
Targeted Metabolomics Identifies Plasma Biomarkers in Mice with Metabolically Heterogeneous Melanoma Xenografts

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
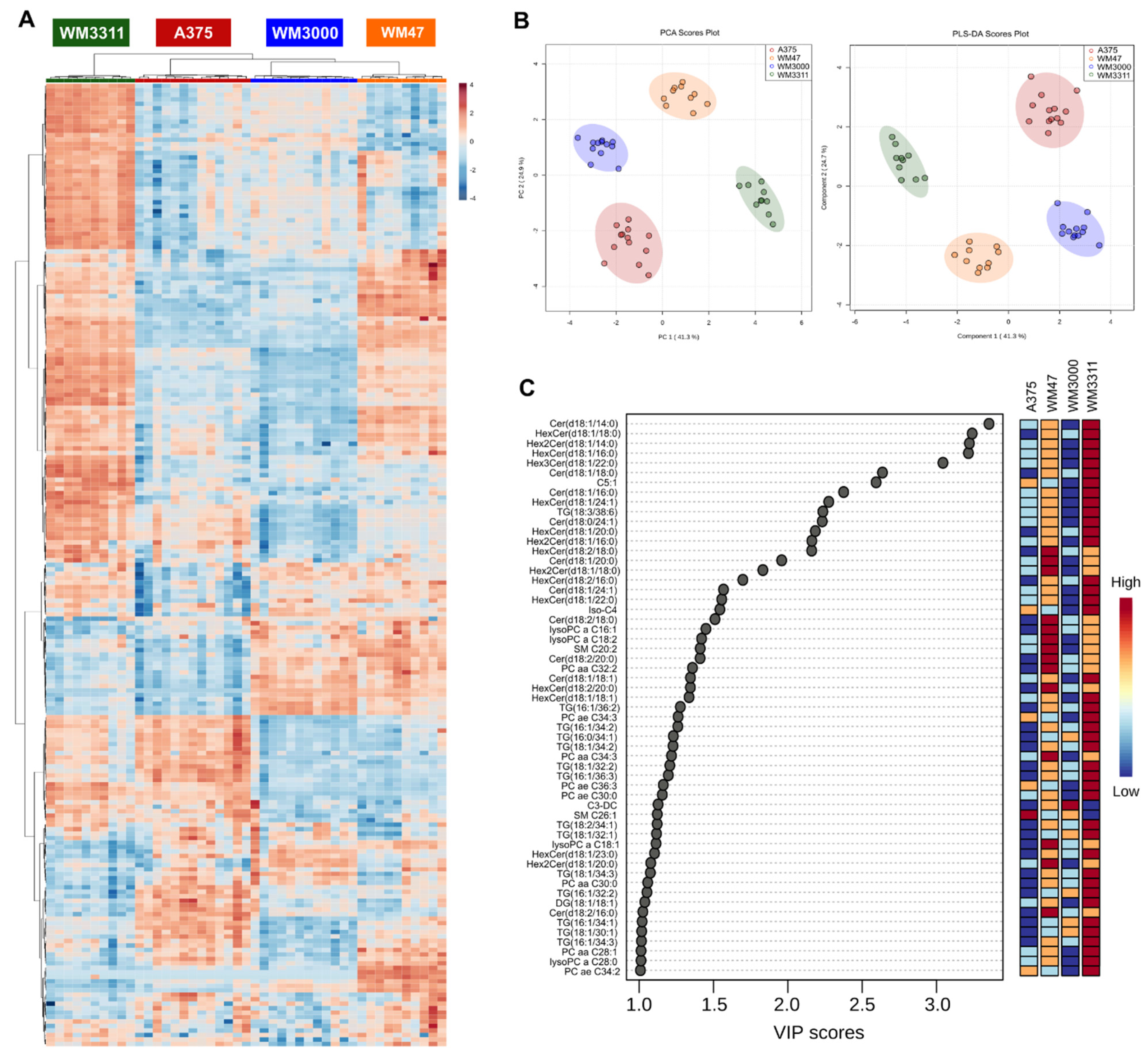
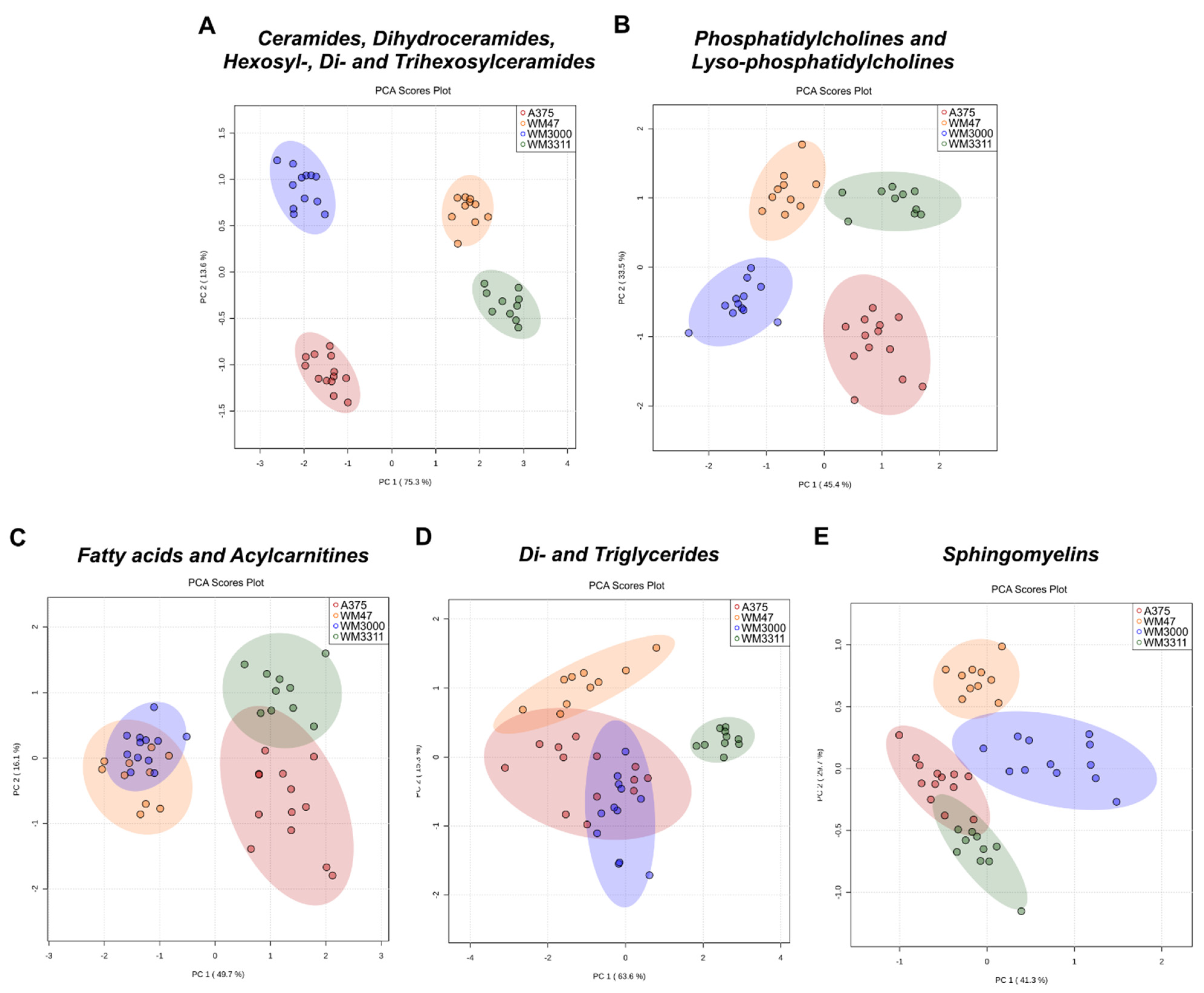
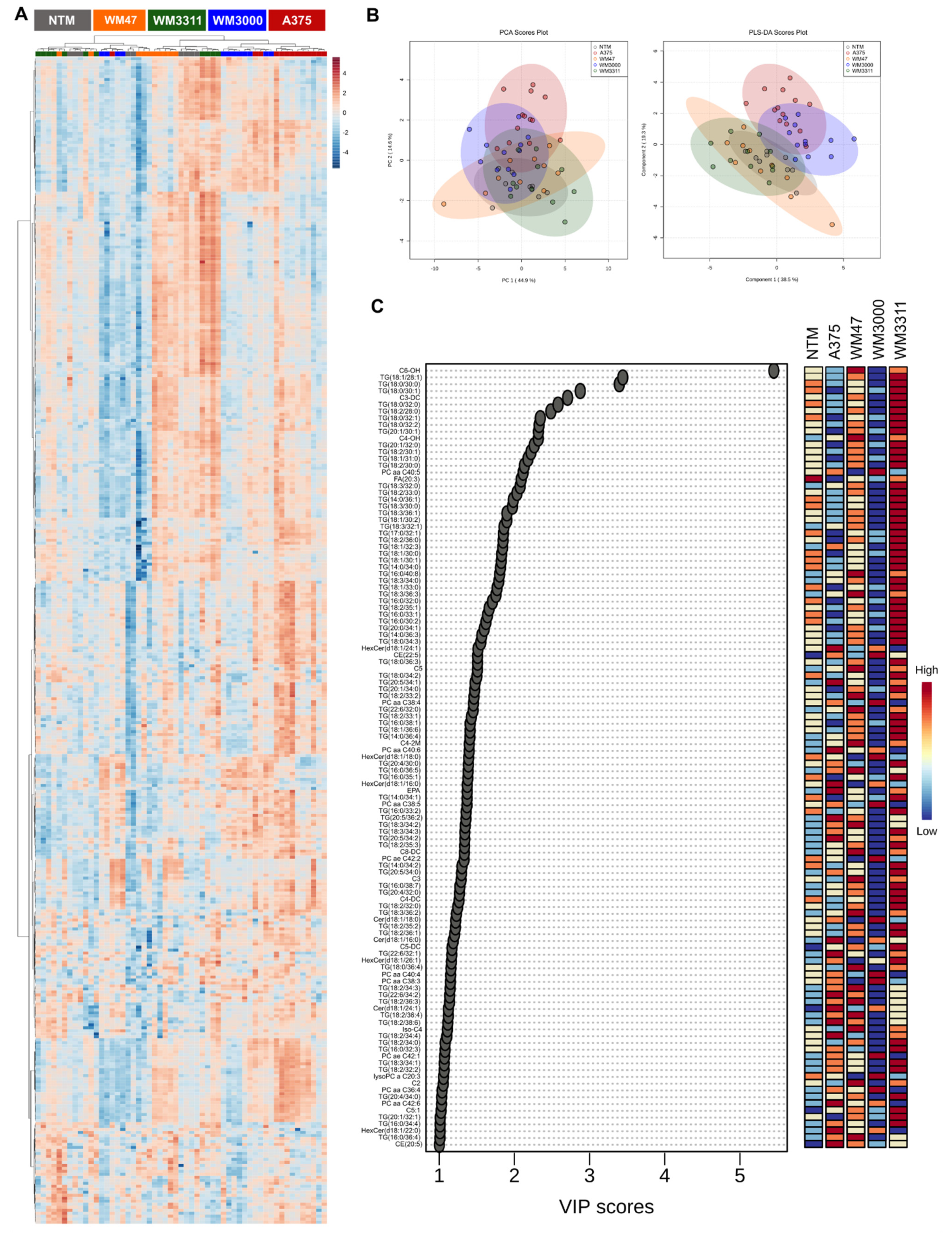
2.1. Metabolite Profiling in Xenograft Tissue
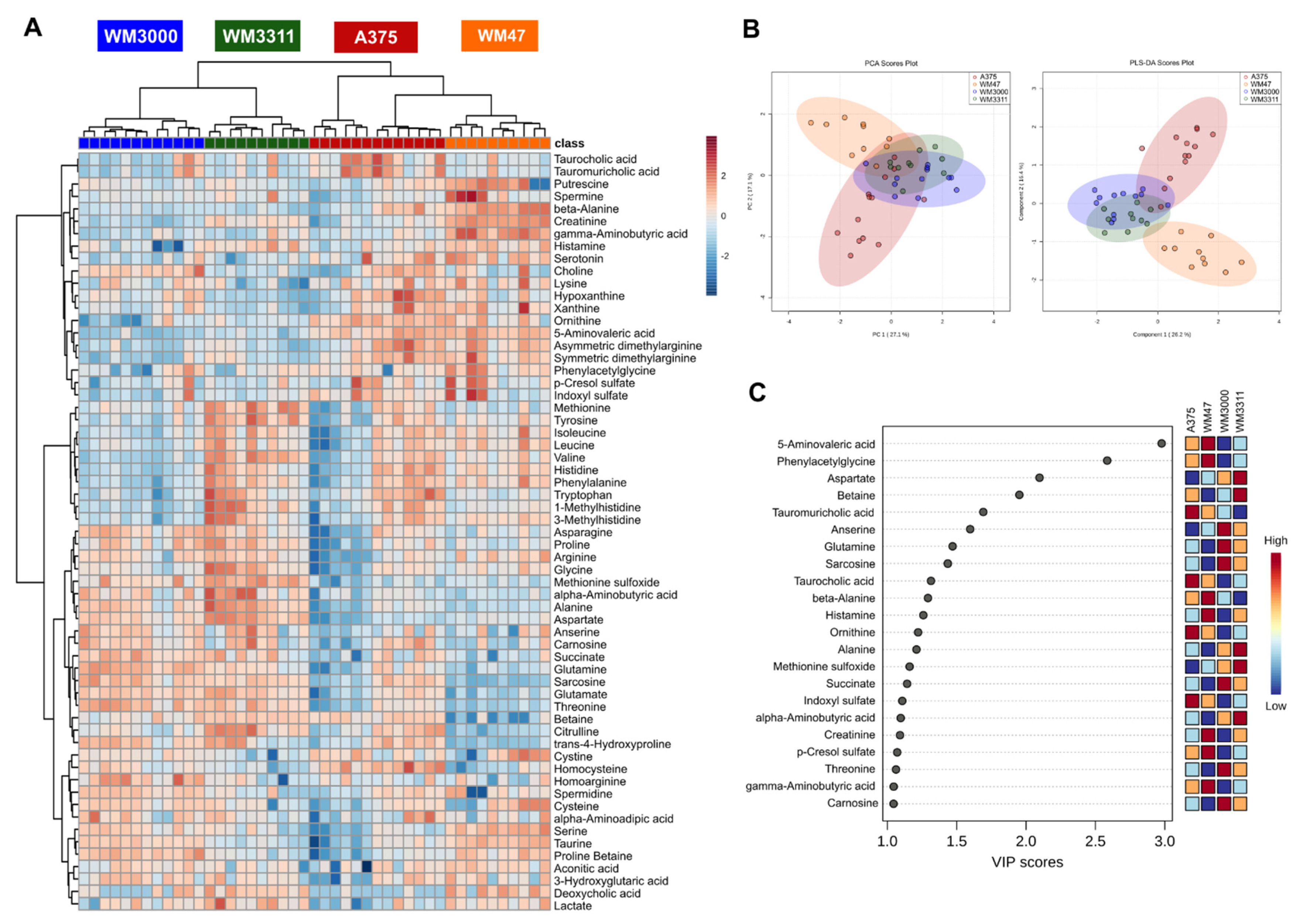
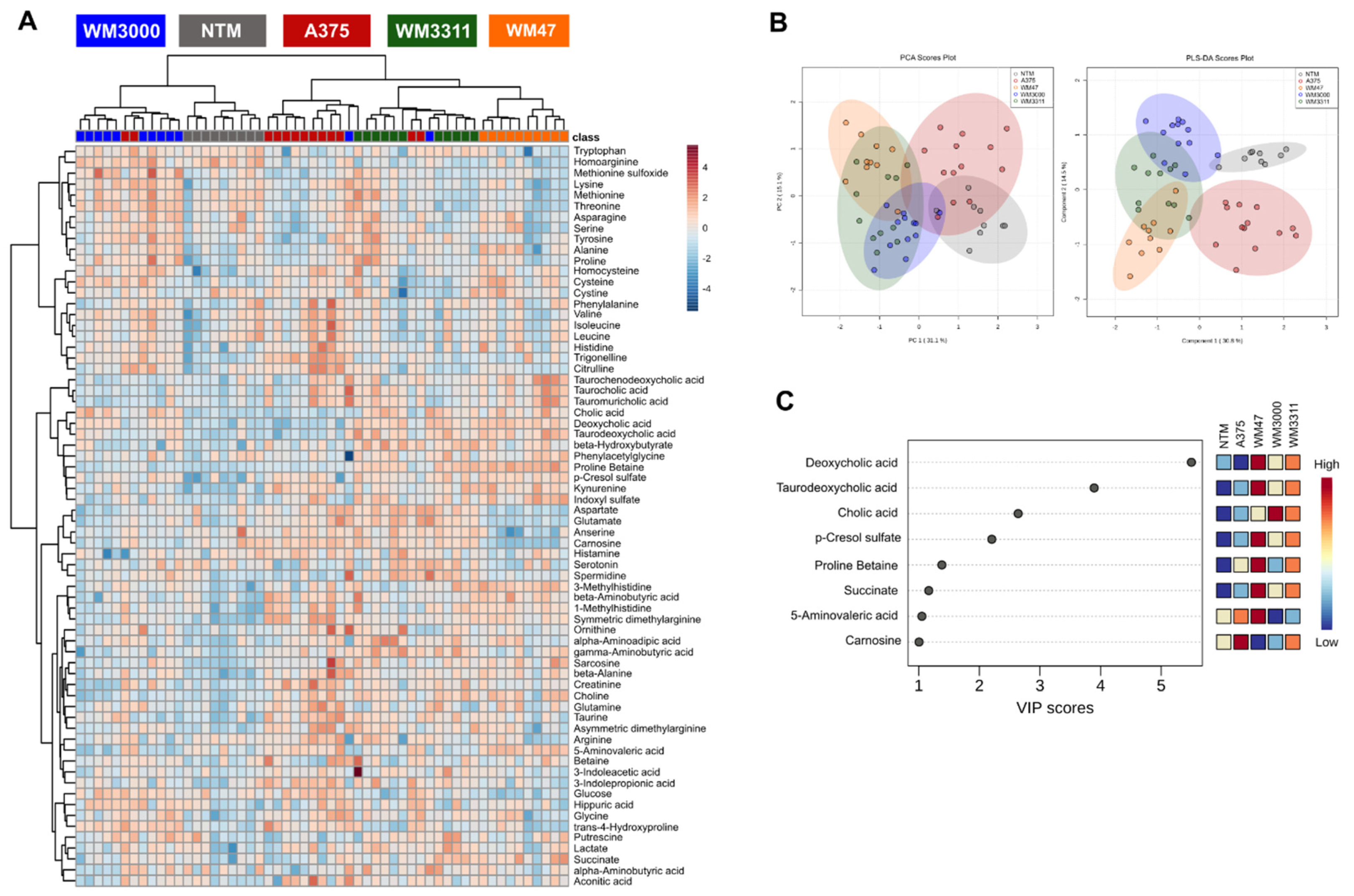
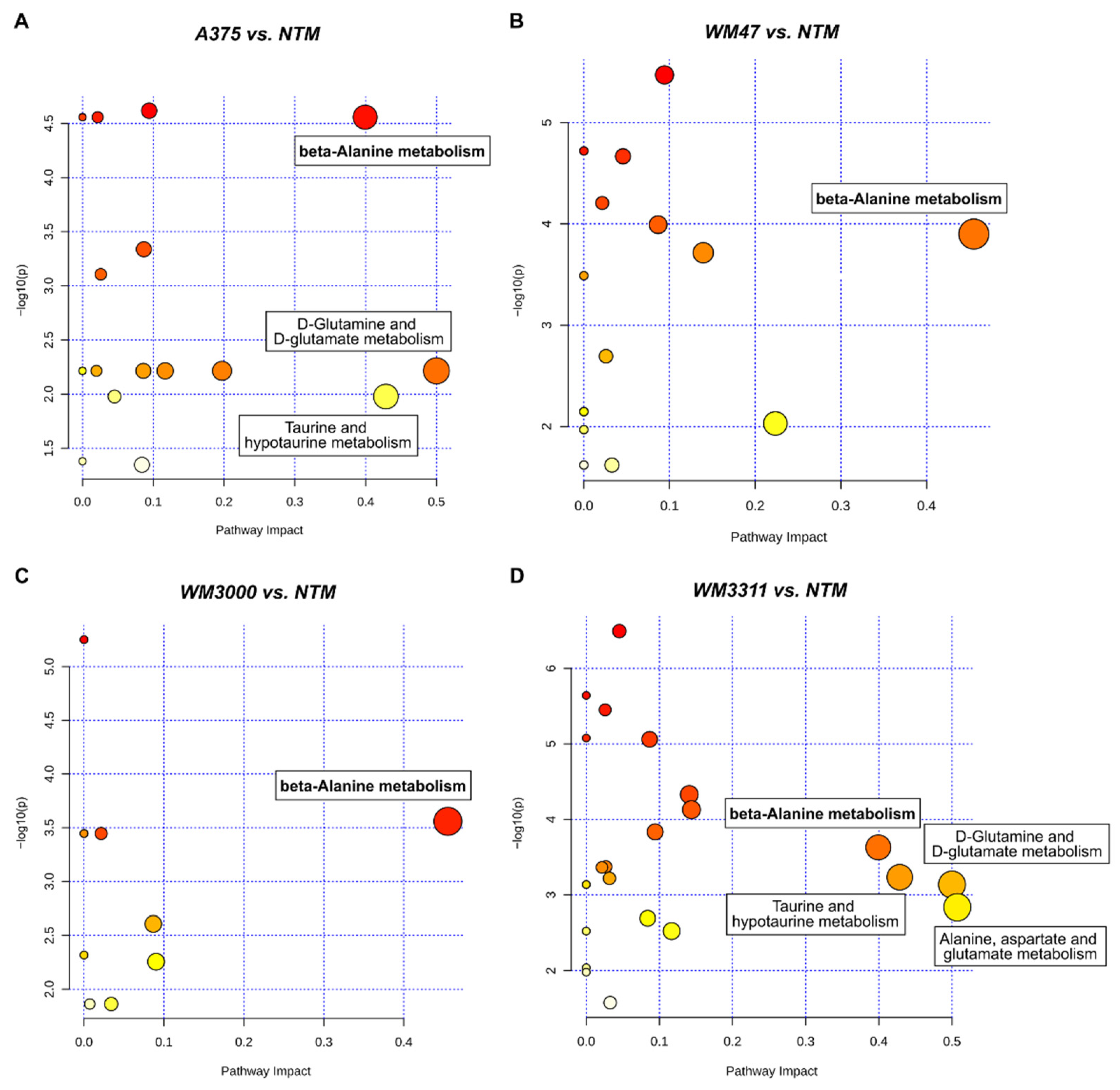
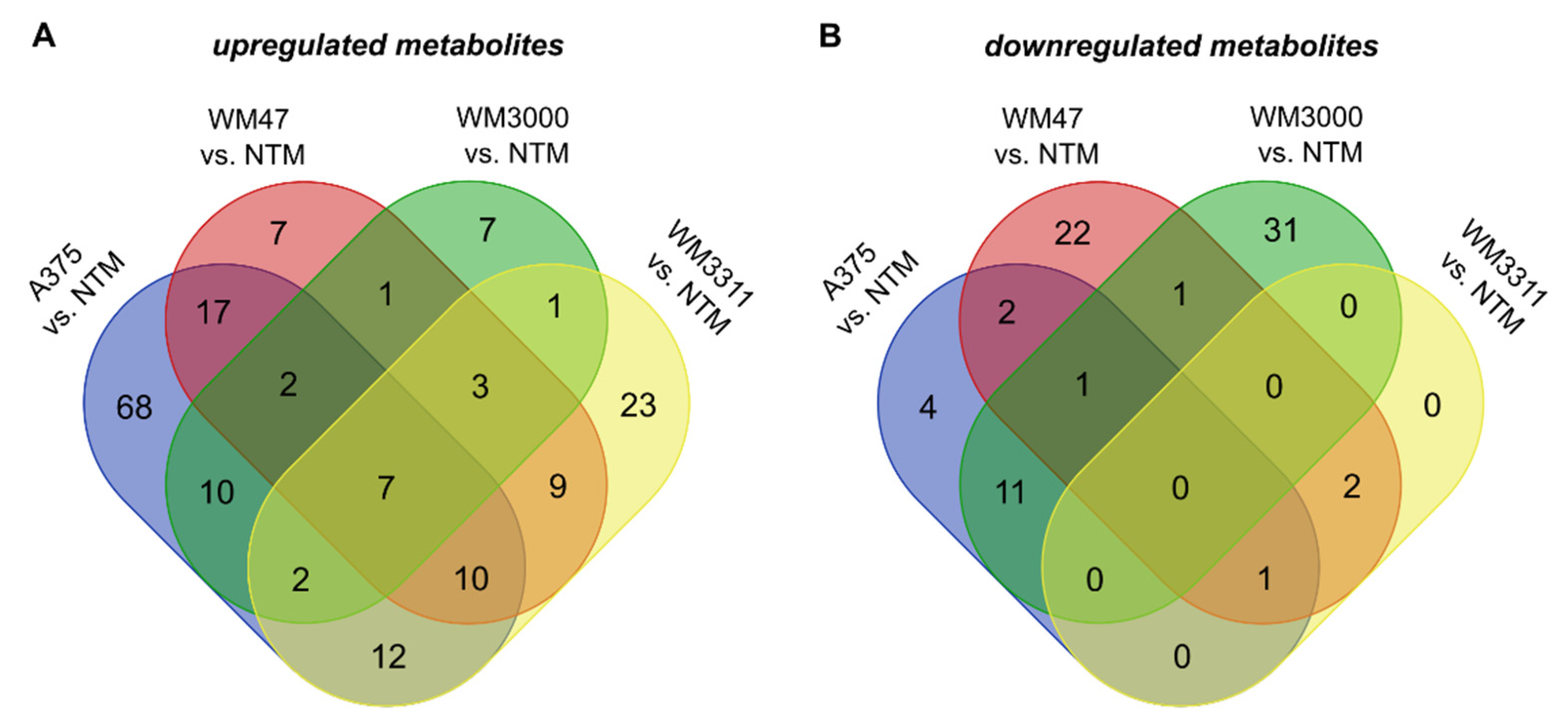
2.2. Metabolite Profiling in Plasma
2.3. Biomarker Analysis
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Animals and Sample Collection
4.3. Histology
4.4. Targeted Metabolomics
4.4.1. MxP® Quant 500 Kit
4.4.2. AC Assay
4.5. Data Processing and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Abildgaard, C.; Guldberg, P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol. Med. 2015, 21, 164–171. [Google Scholar] [CrossRef]
- Fischer, G.M.; Vashisht Gopal, Y.N.; McQuade, J.L.; Peng, W.; DeBerardinis, R.J.; Davies, M.A. Metabolic strategies of melanoma cells: Mechanisms, interactions with the tumor microenvironment, and therapeutic implications. Pigment. Cell Melanoma Res. 2018, 31, 11–30. [Google Scholar] [CrossRef]
- Ratnikov, B.I.; Scott, D.A.; Osterman, A.L.; Smith, J.W.; Ronai, Z.A. Metabolic rewiring in melanoma. Oncogene 2017, 36, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. Über den Stoffwechsel der Carcinomzelle. Naturwissenschaften 1924, 50, 1131–1137. [Google Scholar] [CrossRef]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative metabolic flux profiling of melanoma cell lines: Beyond the Warburg effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef] [Green Version]
- Feichtinger, R.G.; Lang, R.; Geilberger, R.; Rathje, F.; Mayr, J.A.; Sperl, W.; Bauer, J.W.; Hauser-Kronberger, C.; Kofler, B.; Emberger, M. Melanoma tumors exhibit a variable but distinct metabolic signature. Exp. Dermatol. 2018, 27, 204–207. [Google Scholar] [CrossRef] [Green Version]
- Aminzadeh-Gohari, S.; Weber, D.D.; Catalano, L.; Feichtinger, R.G.; Kofler, B.; Lang, R. Targeting Mitochondria in Melanoma. Biomolecules 2020, 10, 1395. [Google Scholar] [CrossRef]
- Gopal, Y.N.; Rizos, H.; Chen, G.; Deng, W.; Frederick, D.T.; Cooper, Z.A.; Scolyer, R.A.; Pupo, G.; Komurov, K.; Sehgal, V.; et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1alpha and oxidative phosphorylation in melanoma. Cancer Res. 2014, 74, 7037–7047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef] [PubMed]
- Migneco, G.; Whitaker-Menezes, D.; Chiavarina, B.; Castello-Cros, R.; Pavlides, S.; Pestell, R.G.; Fatatis, A.; Flomenberg, N.; Tsirigos, A.; Howell, A.; et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: Evidence for stromal-epithelial metabolic coupling. Cell Cycle 2010, 9, 2412–2422. [Google Scholar] [CrossRef]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.; de Moura, M.B.; Lin, Y.; Vincent, G.; Thorne, S.; Duncan, L.M.; Hui-Min, L.; Kirkwood, J.M.; Becker, D.; Van Houten, B.; et al. Importance of glycolysis and oxidative phosphorylation in advanced melanoma. Mol. Cancer 2012, 11, 76. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.M.; Yu, H.; Edwards, R.; Chen, L.; Kazianis, S.; Brafford, P.; Acs, G.; Herlyn, M.; Xu, X. Mutant V600E BRAF increases hypoxia inducible factor-1alpha expression in melanoma. Cancer Res. 2007, 67, 3177–3184. [Google Scholar] [CrossRef] [Green Version]
- Parmenter, T.J.; Kleinschmidt, M.; Kinross, K.M.; Bond, S.T.; Li, J.; Kaadige, M.R.; Rao, A.; Sheppard, K.E.; Hugo, W.; Pupo, G.M.; et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014, 4, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Filipp, F.V.; Scott, D.A.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment. Cell Melanoma Res. 2012, 25, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Carrie, L.; Dufau, C.; Nieto, L.; Segui, B.; Levade, T.; Riond, J.; Andrieu-Abadie, N. Lipid metabolic Reprogramming: Role in Melanoma Progression and Therapeutic Perspectives. Cancers 2020, 12, 3147. [Google Scholar] [CrossRef] [PubMed]
- Kapur, P.; Rakheja, D.; Roy, L.C.; Hoang, M.P. Fatty acid synthase expression in cutaneous melanocytic neoplasms. Mod. Pathol 2005, 18, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep. 2001, 2, 282–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.B.; Sarraf, P.; Wright, M.; Yao, K.M.; Mueller, E.; Solanes, G.; Lowell, B.B.; Spiegelman, B.M. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J. Clin. Investig. 1998, 101, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez, J.A. and Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Wu, S.; Naar, A.M. SREBP1-dependent de novo fatty acid synthesis gene expression is elevated in malignant melanoma and represents a cellular survival trait. Sci. Rep. 2019, 9, 10369. [Google Scholar] [CrossRef]
- Clement, E.; Lazar, I.; Attane, C.; Carrie, L.; Dauvillier, S.; Ducoux-Petit, M.; Esteve, D.; Menneteau, T.; Moutahir, M.; Le Gonidec, S.; et al. Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. EMBO J. 2020, 39, e102525. [Google Scholar] [CrossRef]
- Sumantran, V.N.; Mishra, P.; Sudhakar, N. Microarray analysis of differentially expressed genes regulating lipid metabolism during melanoma progression. Indian J. Biochem. Biophys. 2015, 52, 125–131. [Google Scholar]
- Rodrigues, M.F.; Obre, E.; de Melo, F.H.; Santos, G.C., Jr.; Galina, A.; Jasiulionis, M.G.; Rossignol, R.; Rumjanek, F.D.; Amoedo, N.D. Enhanced OXPHOS, glutaminolysis and beta-oxidation constitute the metastatic phenotype of melanoma cells. Biochem. J. 2016, 473, 703–715. [Google Scholar] [CrossRef]
- Yu, Z.; Huang, M.; Clowers, B.H. Comparative metabolite profiling of a metastatic and primary melanoma cell line using untargeted metabolomics: A case study. Clin. Mass Spect. 2018, 10, 16–24. [Google Scholar] [CrossRef]
- Puchades-Carrasco, L.; Pineda-Lucena, A. Metabolomics Applications in Precision Medicine: An Oncological Perspective. Curr Top. Med. Chem. 2017, 17, 2740–2751. [Google Scholar] [PubMed] [Green Version]
- Kaushik, A.K.; DeBerardinis, R.J. Applications of metabolomics to study cancer metabolism. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 2–14. [Google Scholar] [PubMed]
- Roberts, L.D.; Souza, A.L.; Gerszten, R.E.; Clish, C.B. Targeted metabolomics. Curr. Protoc. Mol. Biol. 2012, 30, 30.2.1–30.2.24. [Google Scholar]
- Siskos, A.P.; Jain, P.; Romisch-Margl, W.; Bennett, M.; Achaintre, D.; Asad, Y.; Marney, L.; Richardson, L.; Koulman, A.; Griffin, J.L.; et al. Interlaboratory Reproducibility of a Targeted Metabolomics Platform for Analysis of Human Serum and Plasma. Anal. Chem. 2017, 89, 656–665. [Google Scholar] [PubMed]
- Ribbenstedt, A.; Ziarrusta, H.; Benskin, J.P. Development, characterization and comparisons of targeted and non-targeted metabolomics methods. PLoS ONE 2018, 13, e0207082. [Google Scholar]
- Kus, K.; Kij, A.; Zakrzewska, A.; Jasztal, A.; Stojak, M.; Walczak, M.; Chlopicki, S. Alterations in arginine and energy metabolism, structural and signalling lipids in metastatic breast cancer in mice detected in plasma by targeted metabolomics and lipidomics. Breast Cancer Res. 2018, 20, 148. [Google Scholar]
- Yang, X.H.; Jing, Y.; Wang, S.; Ding, F.; Zhang, X.X.; Chen, S.; Zhang, L.; Hu, Q.G.; Ni, Y.H. Integrated Non-targeted and Targeted Metabolomics Uncovers Amino Acid Markers of Oral Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 426. [Google Scholar]
- Zhang, X.; Zhu, X.; Wang, C.; Zhang, H.; Cai, Z. Non-targeted and targeted metabolomics approaches to diagnosing lung cancer and predicting patient prognosis. Oncotarget 2016, 7, 63437–63448. [Google Scholar]
- Zhu, J.; Djukovic, D.; Deng, L.; Gu, H.; Himmati, F.; Chiorean, E.G.; Raftery, D. Colorectal cancer detection using targeted serum metabolic profiling. J. Proteome Res. 2014, 13, 4120–4130. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar]
- Feng, J.; Isern, N.G.; Burton, S.D.; Hu, J.Z. Studies of Secondary Melanoma on C57BL/6J Mouse Liver Using 1H NMR Metabolomics. Metabolites 2013, 3, 1011–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Hu, M.; Liu, M.; Hu, J.Z. Metastatic Melanoma Induced Metabolic Changes in C57BL/6J Mouse Stomach Measured by 1H NMR Spectroscopy. Metabolomics 2014, 4, 1000135. [Google Scholar] [PubMed]
- Kim, H.Y.; Lee, H.; Kim, S.H.; Jin, H.; Bae, J.; Choi, H.K. Discovery of potential biomarkers in human melanoma cells with different metastatic potential by metabolic and lipidomic profiling. Sci. Rep. 2017, 7, 8864. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.K.; Yeow, C.H. Proton NMR characterization of intact primary and metastatic melanoma cells in 2D & 3D cultures. Biol. Res. 2017, 50, 12. [Google Scholar]
- Fischer, G.M.; Jalali, A.; Kircher, D.A.; Lee, W.C.; McQuade, J.L.; Haydu, L.E.; Joon, A.Y.; Reuben, A.; de Macedo, M.P.; Carapeto, F.C.L.; et al. Molecular Profiling Reveals Unique Immune and Metabolic Features of Melanoma Brain Metastases. Cancer Discov. 2019, 9, 628–645. [Google Scholar] [CrossRef] [Green Version]
- Kosmopoulou, M.; Giannopoulou, A.F.; Iliou, A.; Benaki, D.; Panagiotakis, A.; Velentzas, A.D.; Konstantakou, E.G.; Papassideri, I.S.; Mikros, E.; Stravopodis, D.J.; et al. Human Melanoma-Cell Metabolic Profiling: Identification of Novel Biomarkers Indicating Metastasis. Int. J. Mol. Sci. 2020, 21, 2436. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Tasdogan, A.; Huang, F.; Hu, Z.; Morrison, S.J.; DeBerardinis, R.J. The abundance of metabolites related to protein methylation correlates with the metastatic capacity of human melanoma xenografts. Sci. Adv. 2017, 3, eaao5268. [Google Scholar] [CrossRef] [Green Version]
- Lligona Trulla, L.; Magistrelli, A.; Salmona, M.; Tacconi, M.T. Phospholipid composition, phosphoinositide metabolism and metastatic capacity in murine melanoma B16 variants at different stages of growth. Melanoma Res. 1992, 2, 235–240. [Google Scholar]
- Muqaku, B.; Eisinger, M.; Meier, S.M.; Tahir, A.; Pukrop, T.; Haferkamp, S.; Slany, A.; Reichle, A.; Gerner, C. Multi-omics Analysis of Serum Samples Demonstrates Reprogramming of Organ Functions Via Systemic Calcium Mobilization and Platelet Activation in Metastatic Melanoma. Mol. Cell Proteomics 2017, 16, 86–99. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, F.; Gardiner, J.M. Membrane lipids and enzymes of cultured high- and low-metastatic B16 melanoma variants. Cancer Res. 1984, 44, 3262–3269. [Google Scholar]
- Wang, X.; Hu, M.; Feng, J.; Liu, M.; Hu, J.Z. (1)H NMR Metabolomics Study of Metastatic Melanoma in C57BL/6J Mouse Spleen. Metabolomics 2014, 10, 1129–1144. [Google Scholar] [CrossRef] [PubMed]
- Santana-Filho, A.P.; Jacomasso, T.; Riter, D.S.; Barison, A.; Iacomini, M.; Winnischofer, S.M.; Sassaki, G.L. NMR metabolic fingerprints of murine melanocyte and melanoma cell lines: Application to biomarker discovery. Sci. Rep. 2017, 7, 42324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soumoy, L.; Schepkens, C.; Krayem, M.; Najem, A.; Tagliatti, V.; Ghanem, G.E.; Saussez, S.; Colet, J.M.; Journe, F. Metabolic Reprogramming in Metastatic Melanoma with Acquired Resistance to Targeted Therapies: Integrative Metabolomic and Proteomic Analysis. Cancers 2020, 12, 1323. [Google Scholar]
- Cernei, N.; Heger, Z.; Gumulec, J.; Zitka, O.; Masarik, M.; Babula, P.; Eckschlager, T.; Stiborova, M.; Kizek, R.; Adam, V. Sarcosine as a potential prostate cancer biomarker--a review. Int. J. Mol. Sci. 2013, 14, 13893–13908. [Google Scholar] [CrossRef] [Green Version]
- Pacik, D.; Plevova, M.; Urbanova, L.; Lackova, Z.; Strmiska, V.; Necas, A.; Heger, Z.; Adam, V. Identification of Sarcosine as a Target Molecule for the Canine Olfactory Detection of Prostate Carcinoma. Sci. Rep. 2018, 8, 4958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef] [Green Version]
- Ang, J.E.; Pal, A.; Asad, Y.J.; Henley, A.T.; Valenti, M.; Box, G.; de Haven Brandon, A.; Revell, V.L.; Skene, D.J.; Venturi, M.; et al. Modulation of Plasma Metabolite Biomarkers of the MAPK Pathway with MEK Inhibitor RO4987655: Pharmacodynamic and Predictive Potential in Metastatic Melanoma. Mol. Cancer Ther. 2017, 16, 2315–2323. [Google Scholar] [CrossRef] [Green Version]
- Bayci, A.W.L.; Baker, D.A.; Somerset, A.E.; Turkoglu, O.; Hothem, Z.; Callahan, R.E.; Mandal, R.; Han, B.; Bjorndahl, T.; Wishart, D.; et al. Metabolomic identification of diagnostic serum-based biomarkers for advanced stage melanoma. Metabolomics 2018, 14, 105. [Google Scholar] [CrossRef]
- Budczies, J.; Brockmoller, S.F.; Muller, B.M.; Barupal, D.K.; Richter-Ehrenstein, C.; Kleine-Tebbe, A.; Griffin, J.L.; Oresic, M.; Dietel, M.; Denkert, C.; et al. Comparative metabolomics of estrogen receptor positive and estrogen receptor negative breast cancer: Alterations in glutamine and beta-alanine metabolism. J. Proteomics 2013, 94, 279–288. [Google Scholar] [CrossRef]
- Trilla-Fuertes, L.; Gamez-Pozo, A.; Diaz-Almiron, M.; Prado-Vazquez, G.; Zapater-Moros, A.; Lopez-Vacas, R.; Nanni, P.; Zamora, P.; Espinosa, E.; Fresno Vara, J.A. Computational metabolism modeling predicts risk of distant relapse-free survival in breast cancer patients. Future Oncol. 2019, 15, 3483–3490. [Google Scholar] [CrossRef]
- Derezinski, P.; Klupczynska, A.; Sawicki, W.; Palka, J.A.; Kokot, Z.J. Amino Acid Profiles of Serum and Urine in Search for Prostate Cancer Biomarkers: A Pilot Study. Int. J. Med. Sci. 2017, 14, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, K.; Han, P.; Song, W.; Wang, Z.; Zhang, F.; Xie, H.; Zhao, W.; Xu, H.; Cai, Y.; Rong, Z.; et al. Plasma metabolomic profiling distinguishes right-sided from left-sided colon cancer. Clin. Chim. Acta 2018, 487, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Funchain, P.; Retuerto, M.; Jurevic, R.J.; Fowler, N.; Burkey, B.; Eng, C.; Ghannoum, M.A. Metabolomic analysis identifies differentially produced oral metabolites, including the oncometabolite 2-hydroxyglutarate, in patients with head and neck squamous cell carcinoma. BBA Clin. 2017, 7, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Vsiansky, V.; Svobodova, M.; Gumulec, J.; Cernei, N.; Sterbova, D.; Zitka, O.; Kostrica, R.; Smilek, P.; Plzak, J.; Betka, J.; et al. Prognostic Significance of Serum Free Amino Acids in Head and Neck Cancers. Cells 2019, 8, 428. [Google Scholar] [CrossRef] [Green Version]
- Hishikawa, D.; Hashidate, T.; Shimizu, T.; Shindou, H. Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J. Lipid Res. 2014, 55, 799–807. [Google Scholar] [CrossRef] [Green Version]
- Parveen, F.; Bender, D.; Law, S.H.; Mishra, V.K.; Chen, C.C.; Ke, L.Y. Role of Ceramidases in Sphingolipid Metabolism and Human Diseases. Cells 2019, 8, 1573. [Google Scholar] [CrossRef] [Green Version]
- Kogot-Levin, A.; Saada, A. Ceramide and the mitochondrial respiratory chain. Biochimie 2014, 100, 88–94. [Google Scholar] [CrossRef]
- Carrie, L.; Virazels, M.; Dufau, C.; Montfort, A.; Levade, T.; Segui, B.; Andrieu-Abadie, N. New Insights into the Role of Sphingolipid Metabolism in Melanoma. Cells 2020, 9, 1967. [Google Scholar] [CrossRef]
- Han, W.S.; Yoo, J.Y.; Youn, S.W.; Kim, D.S.; Park, K.C.; Kim, S.Y.; Kim, K.H. Effects of C2-ceramide on the Malme-3M melanoma cell line. J. Dermatol. Sci. 2002, 30, 10–19. [Google Scholar] [CrossRef]
- Auzenne, E.; Leroux, M.E.; Hu, M.; Pollock, R.E.; Feig, B.; Klostergaard, J. Cytotoxic effects of sphingolipids as single or multi-modality agents on human melanoma and soft tissue sarcoma in vitro. Melanoma Res. 1998, 8, 227–239. [Google Scholar] [CrossRef]
- Charles, A.G.; Han, T.Y.; Liu, Y.Y.; Hansen, N.; Giuliano, A.E.; Cabot, M.C. Taxol-induced ceramide generation and apoptosis in human breast cancer cells. Cancer Chemother. Pharmacol. 2001, 47, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Jin, K.; Huang, S.; Bao, Q.; Shao, Z.; Hu, X.; Ye, J. Liposomal C6 Ceramide Activates Protein Phosphatase 1 to Inhibit Melanoma Cells. PLoS ONE 2016, 11, e0159849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Li, J.; Qiu, Y.; Sun, H. 1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) facilitates curcumin-induced melanoma cell apoptosis by enhancing ceramide accumulation, JNK activation, and inhibiting PI3K/AKT activation. Mol. Cell Biochem. 2012, 361, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Jin, H.; Bae, J.; Choi, H.K. Metabolic and lipidomic investigation of the antiproliferative effects of coronatine against human melanoma cells. Sci. Rep. 2019, 9, 3140. [Google Scholar] [CrossRef]
- Liu, R.; Cao, K.; Tang, Y.; Liu, J.; Li, J.; Chen, J.; Wang, S.; Chen, Z.; Zhou, J. C16:0 ceramide effect on melanoma malignant behavior and glycolysis depends on its intracellular or exogenous location. Am. J. Transl. Res. 2020, 12, 1123–1135. [Google Scholar]
- Henderson, F.; Johnston, H.R.; Badrock, A.P.; Jones, E.A.; Forster, D.; Nagaraju, R.T.; Evangelou, C.; Kamarashev, J.; Green, M.; Fairclough, M.; et al. Enhanced Fatty Acid Scavenging and Glycerophospholipid Metabolism Accompany Melanocyte Neoplasia Progression in Zebrafish. Cancer Res. 2019, 79, 2136–2151. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; DiPaola, R.S.; Mathew, R.; White, E. Metabolic catastrophe as a means to cancer cell death. J. Cell Sci 2007, 120, 379–383. [Google Scholar] [CrossRef] [Green Version]
- Wasinger, C.; Hofer, A.; Spadiut, O.; Hohenegger, M. Amino Acid Signature in Human Melanoma Cell Lines from Different Disease Stages. Sci. Rep. 2018, 8, 6245. [Google Scholar] [CrossRef] [Green Version]
- Kaji, S.; Irino, T.; Kusuhara, M.; Makuuchi, R.; Yamakawa, Y.; Tokunaga, M.; Tanizawa, Y.; Bando, E.; Kawamura, T.; Kami, K.; et al. Metabolomic profiling of gastric cancer tissues identified potential biomarkers for predicting peritoneal recurrence. Gastric Cancer 2020, 23, 874–883. [Google Scholar] [CrossRef]
- Pandurangan, M.; Enkhtaivan, G.; Mistry, B.; Patel, R.V.; Moon, S.; Kim, D.H. beta-Alanine intercede metabolic recovery for amelioration of human cervical and renal tumors. Amino Acids 2017, 49, 1373–1380. [Google Scholar] [CrossRef]
- Vaughan, R.A.; Gannon, N.P.; Garcia-Smith, R.; Licon-Munoz, Y.; Barberena, M.A.; Bisoffi, M.; Trujillo, K.A. beta-alanine suppresses malignant breast epithelial cell aggressiveness through alterations in metabolism and cellular acidity in vitro. Mol. Cancer 2014, 13, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | AUROC | 95% CI | Sensitivity | Specificity | p-Value | Fold Change (Melanoma/NTM) |
|---|---|---|---|---|---|---|
| Tiglylcarnitine (C5:1) | 0.99 | 0.966–1 | 89.13% | 100.00% | 1.58 × 10−11 | 2.88 |
| beta-Alanine | 0.97 | 0.93–1 | 91.30% | 100.00% | 1.76 × 10−7 | 1.80 |
| PC ae C42:4 | 0.95 | 0.886–0.993 | 80.43% | 100.00% | 9.14 × 10−7 | 1.70 |
| Sarcosine | 0.93 | 0.805–1 | 86.96% | 88.89% | 4.46 × 10−5 | 1.93 |
| Hex2Cer (d18:1/16:0) | 0.89 | 0.694–0.995 | 91.30% | 88.89% | 5.24 × 10−4 | 2.33 |
| Hex2Cer (d18:1/20:0) | 0.85 | 0.657–0.979 | 86.96% | 88.89% | 1.97 × 10−3 | 2.08 |
| p-Cresol sulfate | 0.84 | 0.694–0.995 | 97.83% | 77.78% | 6.72 × 10−7 | 3.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weber, D.D.; Thapa, M.; Aminzadeh-Gohari, S.; Redtenbacher, A.-S.; Catalano, L.; Feichtinger, R.G.; Koelblinger, P.; Dallmann, G.; Emberger, M.; Kofler, B.; et al. Targeted Metabolomics Identifies Plasma Biomarkers in Mice with Metabolically Heterogeneous Melanoma Xenografts. Cancers 2021, 13, 434. https://doi.org/10.3390/cancers13030434
Weber DD, Thapa M, Aminzadeh-Gohari S, Redtenbacher A-S, Catalano L, Feichtinger RG, Koelblinger P, Dallmann G, Emberger M, Kofler B, et al. Targeted Metabolomics Identifies Plasma Biomarkers in Mice with Metabolically Heterogeneous Melanoma Xenografts. Cancers. 2021; 13(3):434. https://doi.org/10.3390/cancers13030434
Chicago/Turabian StyleWeber, Daniela D., Maheshwor Thapa, Sepideh Aminzadeh-Gohari, Anna-Sophia Redtenbacher, Luca Catalano, René G. Feichtinger, Peter Koelblinger, Guido Dallmann, Michael Emberger, Barbara Kofler, and et al. 2021. "Targeted Metabolomics Identifies Plasma Biomarkers in Mice with Metabolically Heterogeneous Melanoma Xenografts" Cancers 13, no. 3: 434. https://doi.org/10.3390/cancers13030434
APA StyleWeber, D. D., Thapa, M., Aminzadeh-Gohari, S., Redtenbacher, A. -S., Catalano, L., Feichtinger, R. G., Koelblinger, P., Dallmann, G., Emberger, M., Kofler, B., & Lang, R. (2021). Targeted Metabolomics Identifies Plasma Biomarkers in Mice with Metabolically Heterogeneous Melanoma Xenografts. Cancers, 13(3), 434. https://doi.org/10.3390/cancers13030434