
Advances in Pancreatic Ductal Adenocarcinoma Treatment



Abstract
:Simple Summary
Abstract
1. Introduction
2. Surgical Approaches
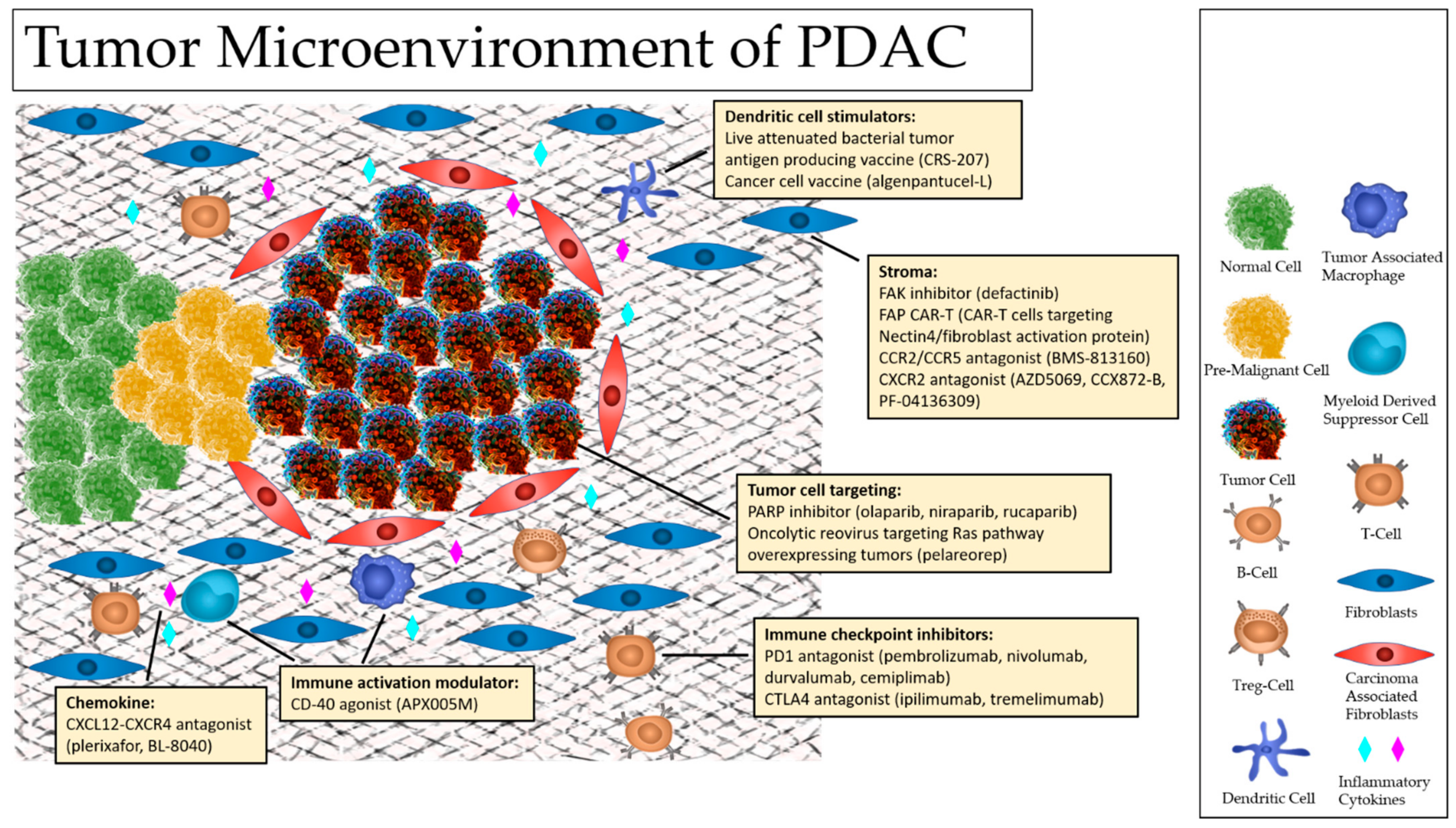
3. The TME of Pancreatic Caner
4. Immunotherapy
5. Targeted Therapies
6. Stromal Targeting
7. Combinatorial Systemic Strategies
8. Prospective and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Key Statistics for Pancreatic Cancer. Available online: https://www.cancer.org/cancer/pancreatic-cancer/about/key-statistics.html (accessed on 18 September 2020).
- Cancer of the Pancreas—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/pancreas.html (accessed on 18 September 2020).
- Callahan, M.K.; Flaherty, C.R.; Postow, M.A. Checkpoint Blockade for the Treatment of Advanced Melanoma. In Melanoma; Kaufman, H.L., Mehnert, J.M., Eds.; Cancer Treatment and Research; Springer International Publishing: Cham, Switzerland, 2016; Volume 167, pp. 231–250. ISBN 978-3-319-22538-8. [Google Scholar]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Kunk, P.R.; Bauer, T.W.; Slingluff, C.L.; Rahma, O.E. From Bench to Bedside a Comprehensive Review of Pancreatic Cancer Immunotherapy. J. Immunother. Cancer 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, J.; Jabbour, S.K.; Aisner, J. Current State of Immunotherapy for Non-Small Cell Lung Cancer. Transl. Lung Cancer Res. 2007, 6, 196–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Osipov, A.; Lim, S.J.; Popovic, A.; Azad, N.S.; Laheru, D.A.; Zheng, L.; Jaffee, E.M.; Wang, H.; Yarchoan, M. Tumor Mutational Burden, Toxicity, and Response of Immune Checkpoint Inhibitors Targeting PD(L)1, CTLA-4, and Combination: A Meta-Regression Analysis. Clin. Cancer Res. 2020, 26, 4842–4851. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Hackert, T.; Strobel, O.; Büchler, M.W. Technical Advances in Surgery for Pancreatic Cancer. Br. J. Surg. 2021, 108, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Tummala, P.; Howard, T.; Agarwal, B. Dramatic Survival Benefit Related to R0 Resection of Pancreatic Adenocarcinoma in Patients with Tumor ≤ 25 Mm in Size and ≤1 Involved Lymph Nodes. Clin. Transl. Gastroenterol. 2013, 4, e33. [Google Scholar] [CrossRef] [PubMed]
- Strobel, O.; Hank, T.; Hinz, U.; Bergmann, F.; Schneider, L.; Springfeld, C.; Jäger, D.; Schirmacher, P.; Hackert, T.; Büchler, M.W. Pancreatic Cancer Surgery: The New R-Status Counts. Ann. Surg. 2017, 265, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Malinka, T.; Timmermann, L.; Klein, F.; Geisel, D.; Pratschke, J.; Bahra, M. Is There a Role for the Appleby Procedure in 2020? Results from a Matched-Pair-Analysis. Anticancer. Res. 2020, 40, 387–392. [Google Scholar] [CrossRef]
- Xu, J.; Tian, X.; Chen, Y.; Ma, Y.; Liu, C.; Tian, L.; Wang, J.; Dong, J.; Cui, D.; Wang, Y.; et al. Total Mesopancreas Excision for the Treatment of Pancreatic Head Cancer. J. Cancer 2017, 8, 3575–3584. [Google Scholar] [CrossRef] [Green Version]
- Safi, S.-A.; Haeberle, L.; Fluegen, G.; Lehwald-Tywuschik, N.; Krieg, A.; Keitel, V.; Luedde, T.; Esposito, I.; Rehders, A.; Knoefel, W.T. Mesopancreatic Excision for Pancreatic Ductal Adenocarcinoma Improves Local Disease Control and Survival. Pancreatology 2021, 21, 787–795. [Google Scholar] [CrossRef]
- Hirota, M.; Sugita, H.; Honda, S.; Tanaka, H.; Tashima, R.; Daitoku, N.; Akiyama, T.; Komori, H.; Taki, K.; Kitamura, F.; et al. No-Touch Total Mesopancreas Excision for Pancreatic Head Cancer. JOP J. Pancreas 2017, 18, 216. [Google Scholar]
- Martin, R.C.G. Use of Irreversible Electroporation in Unresectable Pancreatic Cancer. Hepatobiliary Surg. Nutr. 2015, 4, 211–215. [Google Scholar] [CrossRef]
- Centonze, L.; Sandro, S.D.; Lauterio, A.; Carlis, R.D.; Botta, F.; Mariani, A.; Bagnardi, V.; Carlis, L.D. The Impact of Sarcopenia on Postoperative Course Following Pancreatoduodenectomy: Single-Center Experience of 110 Consecutive Cases. DSU 2020, 37, 312–320. [Google Scholar] [CrossRef]
- Hendifar, A.; Osipov, A.; Khanuja, J.; Nissen, N.; Naziri, J.; Yang, W.; Li, Q.; Tuli, R. Influence of Body Mass Index and Albumin on Perioperative Morbidity and Clinical Outcomes in Resected Pancreatic Adenocarcinoma. PLoS ONE 2016, 11, e0152172. [Google Scholar] [CrossRef]
- Son, B.; Lee, S.; Youn, H.; Kim, E.; Kim, W.; Youn, B. The Role of Tumor Microenvironment in Therapeutic Resistance. Oncotarget 2017, 8, 3933–3945. [Google Scholar] [CrossRef] [Green Version]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Jaffee, E.M. Vaccine Therapy and Immunotherapy for Pancreatic Cancer. In Pancreatic Cancer; Neoptolemos, J., Urrutia, R., Abbruzzese, J., Büchler, M.W., Eds.; Springer: New York, NY, USA, 2017; pp. 1–45. ISBN 978-1-4939-6631-8. [Google Scholar]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the Immune Reaction to Pancreatic Cancer from Inception to Invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [Green Version]
- Ikemoto, T.; Yamaguchi, T.; Morine, Y.; Imura, S.; Soejima, Y.; Fujii, M.; Maekawa, Y.; Yasutomo, K.; Shimada, M. Clinical Roles of Increased Populations of Foxp3+ CD4+ T Cells in Peripheral Blood from Advanced Pancreatic Cancer Patients. Pancreas 2006, 33, 386–390. [Google Scholar] [CrossRef]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ Regulatory T Cells Increases during the Progression of Pancreatic Ductal Adenocarcinoma and Its Premalignant Lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Tao, X.; Xia, S.; Guo, F.; Pan, C.; Xiang, H.; Shang, D. T Lymphocytes: A Promising Immunotherapeutic Target for Pancreatitis and Pancreatic Cancer? Front. Oncol. 2020, 10, 382. [Google Scholar] [CrossRef] [Green Version]
- Kiss, M.; Van Gassen, S.; Movahedi, K.; Saeys, Y.; Laoui, D. Myeloid Cell Heterogeneity in Cancer: Not a Single Cell Alike. Cell. Immunol. 2018, 330, 188–201. [Google Scholar] [CrossRef]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The Role of Myeloid Cells in Cancer Therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Immune Responses to Malignancies. J. Allergy Clin. Immunol. 2010, 125, S272–S283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, M.B.; Danielsen, A.V.; Hamilton-Dutoit, S.J.; Bendix, K.; Nørgaard, P.; Møller, M.B.; Steiniche, T.; d’Amore, F. High Intratumoral Macrophage Content Is an Adverse Prognostic Feature in Anaplastic Large Cell Lymphoma. Histopathology 2014, 65, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-Induced Myeloid Deviation: When Myeloid-Derived Suppressor Cells Meet Tumor-Associated Macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Li, Q.; Qin, L.; Zhao, S.; Wang, J.; Chen, X. Transition of Tumor-Associated Macrophages from MHC Class IIhi to MHC Class IIlow Mediates Tumor Progression in Mice. BMC Immunol. 2011, 12, 43. [Google Scholar] [CrossRef]
- Prada, C.E.; Jousma, E.; Rizvi, T.A.; Wu, J.; Dunn, R.S.; Mayes, D.A.; Cancelas, J.A.; Dombi, E.; Kim, M.-O.; West, B.L.; et al. Neurofibroma-Associated Macrophages Play Roles in Tumor Growth and Response to Pharmacological Inhibition. Acta Neuropathol. 2013, 125, 159–168. [Google Scholar] [CrossRef]
- De Palma, M.; Lewis, C.E. Macrophage Regulation of Tumor Responses to Anticancer Therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Allegrezza, M.J.; Conejo-Garcia, J.R. Targeted Therapy and Immunosuppression in the Tumor Microenvironment. Trends Cancer 2017, 3, 19–27. [Google Scholar] [CrossRef]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [Green Version]
- Osipov, A.; Murphy, A.; Zheng, L. From immune checkpoints to vaccines: The past, present and future of cancer immunotherapy. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2019; Volume 143, pp. 63–144. ISBN 978-0-12-817022-9. [Google Scholar]
- Mahadevan, D.; Von Hoff, D.D. Tumor-Stroma Interactions in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2007, 6, 1186–1197. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H.; Domchek, S.M.; Clark, A.S. Immunotherapy for Breast Cancer: What Are We Missing? Clin. Cancer Res. 2017, 23, 2640–2646. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H.; LoRusso, P.M.; Khalil, M.; Gartner, E.M.; Khaira, D.; Soulieres, D.; Dorazio, P.; Trosko, J.A.; Ruter, J.; Mariani, G.L.; et al. Tremelimumab in Combination with Exemestane in Patients with Advanced Breast Cancer and Treatment-Associated Modulation of Inducible Costimulator Expression on Patient T Cells. Clin. Cancer Res. 2010, 16, 3485–3494. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Der, C.J.; Cox, A.D. The Role of Wild Type RAS Isoforms in Cancer. Semin. Cell Dev. Biol. 2016, 58, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in Developmental Disorders and Cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef]
- Dias Carvalho, P.; Guimarães, C.F.; Cardoso, A.P.; Mendonça, S.; Costa, Â.M.; Oliveira, M.J.; Velho, S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018, 78, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Testorelli, C.; Bussini, S.; De Filippi, R.; Marelli, O.; Orlando, L.; Greiner, J.W.; Grohmann, U.; Tentori, L.; Giuliani, A.; Bonmassar, E.; et al. Dacarbazine-Induced Immunogenicity of a Murine Leukemia Is Attenuated in Cells Transfected with Mutated K-Ras Gene. J. Exp. Clin. Cancer Res. 1997, 16, 15–22. [Google Scholar]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype Tunes Pancreatic Ductal Adenocarcinoma Tissue Tension to Induce Matricellular Fibrosis and Tumor Progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef] [Green Version]
- Sato, N.; Kohi, S.; Hirata, K.; Goggins, M. Role of Hyaluronan in Pancreatic Cancer Biology and Therapy: Once Again in the Spotlight. Cancer Sci. 2016, 107, 569–575. [Google Scholar] [CrossRef]
- Zhang, L.; Li, J.; Zong, L.; Chen, X.; Chen, K.; Jiang, Z.; Nan, L.; Li, X.; Li, W.; Shan, T.; et al. Reactive Oxygen Species and Targeted Therapy for Pancreatic Cancer. Oxidative Med. Cell. Longev. 2016, 2016, e1616781. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.; Radhakrishnan, P. Tumor-Stromal Crosstalk in Pancreatic Cancer and Tissue Fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef]
- Liao, Z.; Chua, D.; Tan, N.S. Reactive Oxygen Species: A Volatile Driver of Field Cancerization and Metastasis. Mol. Cancer 2019, 18, 65. [Google Scholar] [CrossRef]
- Arcucci, A.; Ruocco, M.R.; Granato, G.; Sacco, A.M.; Montagnani, S. Cancer: An Oxidative Crosstalk between Solid Tumor Cells and Cancer Associated Fibroblasts. Bio. Med. Res. Int. 2016, 2016, e4502846. [Google Scholar] [CrossRef] [Green Version]
- Mehlen, P.; Puisieux, A. Metastasis: A Question of Life or Death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Kim, S.-H.; Turnbull, J.; Guimond, S. Extracellular Matrix and Cell Signalling: The Dynamic Cooperation of Integrin, Proteoglycan and Growth Factor Receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Digiacomo, G.; Tusa, I.; Bacci, M.; Cipolleschi, M.G.; Dello Sbarba, P.; Rovida, E. Fibronectin Induces Macrophage Migration through a SFK-FAK/CSF-1R Pathway. Cell Adhes. Migr. 2017, 11, 327–337. [Google Scholar] [CrossRef]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A.; Gómez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK Controls Chemokine Transcription, Tregs, and Evasion of Anti-Tumor Immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Yang, G.; Zhou, W.; Qiu, J.; Chen, G.; Luo, W.; Zhao, F.; You, L.; Zheng, L.; Zhang, T.; et al. Targeting Hypoxic Tumor Microenvironment in Pancreatic Cancer. J. Hematol. Oncol. 2021, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T. Regulatory T Cells: How Do They Suppress Immune Responses? Int. Immunol. 2009, 21, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Wahl, S.M. Engagement of Cytotoxic T Lymphocyte–Associated Antigen 4 (CTLA-4) Induces Transforming Growth Factor β (TGF-β) Production by Murine CD4+ T Cells. J. Exp. Med. 1998, 188, 1849–1857. [Google Scholar] [CrossRef] [Green Version]
- Chuang, E.; Fisher, T.S.; Morgan, R.W.; Robbins, M.D.; Duerr, J.M.; Vander Heiden, M.G.; Gardner, J.P.; Hambor, J.E.; Neveu, M.J.; Thompson, C.B. The CD28 and CTLA-4 Receptors Associate with the Serine/Threonine Phosphatase PP2A. Immunity 2000, 13, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Lao, C.D.; et al. Nivolumab versus Chemotherapy in Patients with Advanced Melanoma Who Progressed after Anti-CTLA-4 Treatment (CheckMate 037): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 Ligands, and Other Features of the Tumor Immune Microenvironment with Response to Anti-PD-1 Therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [Green Version]
- Clark, C.E.; Beatty, G.L.; Vonderheide, R.H. Immunosurveillance of Pancreatic Adenocarcinoma: Insights from Genetically Engineered Mouse Models of Cancer. Cancer Lett. 2009, 279, 37. [Google Scholar] [CrossRef]
- Von Bernstorff, W.; Voss, M.; Freichel, S.; Schmid, A.; Vogel, I.; Jöhnk, C.; Henne-Bruns, D.; Kremer, B.; Kalthoff, H. Systemic and Local Immunosuppression in Pancreatic Cancer Patients. Clin. Cancer Res. 2001, 7, 925s–932s. [Google Scholar]
- Rizvi, N.A.; Cho, B.C.; Reinmuth, N.; Lee, K.H.; Luft, A.; Ahn, M.-J.; van den Heuvel, M.M.; Cobo, M.; Vicente, D.; Smolin, A.; et al. Durvalumab With or Without Tremelimumab vs Standard Chemotherapy in First-Line Treatment of Metastatic Non–Small Cell Lung Cancer: The MYSTIC Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 661. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, M.H.; O’Reilly, E.M.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Rahma, O.E.; Fisher, G.A.; Lyman, J.P.; Cabanski, C.R.; Karakunnel, J.J.; et al. Gemcitabine (Gem) and Nab-Paclitaxel (NP) ± Nivolumab (Nivo) ± CD40 Agonistic Monoclonal Antibody APX005M (Sotigalimab), in Patients (Pts) with Untreated Metastatic Pancreatic Adenocarcinoma (MPDAC): Phase (Ph) 2 Final Results. J. Clin. Oncol. 2021, 39, 4019. [Google Scholar] [CrossRef]
- Lutz, E.R.; Kinkead, H.; Jaffee, E.M.; Zheng, L. Priming the Pancreatic Cancer Tumor Microenvironment for Checkpoint-Inhibitor Immunotherapy. OncoImmunology 2014, 3, e962401. [Google Scholar] [CrossRef] [Green Version]
- Jaffee, E.M.; Schutte, M.; Gossett, J.; Morsberger, L.A.; Adler, A.J.; Thomas, M.; Greten, T.F.; Hruban, R.H.; Yeo, C.J.; Griffin, C.A. Development and Characterization of a Cytokine-Secreting Pancreatic Adenocarcinoma Vaccine from Primary Tumors for Use in Clinical Trials. Cancer J. Sci. Am. 1998, 4, 194–203. [Google Scholar]
- Jaffee, E.M.; Hruban, R.H.; Biedrzycki, B.; Laheru, D.; Schepers, K.; Sauter, P.R.; Goemann, M.; Coleman, J.; Grochow, L.; Donehower, R.C.; et al. Novel Allogeneic Granulocyte-Macrophage Colony-Stimulating Factor-Secreting Tumor Vaccine for Pancreatic Cancer: A Phase I Trial of Safety and Immune Activation. J. Clin. Oncol. 2001, 19, 145–156. [Google Scholar] [CrossRef]
- Laheru, D.; Lutz, E.; Burke, J.; Biedrzycki, B.; Solt, S.; Onners, B.; Tartakovsky, I.; Nemunaitis, J.; Le, D.; Sugar, E.; et al. Allogeneic Granulocyte Macrophage Colony-Stimulating Factor-Secreting Tumor Immunotherapy Alone or in Sequence with Cyclophosphamide for Metastatic Pancreatic Cancer: A Pilot Study of Safety, Feasibility, and Immune Activation. Clin. Cancer Res. 2008, 14, 1455–1463. [Google Scholar] [CrossRef] [Green Version]
- Eric, L.; Yeo, C.J.; Lillemoe, K.D.; Biedrzycki, B.; Kobrin, B.; Herman, J.; Sugar, E.; Piantadosi, S.; Cameron, J.L.; Solt, S.; et al. A Lethally Irradiated Allogeneic Granulocyte-Macrophage Colony Stimulating Factor-Secreting Tumor Vaccine for Pancreatic Adenocarcinoma: A Phase II Trial of Safety, Efficacy, and Immune Activation. Ann. Surg. 2011, 253, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Soares, K.C.; Rucki, A.A.; Wu, A.A.; Olino, K.; Xiao, Q.; Chai, Y.; Wamwea, A.; Bigelow, E.; Lutz, E.; Liu, L.; et al. PD-1/PD-L1 Blockade Together with Vaccine Therapy Facilitates Effector T-Cell Infiltration into Pancreatic Tumors. J. Immunother. 2015, 38, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of Ipilimumab in Combination with Allogeneic Pancreatic Tumor Cells Transfected with a GM-CSF Gene in Previously Treated Pancreatic Cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Hardacre, J.M.; Mulcahy, M.; Small, W.; Talamonti, M.; Obel, J.; Krishnamurthi, S.; Rocha-Lima, C.S.; Safran, H.; Lenz, H.-J.; Chiorean, E.G. Addition of Algenpantucel-L Immunotherapy to Standard Adjuvant Therapy for Pancreatic Cancer: A Phase 2 Study. J. Gastrointest Surg. 2013, 17, 94–101. [Google Scholar] [CrossRef]
- Inc, L.P. New Link Genetics Announces Results from Phase 3 IMPRESS Trial of Algenpantucel-L for Patients with Resected Pancreatic Cancer. Available online: https://www.globenewswire.com/news-release/2016/05/09/837878/15114/en/NewLink-Genetics-Announces-Results-from-Phase-3-IMPRESS-Trial-of-Algenpantucel-L-for-Patients-with-Resected-Pancreatic-Cancer.html (accessed on 30 June 2021).
- Gjertsen, M.K.; Bakka, A.; Breivik, J.; Saeterdal, I.; Gedde-Dahl, T.; Stokke, K.T.; Sølheim, B.G.; Egge, T.S.; Søreide, O.; Thorsby, E.; et al. Ex Vivo Ras Peptide Vaccination in Patients with Advanced Pancreatic Cancer: Results of a Phase I/II Study. Int. J. Cancer 1996, 65, 450–453. [Google Scholar] [CrossRef]
- Gjertsen, M.K.; Buanes, T.; Rosseland, A.R.; Bakka, A.; Gladhaug, I.; Søreide, O.; Eriksen, J.A.; Møller, M.; Baksaas, I.; Lothe, R.A.; et al. Intradermal Ras Peptide Vaccination with Granulocyte-Macrophage Colony-Stimulating Factor as Adjuvant: Clinical and Immunological Responses in Patients with Pancreatic Adenocarcinoma. Int. J. Cancer 2001, 92, 441–450. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chapman, P.B.; Feilchenfeldt, J.; Brennan, M.F.; Capanu, M.; Gansukh, B.; Jacobs, G.; Levin, A.; Neville, D.; Kelsen, D.P.; et al. Targeting Mutated K-Ras in Pancreatic Adenocarcinoma Using an Adjuvant Vaccine. Am. J. Clin. Oncol. 2011, 34, 321–325. [Google Scholar] [CrossRef]
- Gjertsen, M.K.; Bakka, A.; Breivik, J.; Saeterdal, I.; Solheim, B.G.; Søreide, O.; Thorsby, E.; Gaudernack, G. Vaccination with Mutant Ras Peptides and Induction of T-Cell Responsiveness in Pancreatic Carcinoma Patients Carrying the Corresponding RAS Mutation. Lancet 1995, 346, 1399–1400. [Google Scholar] [CrossRef]
- Foley, K.; Kim, V.; Jaffee, E.; Zheng, L. Current Progress in Immunotherapy for Pancreatic Cancer. Cancer Lett. 2016, 381, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Brockstedt, D.G.; Nir-Paz, R.; Hampl, J.; Mathur, S.; Nemunaitis, J.; Sterman, D.H.; Hassan, R.; Lutz, E.; Moyer, B.; et al. A Live-Attenuated Listeria Vaccine (ANZ-100) and a Live-Attenuated Listeria Vaccine Expressing Mesothelin (CRS-207) for Advanced Cancers: Phase I Studies of Safety and Immune Induction. Clin. Cancer Res. 2012, 18, 858–868. [Google Scholar] [CrossRef] [Green Version]
- Brockstedt, D.G.; Giedlin, M.A.; Leong, M.L.; Bahjat, K.S.; Gao, Y.; Luckett, W.; Liu, W.; Cook, D.N.; Portnoy, D.A.; Dubensky, T.W. Listeria-Based Cancer Vaccines That Segregate Immunogenicity from Toxicity. Proc. Natl. Acad. Sci. USA 2004, 101, 13832–13837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and Survival with GVAX Pancreas Prime and Listeria Monocytogenes-Expressing Mesothelin (CRS-207) Boost Vaccines for Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Mo, F.; Shou, J.; Wang, H.; Luo, K.; Zhang, S.; Han, N.; Li, H.; Ye, S.; Zhou, Z.; et al. A Pan-Cancer Clinical Study of Personalized Neoantigen Vaccine Monotherapy in Treating Patients with Various Types of Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 4511–4520. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chen, S.; Mo, F.; Chen, Z.; Jiang, J.; Zhang, S.; Han, N.; Xu, Y.; Ma, D.; Wang, H.; et al. A Peptide-Based Neoantigen Vaccine for Patients with Advanced Pancreatic Cancer Refractory to Standard Treatment. JCO 2021, 39, e14563. [Google Scholar] [CrossRef]
- Cheng, B.; Yuan, W.-E.; Su, J.; Liu, Y.; Chen, J. Recent Advances in Small Molecule Based Cancer Immunotherapy. Eur. J. Med. Chem. 2018, 157, 582–598. [Google Scholar] [CrossRef]
- Shoushtari, A.N.; Friedman, C.F.; Navid-Azarbaijani, P.; Postow, M.A.; Callahan, M.K.; Momtaz, P.; Panageas, K.S.; Wolchok, J.D.; Chapman, P.B. Measuring Toxic Effects and Time to Treatment Failure for Nivolumab Plus Ipilimumab in Melanoma. JAMA Oncol. 2018, 4, 98–101. [Google Scholar] [CrossRef]
- Dancey, J.; Sausville, E.A. Issues and Progress with Protein Kinase Inhibitors for Cancer Treatment. Nat. Rev. Drug. Discov. 2003, 2, 296–313. [Google Scholar] [CrossRef]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Lim, S.-T.S. Nuclear FAK: A New Mode of Gene Regulation from Cellular Adhesions. Mol. Cells 2013, 36, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting Focal Adhesion Kinase Renders Pancreatic Cancers Responsive to Checkpoint Immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Stokes, J.B.; Adair, S.J.; Slack-Davis, J.K.; Walters, D.M.; Tilghman, R.W.; Hershey, E.D.; Lowrey, B.; Thomas, K.S.; Bouton, A.H.; Hwang, R.F.; et al. Inhibition of Focal Adhesion Kinase by PF-562271 Inhibits the Growth and Metastasis of Pancreatic Cancer Concomitant with Altering the Tumor Microenvironment. Mol. Cancer Ther. 2011, 10, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, R.C.; Wicha, M.S. Stem Cells in Normal Development and Cancer. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2010; Volume 95, pp. 113–158. ISBN 978-0-12-385071-3. [Google Scholar]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer Stem Cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [Green Version]
- Wang-Gillam, A.; McWilliams, R.; Lockhart, A.C.; Tan, B.R.; Suresh, R.; Lim, K.-H.; Pedersen, K.S.; Trikalinos, N.; Aranha, O.; Park, H.; et al. Abstract CT118: Phase I Study of Defactinib Combined with Pembrolizumab and Gemcitabine in Patients with Advanced Cancer: Experiences of Pancreatic Ductal Adenocarcinoma (PDAC) Patients. Cancer Res. 2020, 80, CT118. [Google Scholar] [CrossRef]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting Tumor-Infiltrating Macrophages Decreases Tumor-Initiating Cells, Relieves Immunosuppression, and Improves Chemotherapeutic Responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [Green Version]
- Janson, C.; Jung, H.; Ertl, L.; Liu, S.; Dang, T.; Zeng, Y.; Krasinski, A.; McMahon, J.; Zhang, P.; Charo, I.; et al. Abstract 5655: Inhibition of CCR2 Potentiates Checkpoint Inhibitor Immunotherapy in Murine Model of Pancreatic Cancer. In Proceedings of the Immunology, American Association for Cancer Research, Washington, DC, USA, 1–5 July 2017; p. 5655. [Google Scholar]
- Linehan, D.; Noel, M.S.; Hezel, A.F.; Wang-Gillam, A.; Eskens, F.; Sleijfer, S.; Desar, I.M.E.; Erdkamp, F.; Wilmink, J.; Diehl, J.; et al. Overall Survival in a Trial of Orally Administered CCR2 Inhibitor CCX872 in Locally Advanced/Metastatic Pancreatic Cancer: Correlation with Blood Monocyte Counts. JCO 2018, 36, 92. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Nywening, T.M.; Sanford, D.E.; Lockhart, A.C.; Suresh, R.; Tan, B.R.; Lim, K.-H.; Sorscher, S.; Fowler, K.; Amin, M.A.; et al. Phase IB Study of FOLFIRINOX plus PF-04136309 in Patients with Borderline Resectable and Locally Advanced Pancreatic Adenocarcinoma (PC). JCO 2015, 33, 338. [Google Scholar] [CrossRef]
- Noel, M.; O’Reilly, E.M.; Wolpin, B.M.; Ryan, D.P.; Bullock, A.J.; Britten, C.D.; Linehan, D.C.; Belt, B.A.; Gamelin, E.C.; Ganguly, B.; et al. Phase 1b Study of a Small Molecule Antagonist of Human Chemokine (C-C Motif) Receptor 2 (PF-04136309) in Combination with Nab-Paclitaxel/Gemcitabine in First-Line Treatment of Metastatic Pancreatic Ductal Adenocarcinoma. Invest New Drugs 2020, 38, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Hu, P.; Donskov, F.; Wang, G.; Liu, Q.; Du, J. Tumor-Associated Neutrophils as a New Prognostic Factor in Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e98259. [Google Scholar] [CrossRef] [Green Version]
- Chao, T.; Furth, E.E.; Vonderheide, R.H. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-Cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2016, 4, 968–982. [Google Scholar] [CrossRef] [Green Version]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [Green Version]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting Both Tumour-Associated CXCR2+ Neutrophils and CCR2+ Macrophages Disrupts Myeloid Recruitment and Improves Chemotherapeutic Responses in Pancreatic Ductal Adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T. The Underlying Mechanism for the PARP and BRCA Synthetic Lethality: Clearing up the Misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Roberts, N.J.; Norris, A.L.; Petersen, G.M.; Bondy, M.L.; Brand, R.; Gallinger, S.; Kurtz, R.C.; Olson, S.H.; Rustgi, A.K.; Schwartz, A.G.; et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov. 2016, 6, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Shindo, K.; Yu, J.; Suenaga, M.; Fesharakizadeh, S.; Cho, C.; Macgregor-Das, A.; Siddiqui, A.; Witmer, P.D.; Tamura, K.; Song, T.J.; et al. Deleterious Germline Mutations in Patients with Apparently Sporadic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2017, 35, 3382–3390. [Google Scholar] [CrossRef]
- Ferrone, C.R.; Levine, D.A.; Tang, L.H.; Allen, P.J.; Jarnagin, W.; Brennan, M.F.; Offit, K.; Robson, M.E. BRCA Germline Mutations in Jewish Patients with Pancreatic Adenocarcinoma. J. Clin. Oncol. 2009, 27, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib Monotherapy in Patients with Advanced Cancer and a Germline BRCA1/2 Mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients with Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2, 316. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Overall Survival from the Phase 3 POLO Trial: Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. JCO 2021, 39, 378. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.-P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.-Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A Randomised, Double-Blind, Placebo-Controlled Trial of Trametinib, an Oral MEK Inhibitor, in Combination with Gemcitabine for Patients with Untreated Metastatic Adenocarcinoma of the Pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kirane, A.; Huang, H.; Sorrelle, N.B.; Burrows, F.J.; Dellinger, M.T.; Brekken, R.A. Cyclooxygenase-2 Inhibition Potentiates the Efficacy of Vascular Endothelial Growth Factor Blockade and Promotes an Immune Stimulatory Microenvironment in Preclinical Models of Pancreatic Cancer. Mol. Cancer Res. 2019, 17, 348–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, D.; Chen, Z.; Sheng, Z.; Gao, D.; Yan, F.; Ma, T.; Zheng, H.; Hong, M. A Catalase-Loaded Hierarchical Zeolite as an Implantable Nanocapsule for Ultrasound-Guided Oxygen Self-Sufficient Photodynamic Therapy against Pancreatic Cancer. Nanoscale 2018, 10, 17283–17292. [Google Scholar] [CrossRef] [PubMed]
- Blair, A.B.; Kim, V.M.; Muth, S.T.; Saung, M.T.; Lokker, N.; Blouw, B.; Armstrong, T.D.; Jaffee, E.M.; Tsujikawa, T.; Coussens, L.M.; et al. Dissecting the Stromal Signaling and Regulation of Myeloid Cells and Memory Effector T Cells in Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 5351–5363. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.D.; Jiang, X.; Sullivan, K.M.; Jalikis, F.G.; Smythe, K.S.; Abbasi, A.; Vignali, M.; Park, J.O.; Daniel, S.K.; Pollack, S.M.; et al. Mobilization of CD8+ T Cells via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 3934–3945. [Google Scholar] [CrossRef] [Green Version]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 Antagonist, in Combination with Pembrolizumab and Chemotherapy for Pancreatic Cancer: The COMBAT Trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Hofheinz, R.D.; Al-Batran, S.E.; Hartmann, F.; Hartung, G.; Jager, D.; Renner, C.; Tanswell, P.; Kunz, U.; Amelsberg, A.; Kuthan, H.; et al. Stromal Antigen Targeting by a Humanised Monoclonal Antibody: An Early Phase II Trial of Sibrotuzumab in Patients with Metastatic Colorectal Cancer. Onkologie 2003, 26, 44–48. [Google Scholar] [CrossRef]
- Scott, A.M.; Wiseman, G.; Welt, S.; Adjei, A.; Lee, F.-T.; Hopkins, W. A Phase I Dose-Escalation Study of Sibrotuzumab in Patients with Advanced or Metastatic Fibroblast Activation Protein-Positive Cancer. Clin. Cancer Res. 2003, 9, 1639–1647. [Google Scholar]
- Nugent, F.; Cunningham, C.; Barve, M.; Fisher, W.; Patel, H.; Meiri, E.; Oza, Y.; Yang, Z.; Jurkowski, E.; Uprichard, M. Phase 2 Study of Talabostat/Gemcitabine in Stage IV Pancreatic Cancer. J. Clin. Oncol. 2007, 25, 4616. [Google Scholar] [CrossRef]
- Lo, A.; Wang, L.S.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; O’Brien, S.; Evans, R.A.; Bajor, D.J.; Clendenin, C.; et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015, 75, 2800–2810. [Google Scholar] [CrossRef] [Green Version]
- Kakarla, S.; Chow, K.K.; Mata, M.; Shaffer, D.R.; Song, X.T.; Wu, M.F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K.; et al. Antitumor Effects of Chimeric Receptor Engineered Human T Cells Directed to Tumor Stroma. Mol. Ther. 2013, 21, 1611–1620. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Han, H.; Posner, R.G.; Hostetter, G.; Von Hoff, D.D. Targeting the Tumor Microenvironment in Cancer: Why Hyaluronidase Deserves a Second Look. Cancer Discov. 2011, 1, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef] [Green Version]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan Impairs Vascular Function and Drug Delivery in a Mouse Model of Pancreatic Cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients with Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX alone in Patients with Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa with Nab-Paclitaxel Plus Gemcitabine for Patients with Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef]
- Hakim, N.; Patel, R.; Devoe, C.; Saif, M.W. Why HALO 301 Failed and Implications for Treatment of Pancreatic Cancer. Pancreas 2019, 3, e1–e4. [Google Scholar] [CrossRef]
- Sood, A.K.; Coffin, J.E.; Schneider, G.B.; Fletcher, M.S.; DeYoung, B.R.; Gruman, L.M.; Gershenson, D.M.; Schaller, M.D.; Hendrix, M.J. Biological Significance of Focal Adhesion Kinase in Ovarian Cancer: Role in Migration and Invasion. Am. J. Pathol. 2004, 165, 1087–1095. [Google Scholar] [CrossRef]
- Golubovskaya, V.M. Targeting FAK in Human Cancer: From Finding to First Clinical Trials. Front. Biosci. 2014, 19, 687–706. [Google Scholar] [CrossRef] [Green Version]
- Wang-Gillam, A.; Lockhart, A.C.; Tan, B.R.; Suresh, R.; Lim, K.-H.; Ratner, L.; Morton, A.; Huffman, J.; Marquez, S.; Boice, N.; et al. Phase I Study of Defactinib Combined with Pembrolizumab and Gemcitabine in Patients with Advanced Cancer. JCO 2018, 36, 380. [Google Scholar] [CrossRef]
- Alistar, A.; Morris, B.B.; Desnoyer, R.; Klepin, H.D.; Hosseinzadeh, K.; Clark, C.; Cameron, A.; Leyendecker, J.; D’Agostino, R.; Topaloglu, U.; et al. Safety and Tolerability of the First-in-Class Agent CPI-613 in Combination with Modified FOLFIRINOX in Patients with Metastatic Pancreatic Cancer: A Single-Centre, Open-Label, Dose-Escalation, Phase 1 Trial. Lancet Oncol. 2017, 18, 770–778. [Google Scholar] [CrossRef]
- Katayama, E.S.; Hue, J.J.; Bajor, D.L.; Ocuin, L.M.; Ammori, J.B.; Hardacre, J.M.; Winter, J.M. A Comprehensive Analysis of Clinical Trials in Pancreatic Cancer: What Is Coming down the Pike? Oncotarget 2020, 11, 3489–3501. [Google Scholar] [CrossRef]
- FDA Grants Fast Track Designation to CPI-613 in Metastatic Pancreatic Cancer. Available online: https://www.targetedonc.com/view/fda-grants-fast-track-designation-to-cpi-613-in-metastatic-pancreatic-cancer (accessed on 25 October 2021).
- Rafael Pharmaceuticals, Inc. Rafael Pharmaceuticals Provides Update on Pivotal Phase 3 Clinical Trial in Patients with Metastatic Pancreatic Cancer and Interim Analysis of Pivotal Phase 3 Clinical Trial in Patients with Relapsed or Refractory Acute Myeloid Leukemia. Available online: https://www.globenewswire.com/news-release/2021/10/28/2322715/28235/en/Rafael-Pharmaceuticals-Provides-Update-on-Pivotal-Phase-3-Clinical-Trial-in-Patients-with-Metastatic-Pancreatic-Cancer-and-Interim-Analysis-of-Pivotal-Phase-3-Clinical-Trial-in-Pat.html (accessed on 27 August 2021).
- Stone, H.B.; Peters, L.J.; Milas, L. Effect of Host Immune Capability on Radiocurability and Subsequent Transplantability of a Murine Fibrosarcoma. J. Natl. Cancer Inst. 1979, 63, 1229–1235. [Google Scholar]
- Verbrugge, I.; Hagekyriakou, J.; Sharp, L.L.; Galli, M.; West, A.; McLaughlin, N.M.; Duret, H.; Yagita, H.; Johnstone, R.W.; Smyth, M.J.; et al. Radiotherapy Increases the Permissiveness of Established Mammary Tumors to Rejection by Immunomodulatory Antibodies. Cancer Res. 2012, 72, 3163–3174. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 Blockade and Stereotactic Radiation Produce Long-Term Survival in Mice with Intracranial Gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Abstract PO-046: Combining PARP Inhibition with Radiation to Sensitize Homologous Recombination Proficient Pancreatic Cancer to Immunotherapy | Clinical Cancer Research. Available online: https://clincancerres.aacrjournals.org/content/27/8_Supplement/PO-046 (accessed on 13 August 2021).
- Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide alone on Survival in Patients with Glioblastoma: A Randomized Clinical Trial | Neuro-Oncology | JAMA | JAMA Network. Available online: https://jamanetwork.com/journals/jama/fullarticle/2666504 (accessed on 25 October 2021).
- Rivera, F.; Benavides, M.; Gallego, J.; Guillen-Ponce, C.; Lopez-Martin, J.; Küng, M. Tumor Treating Fields in Combination with Gemcitabine or Gemcitabine plus Nab-Paclitaxel in Pancreatic Cancer: Results of the PANOVA Phase 2 Study. Pancreatology 2019, 19, 64–72. [Google Scholar] [CrossRef]


{kind=link}
| Category | Advances |
|---|---|
| Immunotherapy | Anti-CTLA4 Antibodies |
| PD-1/PD-L1 Inhibitors | |
| CD-40 Agonists | |
| TME Modulating Vaccines | |
| Targeted Therapy | FAK pathway Inhibition |
| CCR2/CCR5 Inhibition | |
| PARP Inhibition | |
| Stromal Targeting | CXCR4 Inhibition |
| FAP Inhibition | |
| ECM Targeting | |
| Surgical Strategies | IRE |
| Appleby | |
| Combinatorial Strategies | Radiation + Immunotherapy +/− Chemotherapy |
| Small Molecule Inhibitors + Immunotherapy | |
| Stromal Targeting + Immunotherapy |
| Target: | Agents: | Current Clinical Trial: | Notes: |
|---|---|---|---|
| T cell | Nivolumab + nab-paclitaxel +/− gemcitabine | NCT02309177, phase I, (https://clinicaltrials.gov/ct2/show/NCT02309177), accessed on 27 August 2021 | Nivolumab is an inhibitor of the immune checkpoint PD-1 |
| Durvalumab + SBRT | NCT03245541, phase I/II, (https://clinicaltrials.gov/ct2/show/NCT03245541), accessed on 27 August 2021 | Durvalumab is an inhibitor of the immune checkpoint PD-1 | |
| Nivolumab + ipilimumab + nab-paclitaxel + gemcitabine + SBRT | NCT04247165, phase I/II, (https://clinicaltrials.gov/ct2/show/NCT04247165), accessed on 27 August 2021 | Nivolumab and ipilimumab are inhibitors of the immune checkpoints PD-1 and CTLA4, respectively | |
| Durvalumab + tremelimumab + nab-paclitaxel + gemcitabine | NCT02658214, phase I, (https://clinicaltrials.gov/ct2/show/NCT02658214), accessed on 27 August 2021 | Durvalumab and tremelimumab are inhibitors of the immune checkpoints PD-1 and CTLA4, respectively | |
| Durvalumab and/or tremelimumab + SBRT | NCT02311361, phase I/II, (https://clinicaltrials.gov/ct2/show/NCT02311361), accessed on 27 August 2021 | Durvalumab and tremelimumab are inhibitors of the immune checkpoints PD-1 and CTLA4, respectively | |
| Dendritic cell | CRS-207 + nivolumab + ipilimumab +/− GVAX | NCT03190265, phase II, (https://clinicaltrials.gov/ct2/show/NCT03190265), accessed on 27 August 2021 | CRS-207 is a live-attenuated Listeria monocytogenes engineered to express mesothelin, a tumor-associated antigen, and GVAX is a cancer vaccine composed of whole tumor cells genetically modified to secrete the immune stimulatory cytokine, granulocyte-macrophage colony-stimulating factor (GM-CSF) |
| Algenpantucel-L | NCT00569387, phase II, (https://clinicaltrials.gov/ct2/show/NCT00569387), accessed on 27 August 2021 | Algenpantucel-L is an allogeneic pancreatic cancer vaccine based on the concept of hyperacute rejection and is composed of two human pancreatic ductal adenocarcinoma cell lines (HAPa-1 and HAPa-2) | |
| Algenpantucel-L | NCT03165188, phase II, (https://clinicaltrials.gov/ct2/show/NCT00569387), accessed on 27 August 2021 | ||
| Adjuvant gemcitabine or 5-F- based chemoradiation +/− Algenpantucel-L | NCT01072981, phase III, (https://clinicaltrials.gov/ct2/show/NCT01072981), accessed on 27 August 2021 | ||
| Cancer cell | Olaparib | NCT02184195, phase III, (https://clinicaltrials.gov/ct2/show/NCT02184195), accessed on 27 August 2021 | Olaparib is a PARP inhibitor, used as maintenance in this study for patients with known deleterious or suspected deleterious germline BRCA mutation who did not progress on first line platinum-based chemotherapy |
| Olaparib +/− pembrolizumab | NCT04548752, phase II, (https://clinicaltrials.gov/ct2/show/NCT04548752), accessed on 27 August 2021 | Olaparib is a PARP inhibitor, and pembrolizumab is an inhibitor of the immune checkpoint PD-1 | |
| Niraparib | NCT03601923, phase II, (https://clinicaltrials.gov/ct2/show/NCT03601923), accessed on 27 August 2021 | Niraparib is a PARP inhibitor | |
| Niraparib + ipilimumab or nivolumab | NCT03404960, phase I/II, (https://clinicaltrials.gov/ct2/show/NCT03404960), accessed on 27 August 2021 | ||
| Rucaparib | NCT03140670, phase II, (https://clinicaltrials.gov/ct2/show/NCT03140670), accessed on 27 August 2021 | Rucaparib is a PARP inhibitor, used as maintenance in this study for patients with known deleterious or suspected deleterious germline BRCA1/2 or PALB2 mutation who did not progress on first line platinum-based chemotherapy | |
| Rucaparib | NCT02042378, phase II, (https://clinicaltrials.gov/ct2/show/NCT02042378), accessed on 27 August 2021 | ||
| Pelareorep + pembrolizumab + gemcitabine + irinotecan + leucovorin + 5-FU | NCT02620423, phase I, (https://clinicaltrials.gov/ct2/show/NCT02620423), accessed on 27 August 2021 | Pelareorep is an oncolytic reovirus that acts specifically in tumors with an activated Ras pathway | |
| Myeloid | APX005M + nab-paclitaxel + gemcitabine +/− nivolumab | NCT03214250, phase I/II, (https://clinicaltrials.gov/ct2/show/NCT03214250), accessed on 27 August 2021 | APX005M is a CD-40 agonist |
| Cytokine | Plerixafor + cemiplimab | NCT04177810, phase II, (https://clinicaltrials.gov/ct2/show/NCT04177810), accessed on 27 August 2021 | Plerixafor is an inhibitor of the alpha chemokine receptor CXCR4, and cemiplimab is an inhibitor of the immune checkpoint PD-1 |
| BL-8040 + pembrolizumab +/− liposomal irinotecan/5-FU/leucovorin | NCT02826486, phase II, (https://clinicaltrials.gov/ct2/show/NCT02826486), accessed on 27 August 2021 | BL-8040 is an inhibitor CXCR4 | |
| Stroma | BMS-813160 + nivolumab and/or chemotherapy (irinotecan/5-FU/leucovorin or gemcitabine/nab-paclitaxel) | NCT03184870, phase I/II, (https://clinicaltrials.gov/ct2/show/NCT03184870), accessed on 27 August 2021 | BMS-813160 is a CCR2/CCR5 antagonist |
| Durvalumab + AZD5069 or gemcitabine/nab-paclitaxel | NCT02583477, phase I/II, (https://clinicaltrials.gov/ct2/show/NCT02583477), accessed on 27 August 2021 | AZD5069 is a CXCR2 antagonist that blocks neutrophil migration and reduces circulating neutrophil counts | |
| CCX872-B + oxaliplatin/ irinotecan/5-FU/leucovorin | NCT02345408, phase I, (https://clinicaltrials.gov/ct2/show/NCT02345408), accessed on 27 August 2021 | CCX872-B is a CXCR2 antagonist | |
| PF-04136309 + gemcitabine/nab-paclitaxel | NCT02732938, phase I/II, (https://clinicaltrials.gov/ct2/show/NCT02732938), accessed on 27 August 2021 | PF-04136309 is a CXCR2 antagonist | |
| CAR-T cells targeting Nectin4/FAP | NCT03932565, phase I, (https://clinicaltrials.gov/ct2/show/NCT03932565), accessed on 27 August 2021 | CAR-T cell therapy specifically targeting fibroblast activation protein (FAP) | |
| Defactinib +/− pembrolizumab | NCT03727880, phase I/II, (https://clinicaltrials.gov/ct2/show/NCT03727880), accessed on 27 August 2021 | Defactinib is a focal adhesion kinase (FAK) inhibitor | |
| Defactinib + pembrolizumab + gemcitabine | NCT02546531, phase I/II, (https://clinicaltrials.gov/ct2/show/NCT02546531), accessed on 27 August 2021 | ||
| Defactinib + pembrolizumab | NCT02758587, phase I, (https://www.clinicaltrials.gov/ct2/show/NCT02758587), accessed on 27 August 2021 | ||
| Other | Various agents (Multiple Regimes) | NCT04229004, phase II/III, (https://clinicaltrials.gov/ct2/show/NCT04229004), accessed on 27 August 2021 | Percision Promise |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, E.M.; Thomassian, S.; Gong, J.; Hendifar, A.; Osipov, A. Advances in Pancreatic Ductal Adenocarcinoma Treatment. Cancers 2021, 13, 5510. https://doi.org/10.3390/cancers13215510
Anderson EM, Thomassian S, Gong J, Hendifar A, Osipov A. Advances in Pancreatic Ductal Adenocarcinoma Treatment. Cancers. 2021; 13(21):5510. https://doi.org/10.3390/cancers13215510
Chicago/Turabian StyleAnderson, Eric M., Shant Thomassian, Jun Gong, Andrew Hendifar, and Arsen Osipov. 2021. "Advances in Pancreatic Ductal Adenocarcinoma Treatment" Cancers 13, no. 21: 5510. https://doi.org/10.3390/cancers13215510
APA StyleAnderson, E. M., Thomassian, S., Gong, J., Hendifar, A., & Osipov, A. (2021). Advances in Pancreatic Ductal Adenocarcinoma Treatment. Cancers, 13(21), 5510. https://doi.org/10.3390/cancers13215510