18.03 | 0.124 | 7.67 | 1.21–48.64 | 0.031 |
| Variables | Event-Free Survival | Overall Survival | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p-Value | HR | 95% CI | p-Value | |
| Univariate Analysis | ||||||
| FXIII-A expression pattern | ||||||
| FXIII-A negative | 0.66 | 0.29–1.51 | 0.323 | 0.80 | 0.28–2.24 | 0.665 |
| FXIII-A bright | 1.52 | 0.66–3.47 | 1.26 | 0.45–3.54 | ||
| Age | ||||||
| 1–5 years | 0.52 | 0.24–1.17 | 0.116 | 0.36 | 0.13–1.02 | 0.056 |
| ≥6 years | 1.90 | 0.85–4.24 | 2.75 | 0.98–7.72 | ||
| WBC | ||||||
| <20,000 | 0.49 | 0.21–1.13 | 0.093 | 0.68 | 0.23–1.98 | 0.477 |
| ≥ 20,000 | 2.06 | 0.89–4.76 | 1.48 | 0.50–4.32 | ||
| Sex | ||||||
| Male | 1.21 | 0.53–2.78 | 0.647 | 1.43 | 0.49–4.17 | 0.518 |
| Female | 0.82 | 0.36–1.89 | 0.70 | 0.24–2.05 | ||
| prednisone response | ||||||
| PGR | 0.36 | 0.13–0.96 | 0.041 | 0.28 | 0.09–0.87 | 0.028 |
| PPR | 2.81 | 1.04–7.58 | 3.61 | 1.15–11.35 | ||
| FC risk categories | ||||||
| FLR/FMR | 2.48 | 0.72–8.56 | 0.150 | 1.90 | 0.41–8.80 | 0.411 |
| FLR/FHR | 3.29 | 0.77–13.9 | 0.106 | 3.53 | 0.65–19.28 | 0.145 |
| ALLIC BFM 2009 classification | ||||||
| BFM-HR/BFM-SR | 0.21 | 0.05–0.97 | 0.045 | 0.15 | 0.02–1.22 | 0.076 |
| BFM-HR/BFM-IR | 0.47 | 0.20–1.11 | 0.085 | 0.37 | 0.13–1.04 | 0.060 |
| Genetic risk categories | ||||||
| good/intermediate | 2.14 | 0.89–5.17 | 0.091 | 2.50 | 0.75–8.30 | 0.135 |
| good/high | 6.48 | 1.69–24.84 | 0.006 | 10.5 | 2.33–47.2 | 0.002 |
| B-other | ||||||
| Recurrent genetic abnormalities | 0.61 | 0.27–1.36 | 0.228 | 0.66 | 0.24–1.81 | 0.413 |
| B-other | 1.64 | 0.73–3.67 | 1.53 | 0.55–4.21 | ||
| Multivariate Analysis | ||||||
| FXIII-A bright | 0.54 | 0.22–1.34 | 0.108 | 0.59 | 0.19–1.88 | 0.3751 |
| Genetic risk categories | ||||||
| good/intermediate | 2.33 | 0.87–6.22 | 0.091 | 2.63 | 0.68–10.25 | 0.164 |
| good/high | 4.55 | 1.14–18.15 | 0.032 | 7.44 | 1.54–36.00 | 0.013 |
| FXIII-A Expression Patterns | p Value | OR | 95% CI | |
|---|---|---|---|---|
| FXIII-A Negative | Reference Group (“Base Outcome”) | |||
| FXIII-A dim | ||||
| “B-other” subgroup | 0.009 | 0.49 | 0.28 | 0.84 |
| Sex | 0.851 | 1.05 | 0.61 | 1.81 |
| Age | 0.197 | 0.69 | 0.39 | 1.21 |
| WBC | 0.093 | 1.69 | 0.92 | 3.10 |
| FXIII-A bright | ||||
| “B-other” subgroup | 0.004 | 0.35 | 0.18 | 0.71 |
| Sex | 0.726 | 0.89 | 0.46 | 1.72 |
| Age | 0.581 | 0.82 | 0.41 | 1.64 |
| WBC | 0.393 | 1.38 | 0.66 | 2.92 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kárai, B.; Gyurina, K.; Ujfalusi, A.; Sędek, Ł.; Barna, G.; Jáksó, P.; Svec, P.; Szánthó, E.; Nagy, A.C.; Müller, J.; et al. Expression Patterns of Coagulation Factor XIII Subunit A on Leukemic Lymphoblasts Correlate with Clinical Outcome and Genetic Subtypes in Childhood B-cell Progenitor Acute Lymphoblastic Leukemia. Cancers 2020, 12, 2264. https://doi.org/10.3390/cancers12082264
Kárai B, Gyurina K, Ujfalusi A, Sędek Ł, Barna G, Jáksó P, Svec P, Szánthó E, Nagy AC, Müller J, et al. Expression Patterns of Coagulation Factor XIII Subunit A on Leukemic Lymphoblasts Correlate with Clinical Outcome and Genetic Subtypes in Childhood B-cell Progenitor Acute Lymphoblastic Leukemia. Cancers. 2020; 12(8):2264. https://doi.org/10.3390/cancers12082264
Chicago/Turabian StyleKárai, Bettina, Katalin Gyurina, Anikó Ujfalusi, Łukasz Sędek, Gábor Barna, Pál Jáksó, Peter Svec, Eszter Szánthó, Attila Csaba Nagy, Judit Müller, and et al. 2020. "Expression Patterns of Coagulation Factor XIII Subunit A on Leukemic Lymphoblasts Correlate with Clinical Outcome and Genetic Subtypes in Childhood B-cell Progenitor Acute Lymphoblastic Leukemia" Cancers 12, no. 8: 2264. https://doi.org/10.3390/cancers12082264
APA StyleKárai, B., Gyurina, K., Ujfalusi, A., Sędek, Ł., Barna, G., Jáksó, P., Svec, P., Szánthó, E., Nagy, A. C., Müller, J., Simon, R., Vojczek, Á., Szegedi, I., Tiszlavicz, L. G., Kowalczyk, J. R., Kolenova, A., Kovács, G. T., Szczepański, T., Dworzak, M., ... Kiss, C. (2020). Expression Patterns of Coagulation Factor XIII Subunit A on Leukemic Lymphoblasts Correlate with Clinical Outcome and Genetic Subtypes in Childhood B-cell Progenitor Acute Lymphoblastic Leukemia. Cancers, 12(8), 2264. https://doi.org/10.3390/cancers12082264