
Thiosemicarbazone Derivatives Developed to Overcome COTI-2 Resistance

 ,
,  , , ,
, , ,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Methods for Synthesis
2.1.1. Synthesis of 2-(6,7-Dihydroquinolin-8(5H)-ylidene)-N4,N4-dimethylhydrazine-1-carbothioamide (COTI-NMe2)
2.1.2. Synthesis of N-Cyclohexyl-2-(6,7-dihydroquinolin-8(5H)-ylidene)-N4-methylhydrazine-1-carbothioamide (COTI-NMeCy)
Synthesis of 2-((Cyclohexyl(methyl)carbomothioyl)thio)acetic Acid (1a)
Synthesis of N-Cyclohexyl-N-methylhydrazinecarbothioamide (1b)
Synthesis of N-Cyclohexyl-2-(6,7-dihydroquinolin-8(5H)-ylidene)-N4-methylhydrazine-1-carbothioamide (COTI-NMeCy)
2.1.3. Synthesis of the Cu(II) Complex of COTI-NMe2 (Cu-COTI-NMe2)
2.1.4. Synthesis of the Cu(II) Complex of COTI-NMeCy (Cu-COTI-NMeCy)
2.2. Materials and Methods for Spectrophotometric Investigations
2.3. UV-Visible Spectrophotometric Titrations
2.4. Electron Paramagnetic Resonance Measurements and Evaluation of the Spectra
2.5. Spectrophotometric Kinetic Measurements
2.6. Lipophilicity, Solubility, and Artificial Membrane Permeability Assays
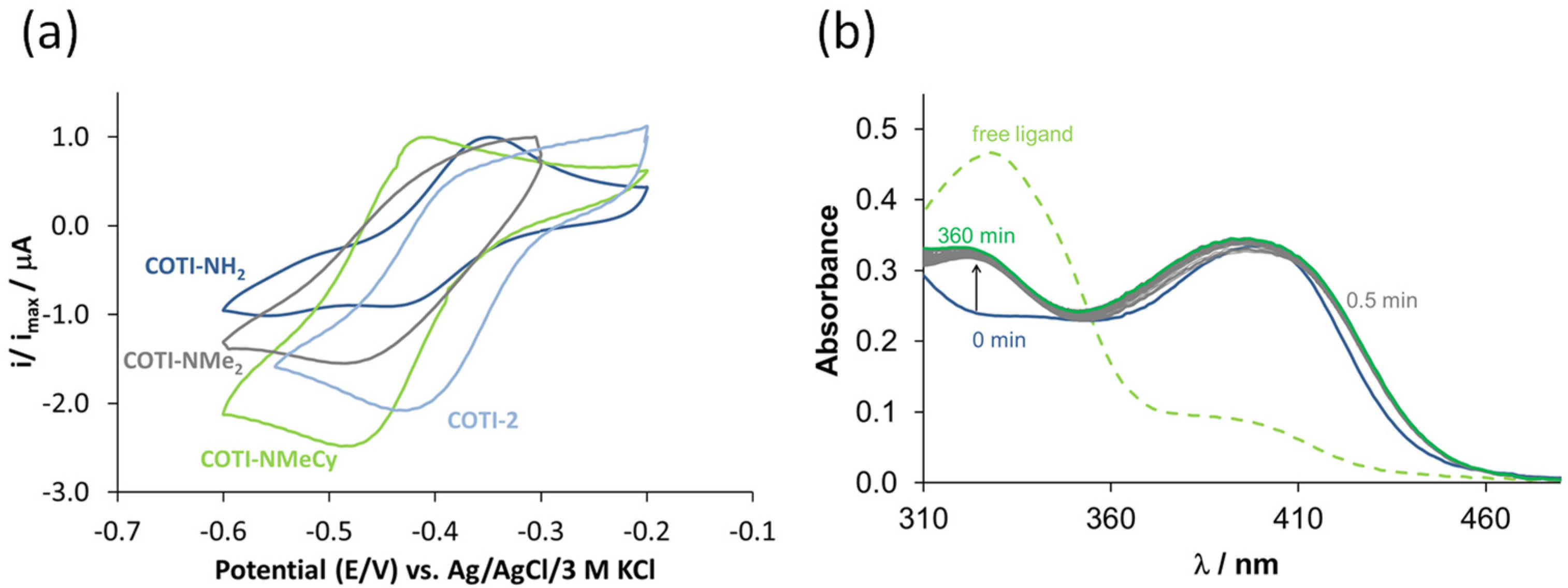
2.7. Cyclic Voltammetry
2.8. Cell Culture Studies
2.9. Viability Assays
2.10. Cell Death Analysis by Flow Cytometry
2.11. Cell Death Analysis by DAPI Staining
2.12. Cell Cycle Analysis by Flow Cytometry
2.13. Detection of Paraptotic Vesicles
2.14. HPLC-MS Cell Uptake Measurements
3. Results and Discussion
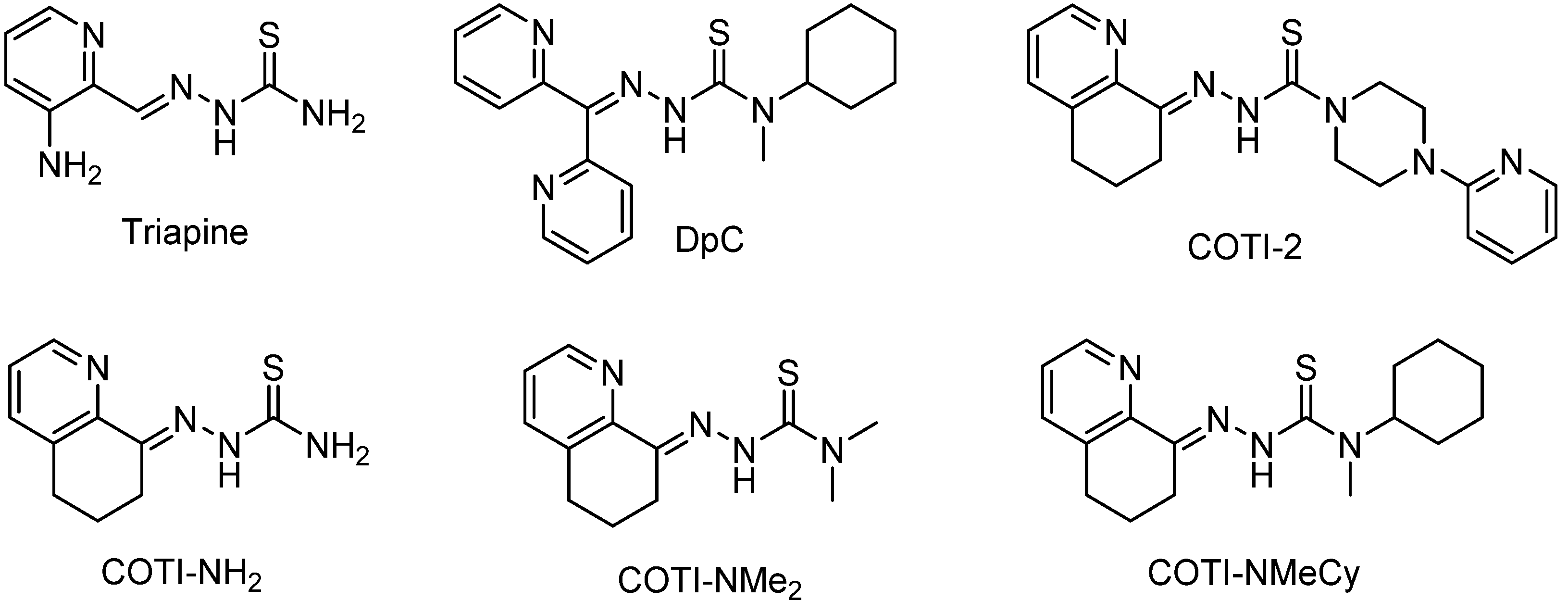
3.1. Syntheses and Chemical Characterization
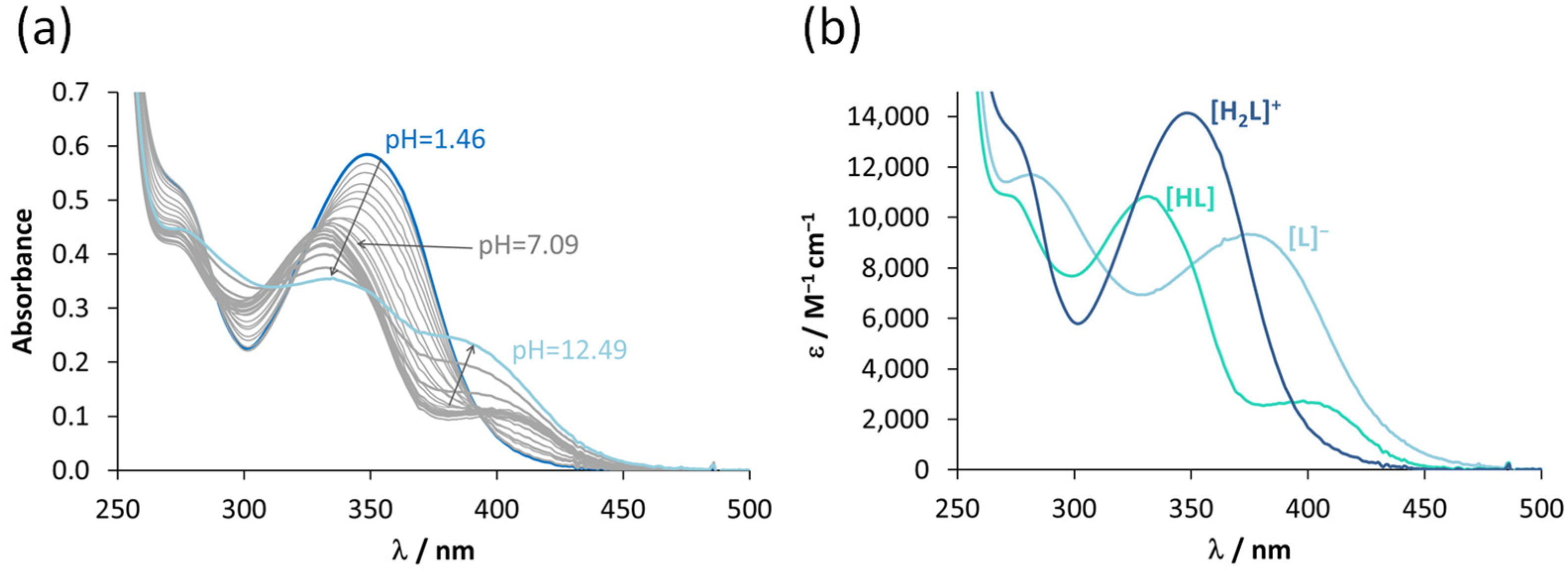
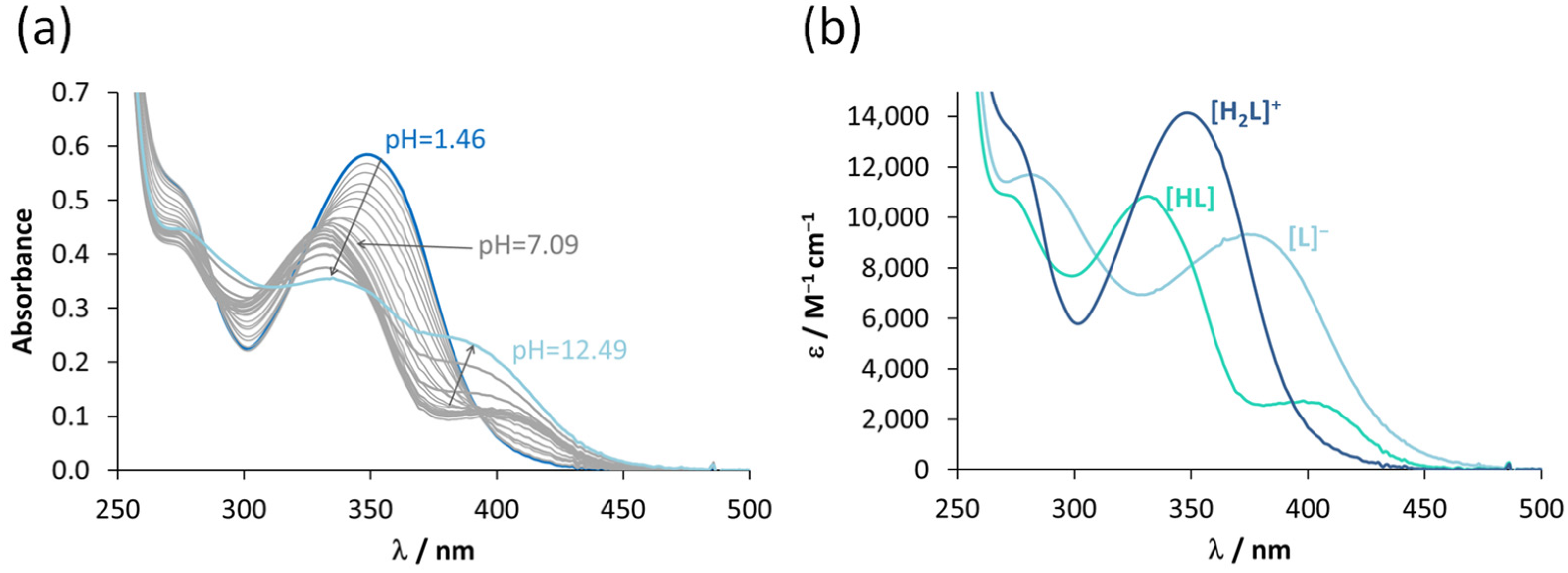
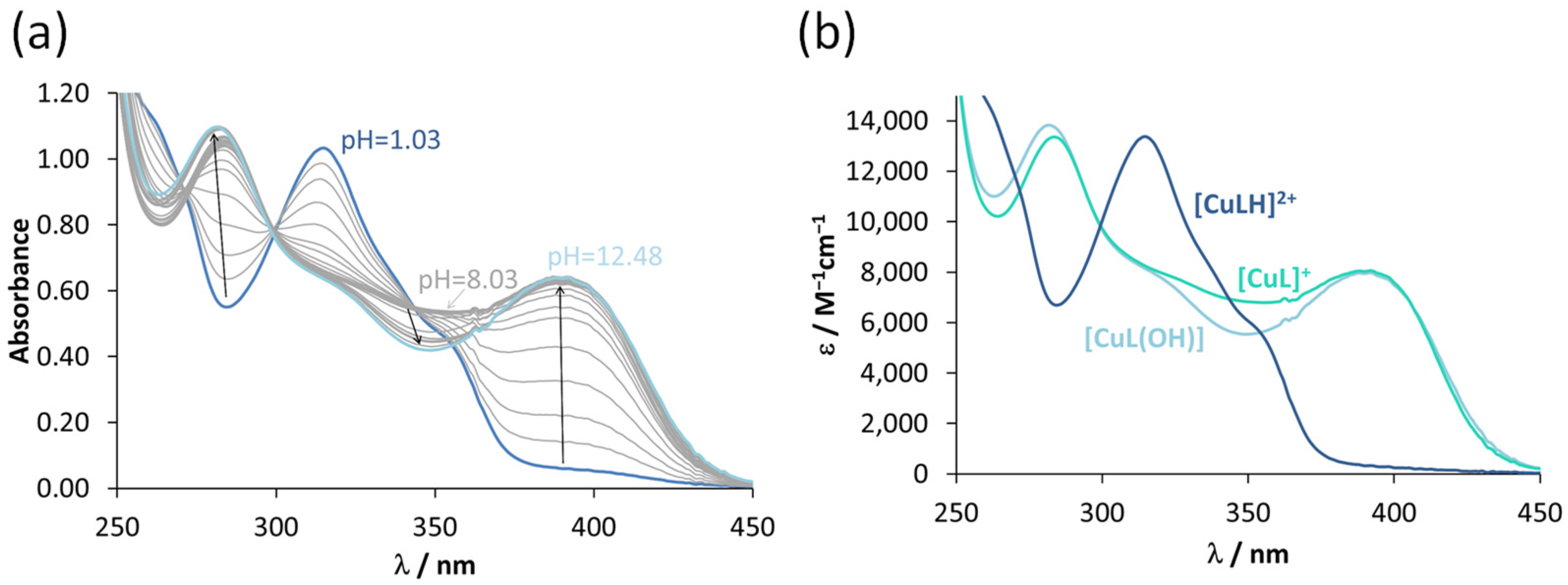
3.2. Characterization of the Solution Chemical Properties
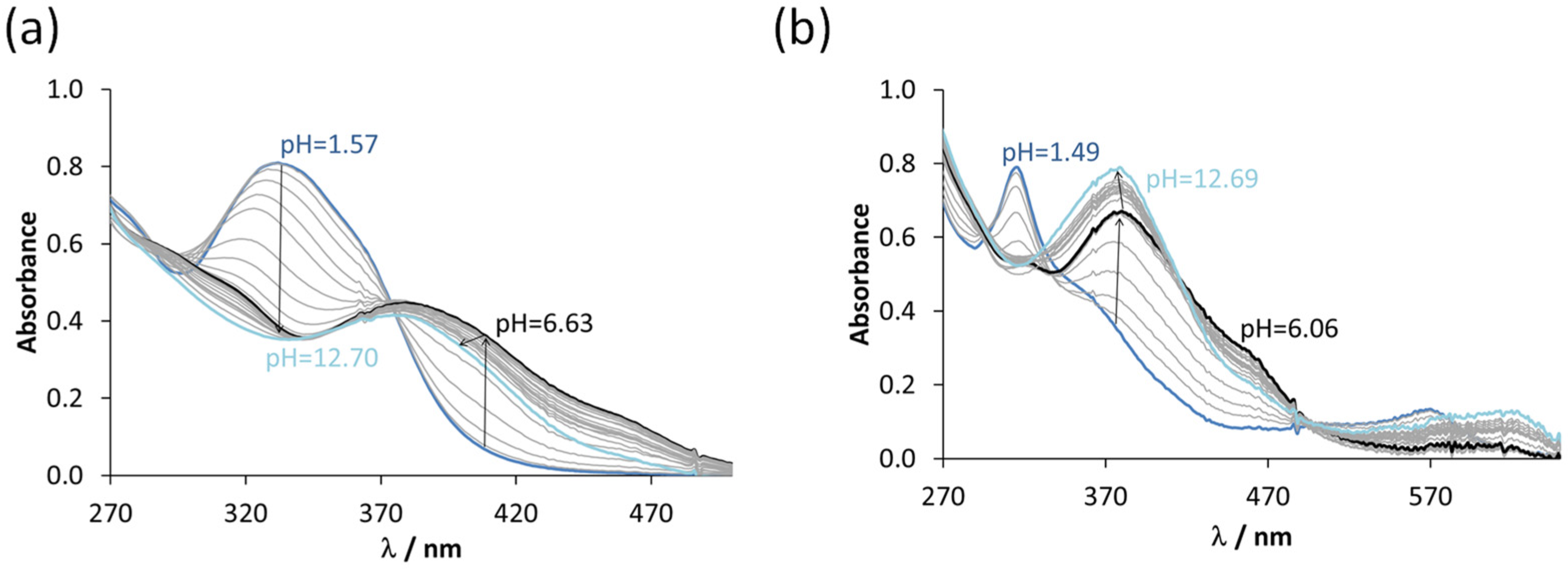
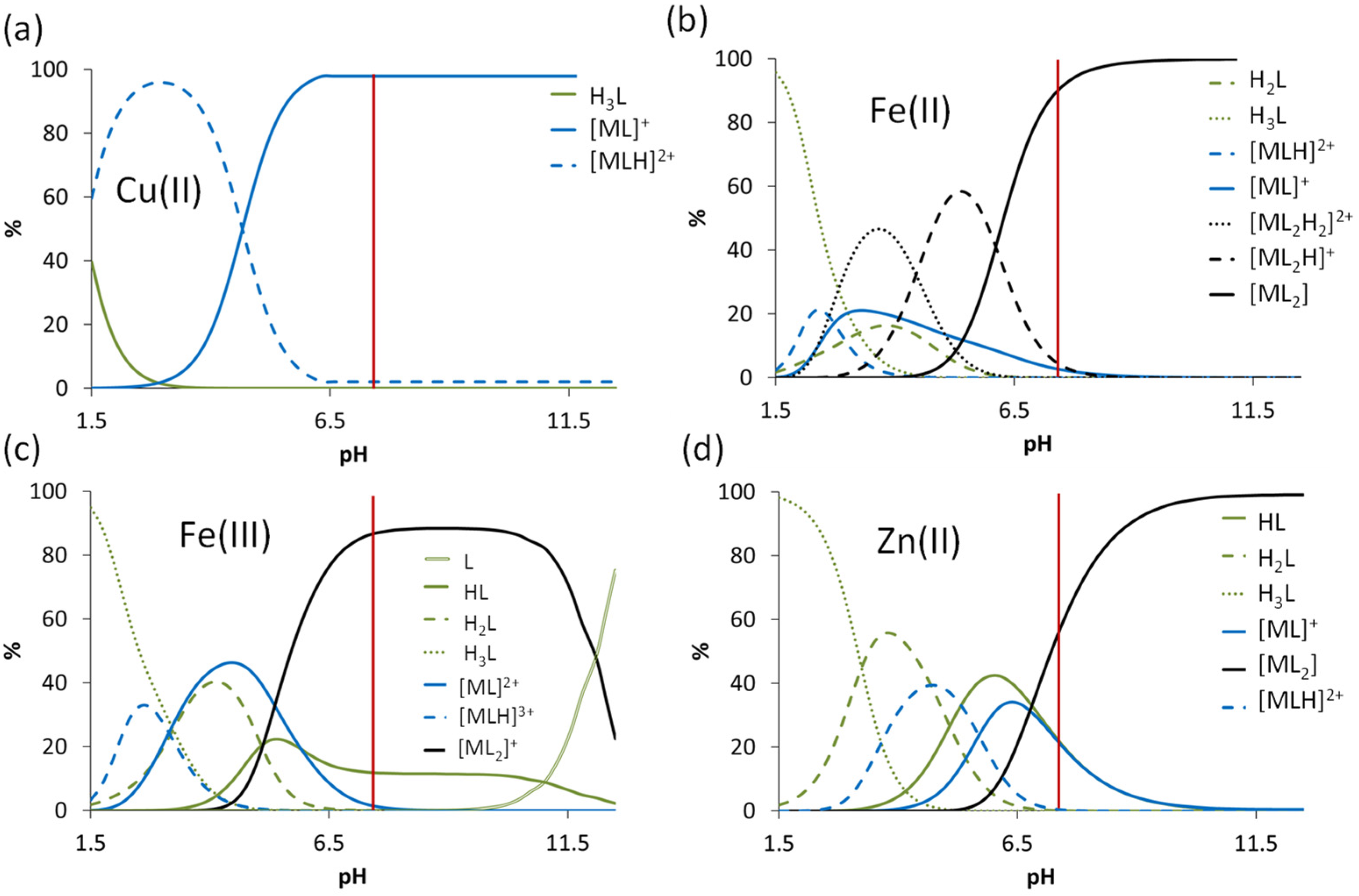
3.3. Interaction of the COTI-2 Derivatives with Iron(II) and Iron(III) Ions
3.4. Interaction of the COTI-2 Derivatives with Copper(II) and Zinc(II)
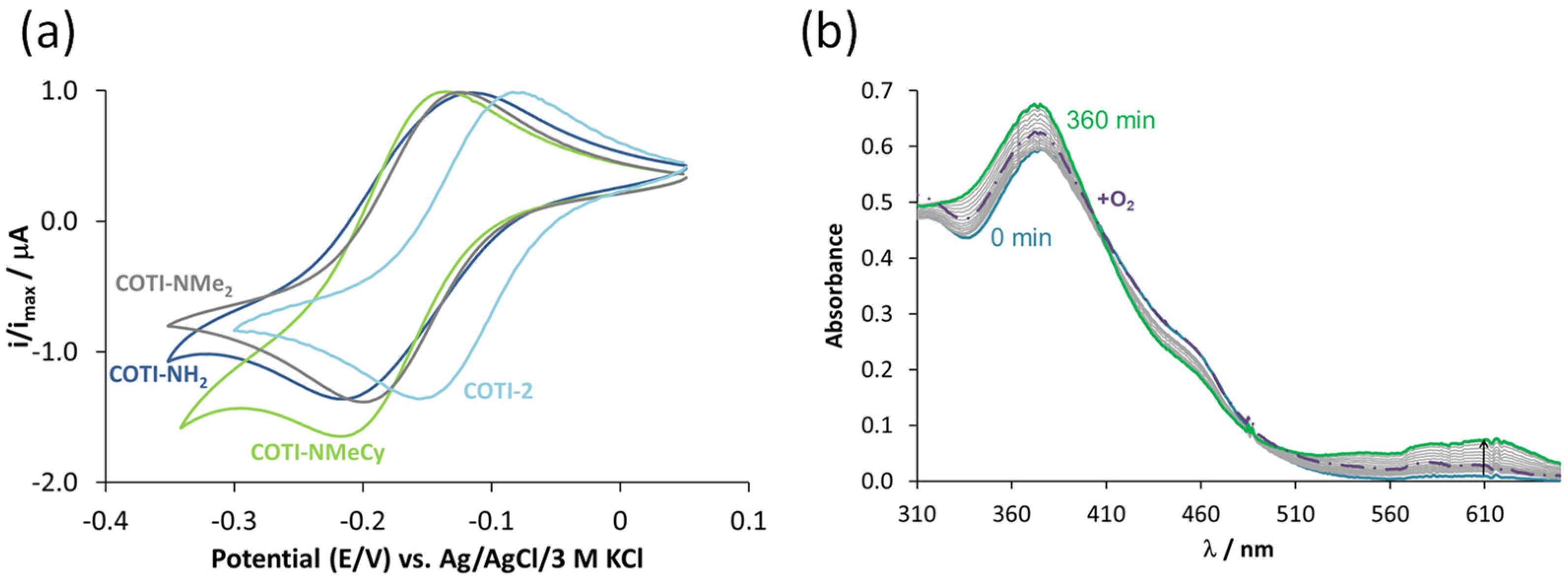
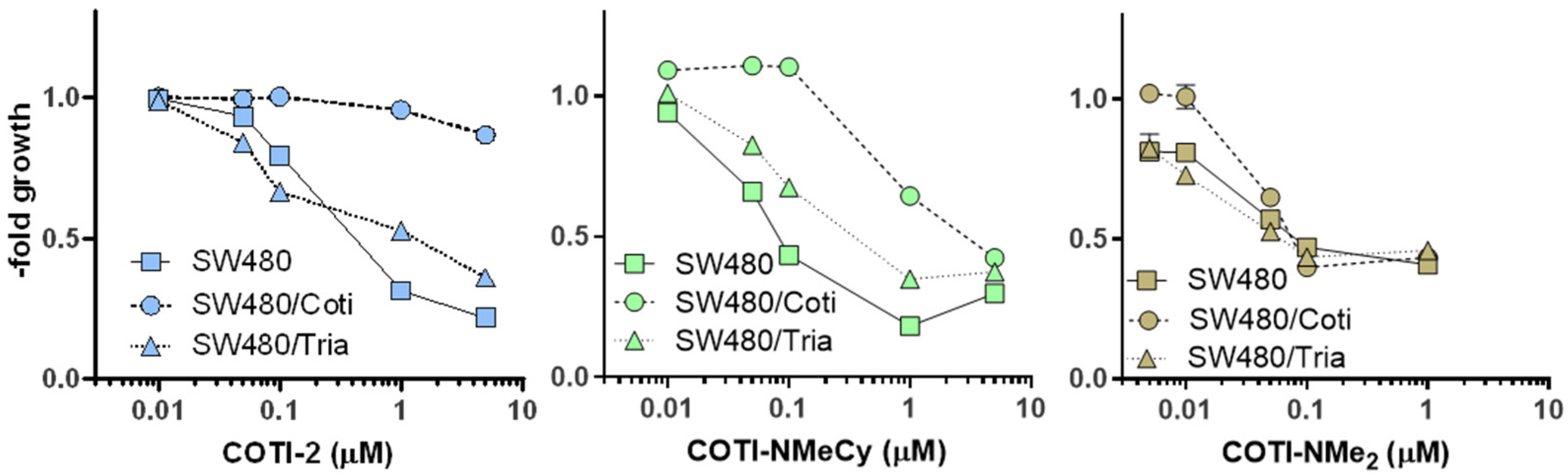
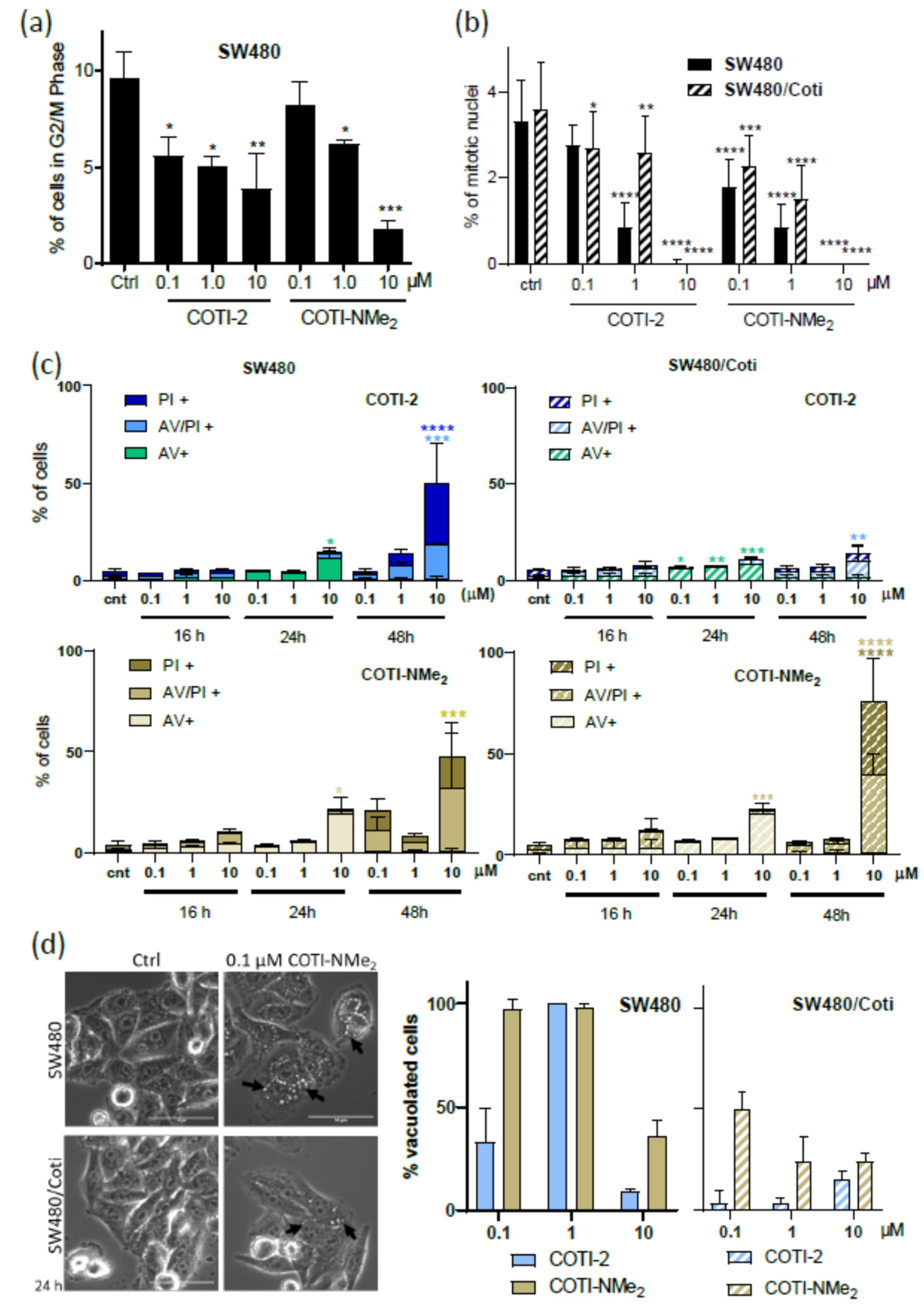
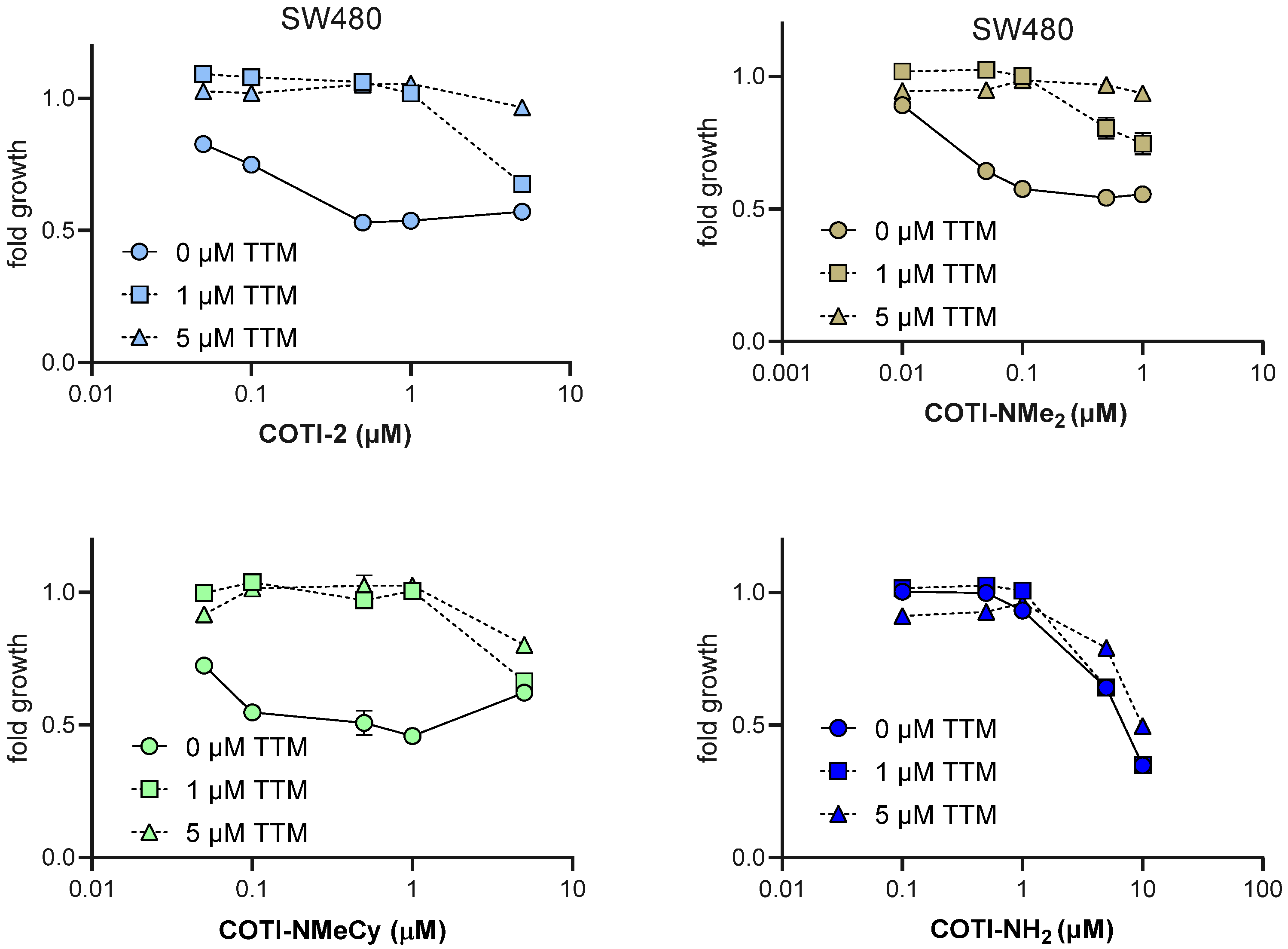
3.5. Anticancer Activity and Thiosemicarbazone Cross-Resistance Profiles of COTI-NMe2 and COTI-NMeCy
3.6. Mode of Action Studies on COTI-NMe2
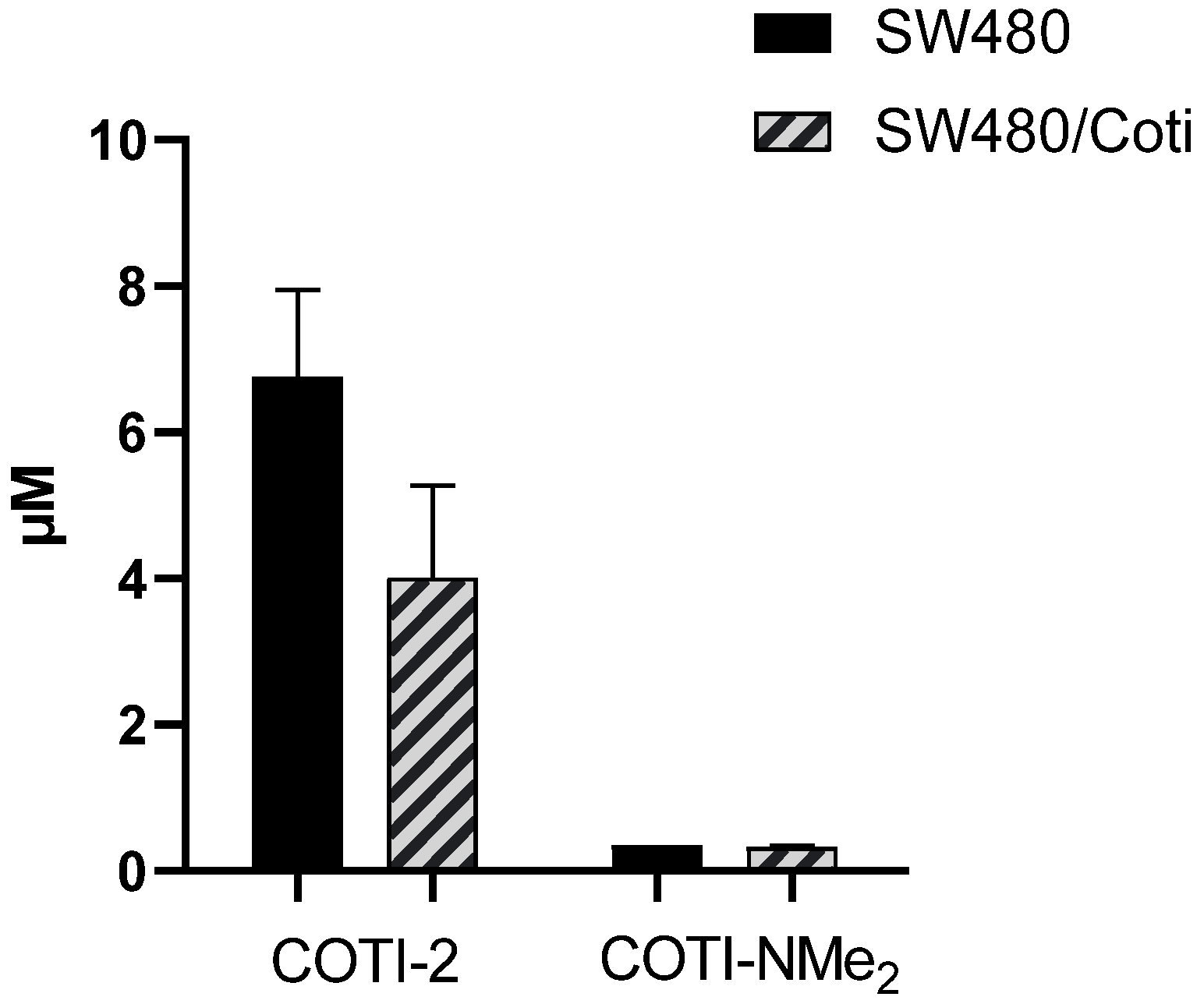
3.7. Cellular Accumulation of COTI-2 and COTI-NMe2 in SW480 and SW480/Coti Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, Y.; Gutierrez, E.; Kovacevic, Z.; Saletta, F.; Obeidy, P.; Suryo Rahmanto, Y.; Richardson, D.R. Iron chelators for the treatment of cancer. Curr. Med. Chem. 2012, 19, 2689–2702. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Mertens, C.; Tomat, E.; Brüne, B. Iron as a Central Player and Promising Target in Cancer Progression. Int. J. Mol. Sci. 2019, 20, 273. [Google Scholar] [CrossRef] [PubMed]
- Miah, A.; Harrington, K.; Nutting, C. Triapine in clinical practice. Eur. J. Clin. Med. Oncol. 2010, 2, 1. [Google Scholar]
- Heffeter, P.; Pape, V.F.S.; Enyedy, É.A.; Keppler, B.K.; Szakacs, G.; Kowol, C.R. Anticancer Thiosemicarbazones: Chemical Properties, Interaction with Iron Metabolism, and Resistance Development. Antioxid. Redox Signal. 2019, 30, 1062–1082. [Google Scholar] [CrossRef] [PubMed]
- Pelivan, K.; Frensemeier, L.; Karst, U.; Koellensperger, G.; Bielec, B.; Hager, S.; Heffeter, P.; Keppler, B.K.; Kowol, C.R. Understanding the metabolism of the anticancer drug Triapine: Electrochemical oxidation, microsomal incubation and in vivo analysis using LC-HRMS. Analyst 2017, 142, 3165–3176. [Google Scholar] [CrossRef]
- Pelivan, K.; Miklos, W.; van Schoonhoven, S.; Koellensperger, G.; Gille, L.; Berger, W.; Heffeter, P.; Kowol, C.R.; Keppler, B.K. Differences in protein binding and excretion of Triapine and its Fe(III) complex. J. Inorg. Biochem. 2016, 160, 61–69. [Google Scholar] [CrossRef]
- Kowol, C.R.; Trondl, R.; Heffeter, P.; Arion, V.B.; Jakupec, M.A.; Roller, A.; Galanski, M.; Berger, W.; Keppler, B.K. Impact of metal coordination on cytotoxicity of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (triapine) and novel insights into terminal dimethylation. J. Med. Chem. 2009, 52, 5032–5043. [Google Scholar] [CrossRef]
- Kowol, C.R.; Miklos, W.; Pfaff, S.; Hager, S.; Kallus, S.; Pelivan, K.; Kubanik, M.; Enyedy, É.A.; Berger, W.; Heffeter, P.; et al. Impact of Stepwise NH2-Methylation of Triapine on the Physicochemical Properties, Anticancer Activity, and Resistance Circumvention. J. Med. Chem. 2016, 59, 6739–6752. [Google Scholar] [CrossRef]
- Guo, Z.L.; Richardson, D.R.; Kalinowski, D.S.; Kovacevic, Z.; Tan-Un, K.C.; Chan, G.C. The novel thiosemicarbazone, di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC), inhibits neuroblastoma growth in vitro and in vivo via multiple mechanisms. J. Hematol. Oncol. 2016, 9, 98. [Google Scholar] [CrossRef]
- Seebacher, N.A.; Richardson, D.R.; Jansson, P.J. A mechanism for overcoming P-glycoprotein-mediated drug resistance: Novel combination therapy that releases stored doxorubicin from lysosomes via lysosomal permeabilization using Dp44mT or DpC. Cell Death Dis. 2016, 7, e2510. [Google Scholar] [CrossRef]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through p53-dependent and -independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Synnott, N.C.; O’Connell, D.; Crown, J.; Duffy, M.J. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res. Treat. 2020, 179, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Hager, S.; Korbula, K.; Bielec, B.; Grusch, M.; Pirker, C.; Schosserer, M.; Liendl, L.; Lang, M.; Grillari, J.; Nowikoysky, K.; et al. The thiosemicarbazone Me2NNMe2 induces paraptosis by disrupting the ER thiol redox homeostasis based on protein disulfide isomerase inhibition. Cell Death Dis. 2018, 9, 1052. [Google Scholar] [CrossRef] [PubMed]
- Hager, S.; Pape, V.F.S.; Posa, V.; Montsch, B.; Uhlik, L.; Szakacs, G.; Toth, S.; Jabronka, N.; Keppler, B.K.; Kowol, C.R.; et al. High Copper Complex Stability and Slow Reduction Kinetics as Key Parameters for Improved Activity, Paraptosis Induction, and Impact on Drug-Resistant Cells of Anticancer Thiosemicarbazones. Antioxid. Redox Signal. 2020, 33, 395–414. [Google Scholar] [CrossRef]
- Nunes, J.H.B.; Hager, S.; Mathuber, M.; Posa, V.; Roller, A.; Enyedy, E.A.; Stefanelli, A.; Berger, W.; Keppler, B.K.; Heffeter, P.; et al. Cancer Cell Resistance Against the Clinically Investigated Thiosemicarbazone COTI-2 Is Based on Formation of Intracellular Copper Complex Glutathione Adducts and ABCC1-Mediated Efflux. J. Med. Chem. 2020, 63, 13719–13732. [Google Scholar] [CrossRef]
- Bryant, B.E.; Fernelius, W.C. Formation Constants of Metal Complexes of Tropolone and its Derivatives. II. Some Alkyltropolones1,2. J. Am. Chem. Soc. 1954, 76, 1696–1697. [Google Scholar] [CrossRef]
- Zékány, L.; Nagypál, I.; Leggett, D. Computational Methods for the Determination of Stability Constants. In Computational Methods for the Determination of Stability Constants, 1st ed.; Leggett, D.J., Ed.; Modern Inorganic Chemistry; Springer US: New York, NY, USA, 1985; pp. 291–353. [Google Scholar]
- Enyedy, É.A.; Nagy, N.V.; Zsigó, É.; Kowol, C.R.; Arion, V.B.; Keppler, B.K.; Kiss, T. Comparative Solution Equilibrium Study of the Interactions of Copper(II), Iron(II) and Zinc(II) with Triapine (3-Aminopyridine-2-carbaldehyde Thiosemicarbazone) and Related Ligands. Eur. J. Inorg. Chem. 2010, 2010, 1717–1728. [Google Scholar] [CrossRef]
- Enyedy, E.A.; Primik, M.F.; Kowol, C.R.; Arion, V.B.; Kiss, T.; Keppler, B.K. Interaction of Triapine and related thiosemicarbazones with iron(III)/(II) and gallium(III): A comparative solution equilibrium study. Dalton Trans. 2011, 40, 5895–5905. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biomed. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Felcman, J.; da Silva, J.J. Complexes of oxovanadium(IV) with polyaminocarboxylic acids. Talanta 1983, 30, 565–570. [Google Scholar] [CrossRef]
- Rockenbauer, A.; Korecz, L. Automatic computer simulations of ESR spectra. Appl. Magn. Reson. 1996, 10, 29–43. [Google Scholar] [CrossRef]
- Chen, X.; Murawski, A.; Patel, K.; Crespi, C.L.; Balimane, P.V. A novel design of artificial membrane for improving the PAMPA model. Pharm. Res. 2008, 25, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Wang, Q.; Sun, Y.; Shen, M.; Li, H.; Duan, Y. A New PAMPA Model Proposed on the Basis of a Synthetic Phospholipid Membrane. PLoS ONE 2015, 10, e0116502. [Google Scholar] [CrossRef]
- Kowol, C.R.; Eichinger, R.; Jakupec, M.A.; Galanski, M.; Arion, V.B.; Keppler, B.K. Effect of metal ion complexation and chalcogen donor identity on the antiproliferative activity of 2-acetylpyridine N,N-dimethyl(chalcogen)semicarbazones. J. Inorg. Biochem. 2007, 101, 1946–1957. [Google Scholar] [CrossRef]
- Pessôa, M.M.B.; Andrade, G.F.S.; Paoli Monteiro, V.R.; Temperini, M.L.A. 2-Formylpyridinethiosemicarbazone and methyl derivatives: Spectroscopic studies. Polyhedron 2001, 20, 3133–3141. [Google Scholar] [CrossRef]
- Enyedy, E.A.; May, N.V.; Pape, V.F.S.; Heffeter, P.; Szakacs, G.; Keppler, B.K.; Kowol, C.R. Complex formation and cytotoxicity of Triapine derivatives: A comparative solution study on the effect of the chalcogen atom and NH-methylation. Dalton Trans. 2020, 49, 16887–16902. [Google Scholar] [CrossRef]
- Enyedy, É.A.; Zsigó, É.; Nagy, N.V.; Kowol, C.R.; Roller, A.; Keppler, B.K.; Kiss, T. Complex-Formation Ability of Salicylaldehyde Thiosemicarbazone towards ZnII, CuII, FeII, FeIII and GaIII Ions. Eur. J. Inorg. Chem. 2012, 2012, 4036–4047. [Google Scholar] [CrossRef]
- Santoro, A.; Calvo, J.S.; Peris-Díaz, M.D.; Krężel, A.; Meloni, G.; Faller, P. The Glutathione/Metallothionein System Challenges the Design of Efficient O(2) -Activating Copper Complexes. Angew. Chem. Int. Ed. 2020, 59, 7830–7835. [Google Scholar] [CrossRef]
- Yu, X.; Blanden, A.R.; Narayanan, S.; Jayakumar, L.; Lubin, D.; Augeri, D.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Small molecule restoration of wildtype structure and function of mutant p53 using a novel zinc-metallochaperone based mechanism. Oncotarget 2014, 5, 8879–8892. [Google Scholar] [CrossRef]
- Yu, X.; Blanden, A.; Tsang, A.T.; Zaman, S.; Liu, Y.; Gilleran, J.; Bencivenga, A.F.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Thiosemicarbazones Functioning as Zinc Metallochaperones to Reactivate Mutant p53. Mol. Pharm. 2017, 91, 567–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanden, A.R.; Yu, X.; Wolfe, A.J.; Gilleran, J.A.; Augeri, D.J.; O’Dell, R.S.; Olson, E.C.; Kimball, S.D.; Emge, T.J.; Movileanu, L.; et al. Synthetic metallochaperone ZMC1 rescues mutant p53 conformation by transporting zinc into cells as an ionophore. Mol. Pharm. 2015, 87, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Miklos, W.; Heffeter, P.; Pirker, C.; Hager, S.; Kowol, C.R.; Schoonhoven, S.V.; Stojanovic, M.; Keppler, B.K.; Berger, W. Loss of phosphodiesterase 4D mediates acquired triapine resistance via Epac-Rap1-Integrin signaling. Oncotarget 2016, 7, 84556. [Google Scholar] [CrossRef] [PubMed]
- Miklos, W.; Pelivan, K.; Kowol, C.R.; Pirker, C.; Dornetshuber-Fleiss, R.; Spitzwieser, M.; Englinger, B.; van Schoonhoven, S.; Cichna-Markl, M.; Koellensperger, G.; et al. Triapine-mediated ABCB1 induction via PKC induces widespread therapy unresponsiveness but is not underlying acquired triapine resistance. Cancer Lett. 2015, 361, 112–120. [Google Scholar] [CrossRef]
- Heffeter, P.; Pirker, C.; Kowol, C.R.; Herrman, G.; Dornetshuber, R.; Miklos, W.; Jungwirth, U.; Koellensperger, G.; Keppler, B.K.; Berger, W. Impact of terminal dimethylation on the resistance profile of alpha-N-heterocyclic thiosemicarbazones. Biochem. Pharm. 2012, 83, 1623–1633. [Google Scholar] [CrossRef]
- Kowol, C.R.; Berger, R.; Eichinger, R.; Roller, A.; Jakupec, M.A.; Schmidt, P.P.; Arion, V.B.; Keppler, B.K. Gallium(III) and iron(III) complexes of alpha-N-heterocyclic thiosemicarbazones: Synthesis, characterization, cytotoxicity, and interaction with ribonucleotide reductase. J. Med. Chem. 2007, 50, 1254–1265. [Google Scholar] [CrossRef]
- Sestak, V.; Stariat, J.; Cermanova, J.; Potuckova, E.; Chladek, J.; Roh, J.; Bures, J.; Jansova, H.; Prusa, P.; Sterba, M.; et al. Novel and potent anti-tumor and anti-metastatic di-2-pyridylketone thiosemicarbazones demonstrate marked differences in pharmacology between the first and second generation lead agents. Oncotarget 2015, 6, 42411–42428. [Google Scholar] [CrossRef]
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Di Domizio, A.; Limonta, P. The emerging role of paraptosis in tumor cell biology: Perspectives for cancer prevention and therapy with natural compounds. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188338. [Google Scholar] [CrossRef]
- Wang, Y.; Wen, X.; Zhang, N.; Wang, L.; Hao, D.; Jiang, X.; He, G. Small-molecule compounds target paraptosis to improve cancer therapy. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 118, 109203. [Google Scholar] [CrossRef]
- Jansson, P.J.; Yamagishi, T.; Arvind, A.; Seebacher, N.; Gutierrez, E.; Stacy, A.; Maleki, S.; Sharp, D.; Sahni, S.; Richardson, D.R. Di-2-pyridylketone 4,4-Dimethyl-3-thiosemicarbazone (Dp44mT) Overcomes Multidrug Resistance by a Novel Mechanism Involving the Hijacking of Lysosomal P-Glycoprotein (Pgp)*. J. Biol. Chem. 2015, 290, 9588–9603. [Google Scholar] [CrossRef]
- Shakya, B.; Yadav, P.N. Thiosemicarbazones as Potent Anticancer Agents and their Modes of Action. Mini Rev. Med. Chem. 2020, 20, 638–661. [Google Scholar] [CrossRef] [PubMed]
- Matesanz, A.I.; Herrero, J.M.; Quiroga, A.G. Chemical and Biological Evaluation of Thiosemicarbazone-Bearing Heterocyclic Metal Complexes. Curr. Top. Med. Chem. 2021, 21, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Merlot, A.M.; Kalinowski, D.S.; Kovacevic, Z.; Jansson, P.J.; Sahni, S.; Huang, M.L.; Lane, D.J.R.; Lok, H.; Richardson, D.R. Exploiting Cancer Metal Metabolism using Anti-Cancer Metal- Binding Agents. Curr. Med. Chem. 2019, 26, 302–322. [Google Scholar] [CrossRef]
- Babak, M.V.; Ahn, D. Modulation of Intracellular Copper Levels as the Mechanism of Action of Anticancer Copper Complexes: Clinical Relevance. Biomedicines 2021, 9, 852. [Google Scholar] [CrossRef]
- de Siqueira, L.R.P.; de Moraes Gomes, P.A.T.; de Lima Ferreira, L.P.; de Melo Rego, M.J.B.; Leite, A.C.L. Multi-target compounds acting in cancer progression: Focus on thiosemicarbazone, thiazole and thiazolidinone analogues. Eur. J. Med. Chem. 2019, 170, 237–260. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T. NUMABS, Numerical Absorption Correction 2002; Rigaku Corporation: Tokyo, Japan, 2002. [Google Scholar]
- CrystalClear, S.M. 1.4. 0; Rigaku/MSC Inc.: The Woodlands, TX, USA, 2008.
- Sheldrick, G.M. ShelXL2013; University of Göttingen: Göttingen, Germany, 2013. [Google Scholar]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L.J. Single-Crystal Structure Validation with the Program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Streek, J.V.D. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Allen, F.H.; Johnson, O.; Shields, G.P.; Smith, B.R.; Towler, M. CIF Applications. XV. EnCIFer: A Program for Viewing, Editing and Visualizing CIFs. J. Appl. Crystallogr. 2004, 37, 335–338. [Google Scholar] [CrossRef]
- Heffeter, P.; Popovic-Bijelic, A.; Saiko, P.; Dornetshuber, R.; Jungwirth, U.; Voevodskaya, N.; Biglino, D.; Jakupec, M.A.; Elbling, L.; Micksche, M.; et al. Ribonucleotide reductase as one important target of [Tris(1,10-phenanthroline)lanthanum(III)] trithiocyanate (KP772). Curr. Cancer Drug Targets 2009, 9, 595–607. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
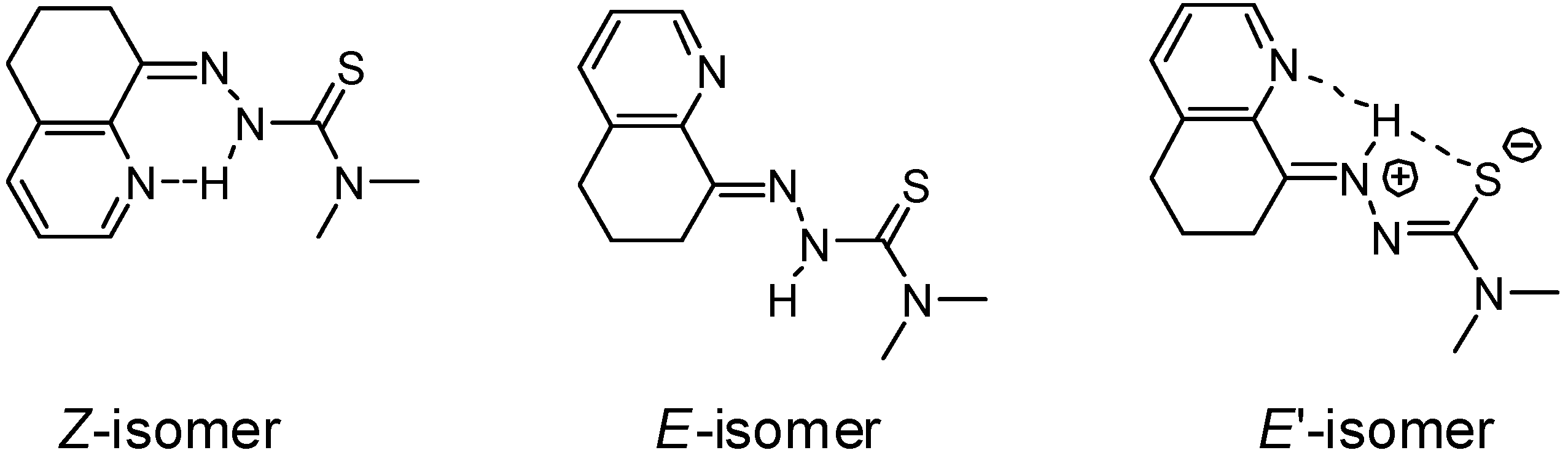{kind=link}
| COTI-2 | COTI-NH2 | COTI-NMe2 | COTI-NMeCy | |
|---|---|---|---|---|
| pKa (H3L a) | 3.23 ± 0.01 | – | – | – |
| pKa (H2L a) | 5.08 ± 0.01 | 4.39 ± 0.01 | 3.65 ± 0.01 | 3.47 ± 0.01 |
| pKa (HL a) | 10.97 ± 0.01 | 12.22 ± 0.01 | 12.09 ± 0.01 | 12.40 ± 0.01 |
| S (calc., μM) b | 5.6 | 954 | 324 | 31 |
| S (meas., μM) | 0.41 | 737 | 417 | 3.9 |
| log D7.4 (calc.) c | +2.99 | +1.43 | +1.88 | +3.68 |
| log D7.4 (meas.) | – | +1.64 ± 0.01 | – | – |
| Peff (×10−6 cm s−1) | – | 6.7 ± 0.7 | 16.0 ± 0.1 | – |
| COTI-2 | COTI-NH2 | COTI-NMe2 | COTI-NMeCy | |
|---|---|---|---|---|
| logβ [Fe(III)LH]3+ | 21.16 ± 0.03 | – | – | – |
| logβ [Fe(III)L]2+ | 18.02 ± 0.03 | 20.00 ± 0.09 | 17.25 ± 0.02 | 17.06 ± 0.02 |
| logβ [Fe(III)L2]+ | 29.98 ± 0.06 | 32.29 ± 0.08 | 32.77 ± 0.03 | 32.20 ± 0.03 |
| logβ [Fe(II)LH]2+ | 21.00 ± 0.03 | – | – | – |
| logβ [Fe(II)L]+ | 18.28 ± 0.06 | 17.36 ± 0.06 | 17.62 ± 0.03 | 18.66 ± 0.03 |
| logβ [Fe(II)L2H2]2+ | 41.18 ± 0.06 | – | – | – |
| logβ [Fe(II)L2H]+ | 36.74 ± 0.09 | – | – | – |
| logβ [Fe(II)L2] | 30.63 ± 0.09 | 31.21 ± 0.09 | 31.33 ± 0.09 | 31.04 ± 0.03 |
| pFe(III) | 18.6 | 18.5 | 19.2 | 18.0 |
| pFe(III)* | 10.6 | 10.4 | 11.1 | 9.9 |
| pFe(II) | 19.3 | 17.4 | 17.8 | 16.9 |
| E’ vs. NHE (V) | +0.11 | +0.06 | +0.06 | +0.05 |
| COTI-2 | COTI-NH2 | COTI-NMe2 | COTI-NMeCy | |
|---|---|---|---|---|
| pKa[CuLH]2+ | 4.66 ± 0.01 | 2.24 ± 0.03 | 1.40 ± 0.02 | 1.26 ± 0.01 |
| pKa[CuL]+ | – | 8.84 ± 0.04 | >8 | >8 |
| logβ5.9′ [CuL]+ a | 13.06 ± 0.02 | 10.59 ± 0.08 | 12.23 ± 0.05 | 12.56 ± 0.09 |
| logβ [CuL]+ b | 18.19 | 16.92 | 18.42 | 19.08 |
| pCu | 15.6 | 13.1 | 14.7 | 15.0 |
| E′ vs. NHE (V) | −0.19 | −0.17 | −0.24 | −0.22 |
| logβ [ZnLH]2+ | 18.54 ± 0.02 | – | – | – |
| logβ [ZnL]+ | 12.85 ± 0.02 | 14.07 ± 0.04 | 13.02 ± 0.04 | 12.94 ± 0.04 |
| logβ [ZnL2] | 23.23 ± 0.08 | 25.42 ± 0.04 | 25.24 ± 0.08 | 26.32 ± 0.09 |
| pZn | 11.9 | 11.6 | 11.7 | 12.1 |
| Compound | SW480 (IC50, µM) a | SW480/Coti (IC50, µM) a | Relative Resistance b | SW480/Tria (IC50, µM) a | Relative Resistance b |
|---|---|---|---|---|---|
| COTI-2 | 0.37 ± 0.17 | 9.51 ± 3.11c | 25.7 *** | 0.41 ± 0.16 c | 1.1 n.s. |
| COTI-NMe2 | 0.04 ± 0.01 | 0.05 ± 0.03 | 1.3 n.s. | 0.04 ± 0.02 | 1.0 n.s. |
| COTI-NMeCy | 0.64 ± 0.10 | 3.98 ± 0.40 | 6.2 ** | 0.71 ± 0.17 | 1.1 n.s. |
| COTI-NH2 c | 7.85 ± 0.92 | 4.39 ± 0.56 | 0.6 *** | >20 | >2.5 |
| Cu-COTI-2 | 0.05 ± 0.02 | 0.46 ± 0.09 | 9.2 ** | n.d. | - |
| Cu-COTI-NMe2 | 0.01 ± 0.01 | 0.03 ± 0.04 | 3.0 n.s. | n.d. | - |
| Cu-COTI-NMeCy | 0.080 ± 0.003 | 0.31 ± 0.03 | 3.9 ** | n.d. | - |
| Zn-COTI-2 | 0.420 ± 0.009 | >10 | >23.4 | n.d. | |
| Triapine c | 0.82 ± 0.18 | 0.81 ± 0.08 | 1.0 n.s. | >50 | >61 |
| Compound | GLC-4 (IC50, µM) a | GLC-4/adr (IC50, µM) a | Relative Resistance b |
|---|---|---|---|
| COTI-2 c | 0.03 ± 0.01 | 0.48 ± 0.15 | 17.3 * |
| COTI-NMe2 | 0.03 ± 0.03 | 0.06 ± 0.03 | 2.0 n.s. |
| COTI-NMeCy | 0.17 ± 0.10 | 0.54 ± 0.15 | 7.8 * |
| COTI-NH2 c | 3.64 ± 0.39 | 3.65 ± 0.41 | 1.0 n.s. |
| Triapine c | 0.65 ± 0.09 | 0.57 ± 0.05 | 0.9 n.s. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pósa, V.; Stefanelli, A.; Nunes, J.H.B.; Hager, S.; Mathuber, M.; May, N.V.; Berger, W.; Keppler, B.K.; Kowol, C.R.; Enyedy, É.A.; et al. Thiosemicarbazone Derivatives Developed to Overcome COTI-2 Resistance. Cancers 2022, 14, 4455. https://doi.org/10.3390/cancers14184455
Pósa V, Stefanelli A, Nunes JHB, Hager S, Mathuber M, May NV, Berger W, Keppler BK, Kowol CR, Enyedy ÉA, et al. Thiosemicarbazone Derivatives Developed to Overcome COTI-2 Resistance. Cancers. 2022; 14(18):4455. https://doi.org/10.3390/cancers14184455
Chicago/Turabian StylePósa, Vivien, Alessia Stefanelli, Julia H. Bormio Nunes, Sonja Hager, Marlene Mathuber, Nóra V. May, Walter Berger, Bernhard K. Keppler, Christian R. Kowol, Éva A. Enyedy, and et al. 2022. "Thiosemicarbazone Derivatives Developed to Overcome COTI-2 Resistance" Cancers 14, no. 18: 4455. https://doi.org/10.3390/cancers14184455
APA StylePósa, V., Stefanelli, A., Nunes, J. H. B., Hager, S., Mathuber, M., May, N. V., Berger, W., Keppler, B. K., Kowol, C. R., Enyedy, É. A., & Heffeter, P. (2022). Thiosemicarbazone Derivatives Developed to Overcome COTI-2 Resistance. Cancers, 14(18), 4455. https://doi.org/10.3390/cancers14184455