


Evaluating Stacked Methylation Markers for Blood-Based Multicancer Detection
 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. The Samples, Datasets, and Preprocessing
2.2. Probe Identification
2.3. Sensitivity, Specificity, and Sample Classification in TCGA Data
2.4. Blood Sample Simulations
2.5. Calculation of Positive Predictive Value
2.6. PCR Assay and Sequencing
- ZNF154, at genomic position Chr19: 58220404-58220705 (+strand)
- Forward primer: 5′-GGTTTTTATTTTAGGTTTGA-3′
- Reverse primer: 5′-AAATCTATAAAAACTACATTACCTAAAATACTCTA-3′
- Amplicon size (incl. primers) 302 bp (20 CpGs)
- TLX1, at genomic position Chr10: 102894992-102895165 (+strand)
- Forward primer: 5′-TTTTTAGTTTAGGTTTTATGGGGTAG-3′
- Reverse primer: 5′-AAAACCATAACTTCCTTTATAACCC-3′
- Amplicon size (incl. primers) 174 bp (13 CpGs)
- GALR1 at genomic position Chr18: 74961979-74962242 (+strand)
- Forward primer: 5′-GGGAGTTTTTTTTGTAGGAGT-3′
- Reverse primer: 5′-AAAACACTAAAATCCCCTTCC-3′
- Amplicon size (incl. primers) 264 bp (27 CpGs)
- -
- Forward adapter: 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′,
- -
- Reverse adapter: 5′-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′.
2.7. EpiClass Procedure to Assess Marker Performance in Plasma Samples
3. Results
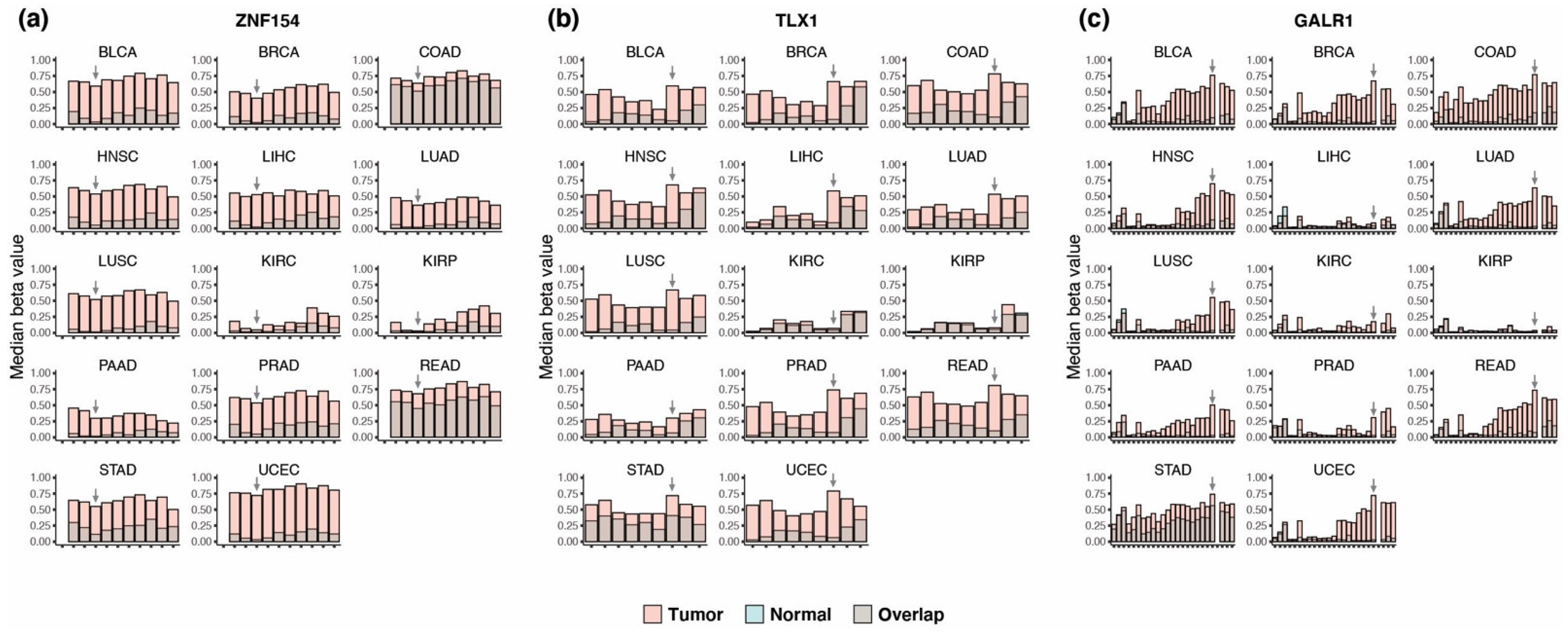
3.1. Discovery of Multi-Cancer Methylation Biomarkers in TCGA Data
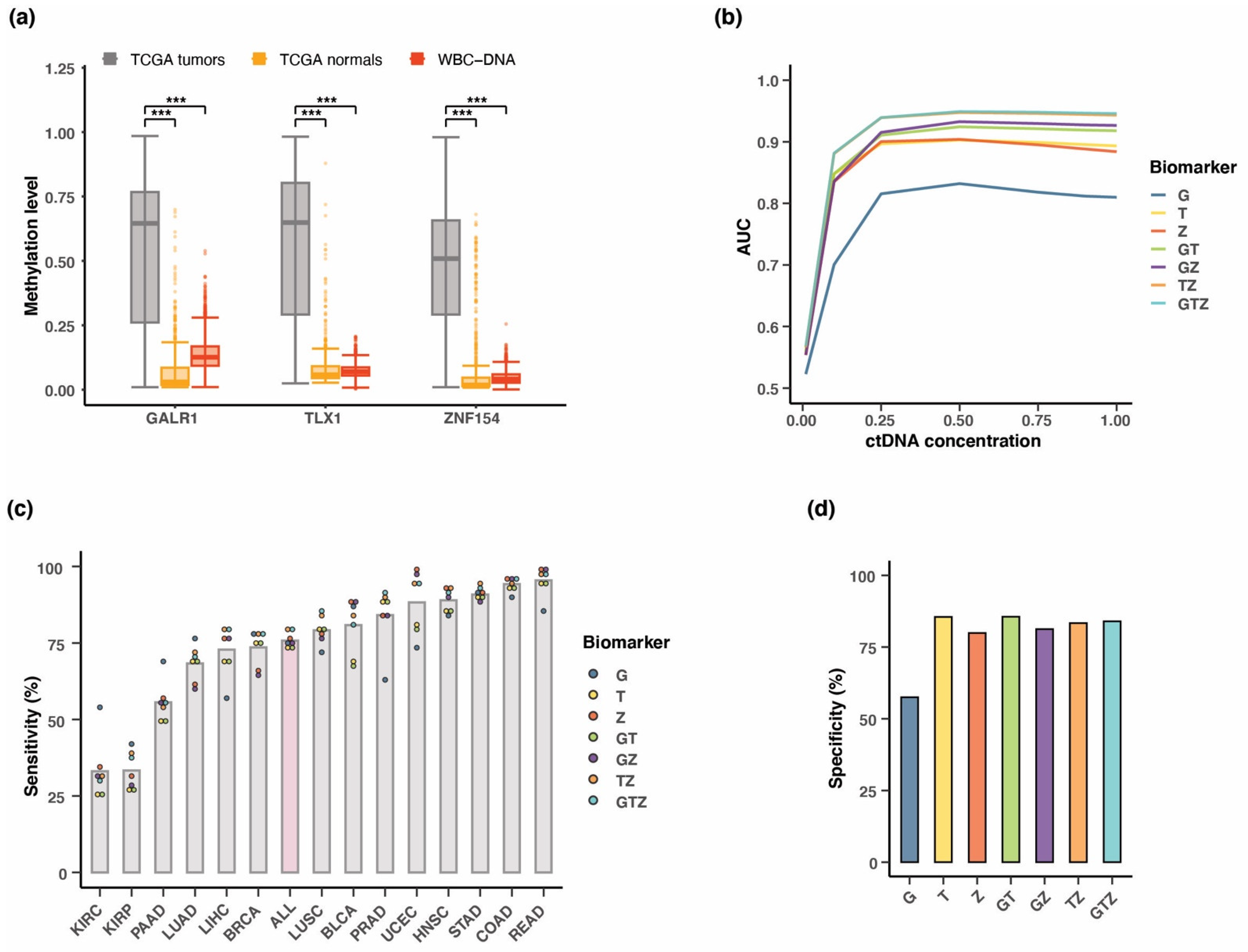
3.2. Methylation at TLX1, GALR1, and ZNF154 in Tumor and Normal Karyotype Cell Lines
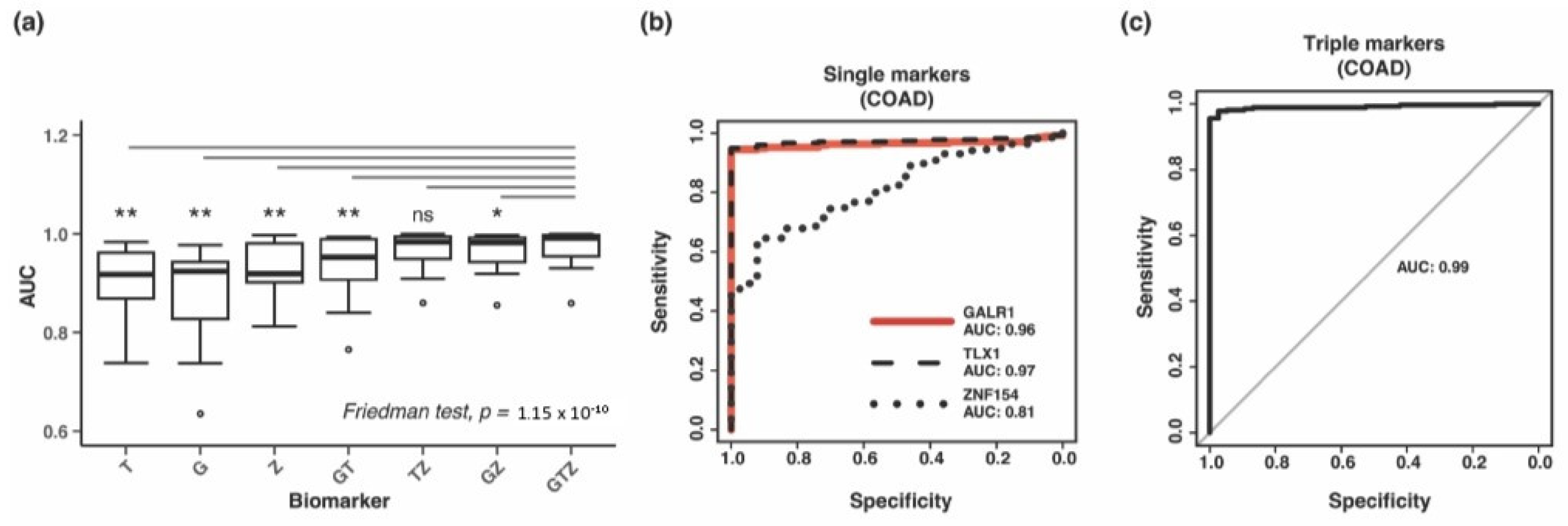
3.3. Performance of the Three Biomarkers Individually
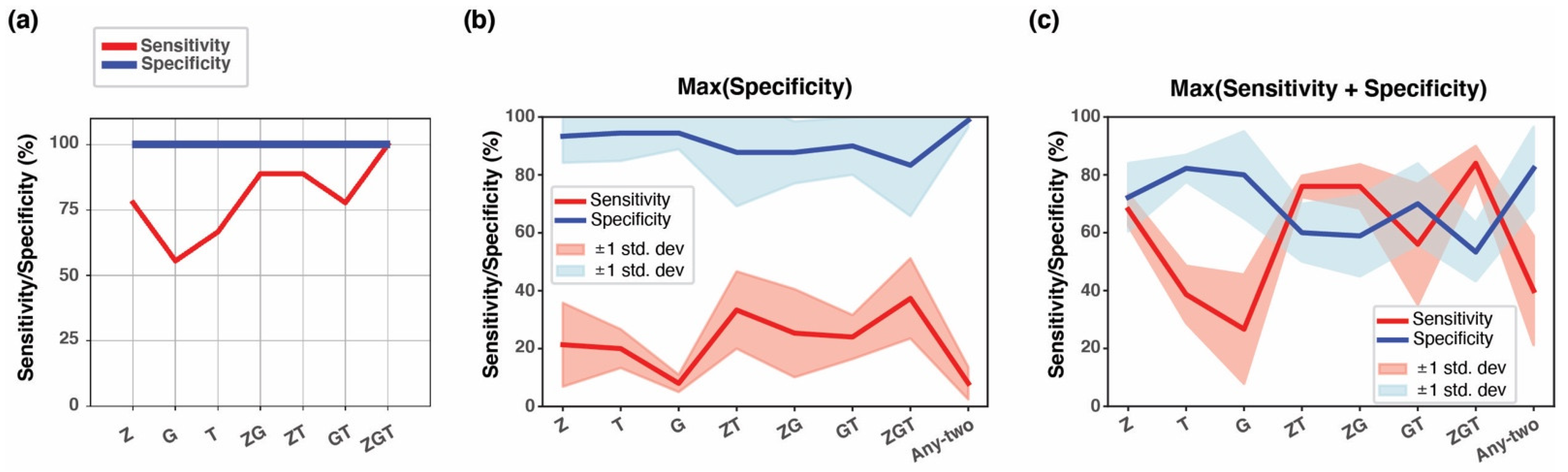
3.4. Combining Methylation Biomarkers into Multi-Marker Assays
3.5. Validation of Three-Marker Combination in Independent Tumor Datasets
3.6. Methylation at TLX1, GALR1, and ZNF154 in Early-Stage Tumors
3.7. Testing Multi-Cancer Assay Performance in Simulated Blood Samples
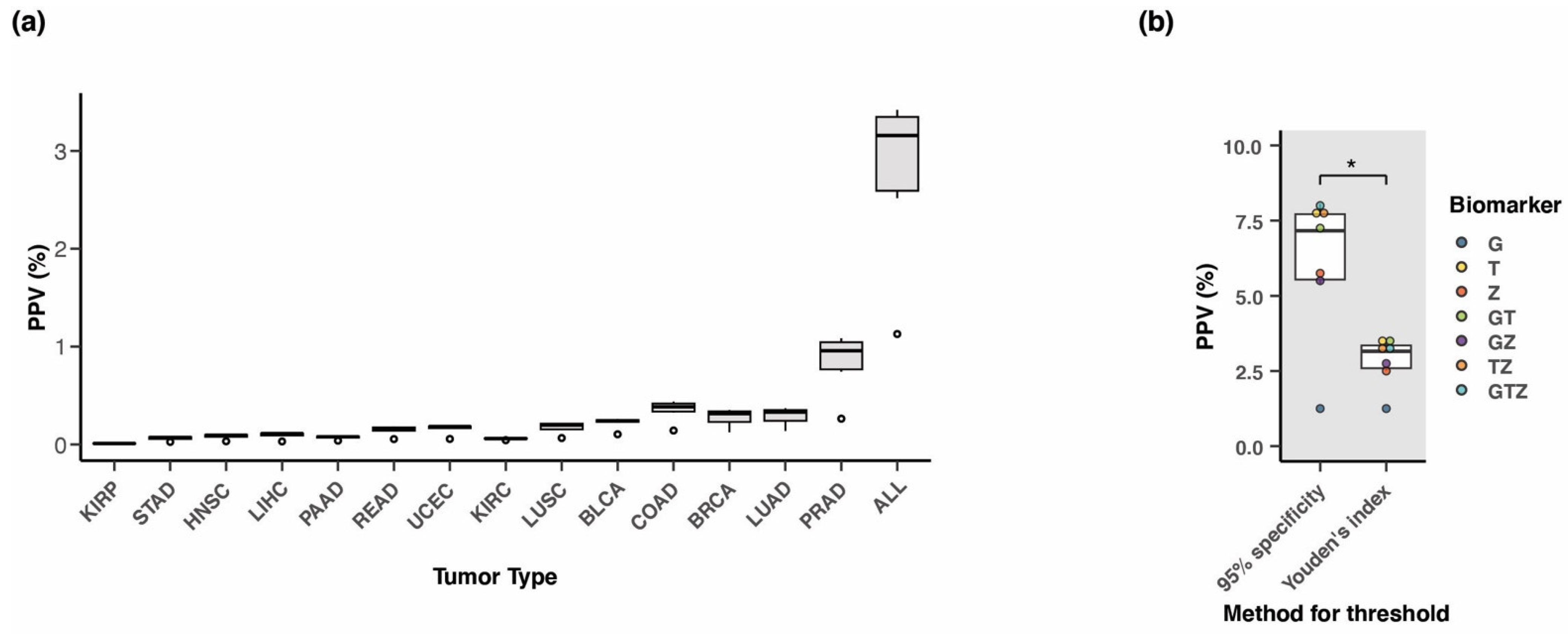
3.8. Positive Predictive Value of the Three-Marker Combination
3.9. Performance of Three-Marker Combination in Cancer Tissue
3.10. Testing of the Three Biomarkers in Patient Plasma Using Whole Genome Bisulfite Sequencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BLCA | bladder urothelial carcinoma; |
| BRCA | breast invasive carcinoma; |
| COAD | colon adenocarcinoma; |
| HNSC | head and neck squamous cell carcinoma; |
| KIRC | kidney renal clear cell carcinoma; |
| KIRP | kidney renal papillary cell carcinoma; |
| LIHC | liver hepatocellular carcinoma; |
| LUAD | lung adenocarcinoma; LUSC, lung squamous cell carcinoma; |
| PAAD | pancreatic adenocarcinoma; |
| PRAD | prostate adenocarcinoma; |
| READ | rectum adenocarcinoma; |
| STAD | stomach adenocarcinoma; |
| UCEC | uterine corpus endometrioid carcinoma. |
References
- Chen, R.C.; Haynes, K.; Du, S.; Barron, J.; Katz, A.J. Association of Cancer Screening Deficit in the United States with the COVID-19 Pandemic. JAMA Oncol. 2021, 7, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Guven, D.C.; Sahin, T.K.; Yildirim, H.C.; Cesmeci, E.; Incesu, F.G.G.; Tahillioglu, Y.; Ucgul, E.; Aksun, M.S.; Gurbuz, S.C.; Aktepe, O.H.; et al. Newly diagnosed cancer and the COVID-19 pandemic: Tumour stage migration and higher early mortality. BMJ Support. Palliat. Care 2021. [Google Scholar] [CrossRef]
- Kufe, D.W.; Pollock, R.E.; Weichselbaum, R.R.; Bast, R.C.; Gansler, T.S.; Holland, J.F.; Frei, E., III. (Eds.) Cancer Medicine, 6th ed.; BC Decker: Hamilton, ON, Canada, 2003. [Google Scholar]
- Smith, R.A.; Andrews, K.S.; Brooks, D.; Fedewa, S.A.; Manassaram-Baptiste, D.; Saslow, D.; Wender, R.C. Cancer screening in the United States, 2019: A review of current American Cancer Society guidelines and current issues in cancer screening. CA Cancer J. Clin. 2019, 69, 184–210. [Google Scholar] [CrossRef] [PubMed]
- US Preventive Services Task Force. Screening for Lung Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2021, 325, 962–970. [Google Scholar] [CrossRef] [PubMed]
- McPhail, S.; Johnson, S.; Greenberg, D.; Peake, M.; Rous, B. Stage at diagnosis and early mortality from cancer in England. Br. J. Cancer 2015, 112 (Suppl. S1), S108–S115. [Google Scholar] [CrossRef] [PubMed]
- Henley, S.J.; King, J.B.; German, R.R.; Richardson, L.C.; Plescia, M. Surveillance of screening-detected cancers (colon and rectum, breast, and cervix)—United States, 2004–2006. Morb. Mortal. Wkly. Rep. Surveill. Summ. 2010, 59, 1–25. [Google Scholar]
- Loud, J.T.; Murphy, J. Cancer Screening and Early Detection in the 21(st) Century. Semin. Oncol. Nurs. 2017, 33, 121–128. [Google Scholar] [CrossRef]
- Wise, J. Diagnosing cancer early is vital, new figures show. BMJ 2016, 353, i3277. [Google Scholar] [CrossRef]
- Alix-Panabieres, C.; Pantel, K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef]
- Watson, I.R.; Takahashi, K.; Futreal, P.A.; Chin, L. Emerging patterns of somatic mutations in cancer. Nat. Rev. Genet. 2013, 14, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, Y.M. Pan-cancer analysis of somatic mutations and transcriptomes reveals common functional gene clusters shared by multiple cancer types. Sci. Rep. 2018, 8, 6041. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ulrich, B.C.; Supplee, J.; Kuang, Y.; Lizotte, P.H.; Feeney, N.B.; Guibert, N.M.; Awad, M.M.; Wong, K.K.; Janne, P.A.; et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4437–4443. [Google Scholar] [CrossRef] [PubMed]
- Shames, D.S.; Girard, L.; Gao, B.; Sato, M.; Lewis, C.M.; Shivapurkar, N.; Jiang, A.; Perou, C.M.; Kim, Y.H.; Pollack, J.R.; et al. A genome-wide screen for promoter methylation in lung cancer identifies novel methylation markers for multiple malignancies. PLoS Med. 2006, 3, e486. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Jensen, T.; Oshiro, M.M.; Watts, G.S.; Kim, C.J.; Futscher, B.W. Agglomerative epigenetic aberrations are a common event in human breast cancer. Cancer Res. 2008, 68, 8616–8625. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Esteller, M. DNA methylation profiling in the clinic: Applications and challenges. Nat. Rev. Genet. 2012, 13, 679–692. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Gotea, V.; Petrykowska, H.M.; Margolin, G.; Krivak, T.C.; DeLoia, J.A.; Bell, D.W.; Elnitski, L. Recurrent patterns of DNA methylation in the ZNF154, CASP8, and VHL promoters across a wide spectrum of human solid epithelial tumors and cancer cell lines. Epigenetics 2013, 8, 1355–1372. [Google Scholar] [CrossRef]
- Margolin, G.; Petrykowska, H.M.; Jameel, N.; Bell, D.W.; Young, A.C.; Elnitski, L. Robust Detection of DNA Hypermethylation of ZNF154 as a Pan-Cancer Locus with in Silico Modeling for Blood-Based Diagnostic Development. J. Mol. Diagn. 2016, 18, 283–298. [Google Scholar] [CrossRef]
- Miller, B.F.; Petrykowska, H.M.; Elnitski, L. Assessing ZNF154 methylation in patient plasma as a multicancer marker in liquid biopsies from colon, liver, ovarian and pancreatic cancer patients. Sci. Rep. 2021, 11, 221. [Google Scholar] [CrossRef]
- Miller, B.F.; Pisanic Ii, T.R.; Margolin, G.; Petrykowska, H.M.; Athamanolap, P.; Goncearenco, A.; Osei-Tutu, A.; Annunziata, C.M.; Wang, T.H.; Elnitski, L. Leveraging locus-specific epigenetic heterogeneity to improve the performance of blood-based DNA methylation biomarkers. Clin. Epigenetics 2020, 12, 154. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Gotea, V.; Margolin, G.; Elnitski, L. Pan-cancer stratification of solid human epithelial tumors and cancer cell lines reveals commonalities and tissue-specific features of the CpG island methylator phenotype. Epigenetics Chromatin 2015, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Liu, J.; Beck, S.; Pan, S.; Capper, D.; Lechner, M.; Thirlwell, C.; Breeze, C.E.; Teschendorff, A.E. A pan-tissue DNA methylation atlas enables in silico decomposition of human tissue methylomes at cell-type resolution. Nat. Methods 2022, 19, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Lehne, B.; Drong, A.W.; Loh, M.; Zhang, W.; Scott, W.R.; Tan, S.T.; Afzal, U.; Scott, J.; Jarvelin, M.R.; Elliott, P.; et al. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol. 2015, 16, 37. [Google Scholar] [CrossRef]
- Terunuma, A.; Putluri, N.; Mishra, P.; Mathé, E.A.; Dorsey, T.H.; Yi, M.; Wallace, T.A.; Issaq, H.J.; Zhou, M.; Killian, J.K.; et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Investig. 2014, 124, 398–412. [Google Scholar] [CrossRef]
- Worsham, M.J.; Chitale, D.; Chen, K.M.; Datta, I.; Divine, G. Cell signaling events differentiate ER-negative subtypes from ER-positive breast cancer. Med. Oncol. 2015, 32, 142. [Google Scholar] [CrossRef]
- Gao, Y.; Jones, A.; Fasching, P.A.; Ruebner, M.; Beckmann, M.W.; Widschwendter, M.; Teschendorff, A.E. The integrative epigenomic-transcriptomic landscape of ER positive breast cancer. Clin. Epigenetics 2015, 7, 126. [Google Scholar] [CrossRef]
- Timp, W.; Bravo, H.C.; McDonald, O.G.; Goggins, M.; Umbricht, C.; Zeiger, M.; Feinberg, A.P.; Irizarry, R.A. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med. 2014, 6, 61. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Schenkel, L.C.; Ainsworth, P.; Lin, H.; Rodenhiser, D.I.; Cutz, J.C.; Sadikovic, B. Genomic DNA Methylation-Derived Algorithm Enables Accurate Detection of Malignant Prostate Tissues. Front. Oncol. 2018, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Li, Q.; Chen, Q.; Zhou, Y.; Park, S.; Lee, G.; Grimes, B.; Krysan, K.; Yu, M.; Wang, W.; et al. CancerLocator: Non-invasive cancer diagnosis and tissue-of-origin prediction using methylation profiles of cell-free DNA. Genome Biol. 2017, 18, 53. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Q.; Kang, S.; Same, M.; Zhou, Y.; Sun, C.; Liu, C.C.; Matsuoka, L.; Sher, L.; Wong, W.H.; et al. CancerDetector: Ultrasensitive and non-invasive cancer detection at the resolution of individual reads using cell-free DNA methylation sequencing data. Nucleic Acids Res. 2018, 46, e89. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.C.; Jiang, P.; Chan, C.W.; Sun, K.; Wong, J.; Hui, E.P.; Chan, S.L.; Chan, W.C.; Hui, D.S.; Ng, S.S.; et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 18761–18768. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Auinger, L.; Speicher, M.R. Cell-Free DNA and Apoptosis: How Dead Cells Inform About the Living. Trends Mol. Med. 2020, 26, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Andrews, P.W.; Damjanov, I.; Simon, D.; Banting, G.S.; Carlin, C.; Dracopoli, N.C.; Føgh, J. Pluripotent embryonal carcinoma clones derived from the human teratocarcinoma cell line Tera-2. Differentiation in vivo and in vitro. Lab. Investig. J. Tech. Methods Pathol. 1984, 50, 147–162. [Google Scholar]
- Sun, K.; Jiang, P.; Chan, K.C.; Wong, J.; Cheng, Y.K.; Liang, R.H.; Chan, W.K.; Ma, E.S.; Chan, S.L.; Cheng, S.H.; et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. USA 2015, 112, E5503–E5512. [Google Scholar] [CrossRef]
- Underhill, H.R.; Kitzman, J.O.; Hellwig, S.; Welker, N.C.; Daza, R.; Baker, D.N.; Gligorich, K.M.; Rostomily, R.C.; Bronner, M.P.; Shendure, J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016, 12, e1006162. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, M.; Singh, S.; Singh, P.; Chauhan, P.; Zaidi, M.A. Tumor markers: A diagnostic tool. Natl. J. Maxillofac. Surg. 2016, 7, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Gonzalez, A.; Cunquero Tomas, A.J.; Calabuig Farinas, S.; Ferrero, M.; Mirda, D.; Sirera, R.; Jantus-Lewintre, E.; Camps, C. A profile on cobas(R) EGFR Mutation Test v2 as companion diagnostic for first-line treatment of patients with non-small cell lung cancer. Expert Rev. Mol. Diagn. 2020, 20, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non-small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef]
- Woodhouse, R.; Li, M.; Hughes, J.; Delfosse, D.; Skoletsky, J.; Ma, P.; Meng, W.; Dewal, N.; Milbury, C.; Clark, T.; et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS ONE 2020, 15, e0237802. [Google Scholar] [CrossRef]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Consortium, C. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, eaan2415. [Google Scholar] [CrossRef]
- Durinck, K.; Van Loocke, W.; Van der Meulen, J.; Van de Walle, I.; Ongenaert, M.; Rondou, P.; Wallaert, A.; de Bock, C.E.; Van Roy, N.; Poppe, B.; et al. Characterization of the genome-wide TLX1 binding profile in T-cell acute lymphoblastic leukemia. Leukemia 2015, 29, 2317–2327. [Google Scholar] [CrossRef]
- Misawa, K.; Ueda, Y.; Kanazawa, T.; Misawa, Y.; Jang, I.; Brenner, J.C.; Ogawa, T.; Takebayashi, S.; Grenman, R.A.; Herman, J.G.; et al. Epigenetic inactivation of galanin receptor 1 in head and neck cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 7604–7613. [Google Scholar] [CrossRef]
- Zhang, W.; Shu, P.; Wang, S.; Song, J.; Liu, K.; Wang, C.; Ran, L. ZNF154 is a promising diagnosis biomarker and predicts biochemical recurrence in prostate cancer. Gene 2018, 675, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Modin, C.; Castano, F.M.; Lamy, P.; Wojdacz, T.K.; Hansen, L.L.; Wiuf, C.; Borre, M.; Dyrskjøt, L.; Orntoft, T.F. Comprehensive genome methylation analysis in bladder cancer: Identification and validation of novel methylated genes and application of these as urinary tumor markers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 5582–5592. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, S.A.; Bashir, M.U.; Yaqinuddin, A. Utility of DNA methylation markers for diagnosing cancer. Int. J. Surg. 2010, 8, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Lissa, D.; Robles, A.I. Methylation analyses in liquid biopsy. Transl. Lung Cancer Res. 2016, 5, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Godsey, J.H.; Silvestro, A.; Barrett, J.C.; Bramlett, K.; Chudova, D.; Deras, I.; Dickey, J.; Hicks, J.; Johann, D.J.; Leary, R.; et al. Generic Protocols for the Analytical Validation of Next-Generation Sequencing-Based ctDNA Assays: A Joint Consensus Recommendation of the BloodPAC’s Analytical Variables Working Group. Clin. Chem. 2020, 66, 1156–1166. [Google Scholar] [CrossRef]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Levin, T.R.; Lavin, P.; Lidgard, G.P.; Ahlquist, D.A.; Berger, B.M. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014, 370, 1287–1297. [Google Scholar] [CrossRef]
- Aberle, D.R.; DeMello, S.; Berg, C.D.; Black, W.C.; Brewer, B.; Church, T.R.; Clingan, K.L.; Duan, F.; Fagerstrom, R.M.; Gareen, I.F.; et al. Results of the two incidence screenings in the National Lung Screening Trial. N. Engl. J. Med. 2013, 369, 920–931. [Google Scholar] [CrossRef]
- Klein, E.A.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N.; et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef]
- Jensen, S.O.; Ogaard, N.; Orntoft, M.W.; Rasmussen, M.H.; Bramsen, J.B.; Kristensen, H.; Mouritzen, P.; Madsen, M.R.; Madsen, A.H.; Sunesen, K.G.; et al. Novel DNA methylation biomarkers show high sensitivity and specificity for blood-based detection of colorectal cancer-a clinical biomarker discovery and validation study. Clin. Epigenetics 2019, 11, 158. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A., Jr.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Fleischhacker, M.; Schmidt, B. Circulating nucleic acids (CNAs) and cancer—A survey. Biochim. Biophys. Acta 2007, 1775, 181–232. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating tumor DNA as a liquid biopsy for cancer. Clin. Chem. 2015, 61, 112–123. [Google Scholar] [CrossRef]
- Bredno, J.; Lipson, J.; Venn, O.; Aravanis, A.M.; Jamshidi, A. Clinical correlates of circulating cell-free DNA tumor fraction. PLoS ONE 2021, 16, e0256436. [Google Scholar] [CrossRef]
- Ahlquist, D.A. Universal cancer screening: Revolutionary, rational, and realizable. NPJ Precis. Oncol. 2018, 2, 23. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Type 1 | #Tumor Samples 2 | #Normal Samples 2 | Threshold 3 | Sensitivity | Specificity | AUC 4 |
|---|---|---|---|---|---|---|
| BLCA | 201 | 20 | 0.93 | 90.5% | 95.0% | 0.967 |
| BRCA | 676 | 96 | 0.90 | 90.5% | 95.8% | 0.979 |
| COAD | 274 | 38 | 0.84 | 96.7% | 100% | 0.992 |
| HNSC | 426 | 50 | 0.80 | 98.6% | 98.0% | 0.996 |
| KIRC | 296 | 160 | 0.65 | 82.4% | 95.6% | 0.931 |
| KIRP | 156 | 45 | 0.70 | 73.7% | 84.4% | 0.859 |
| LIHC | 151 | 50 | 0.43 | 92.1% | 98.0% | 0.950 |
| LUAD | 437 | 32 | 0.98 | 97.3% | 100% | 0.999 |
| LUSC | 359 | 42 | 0.64 | 99.2% | 100% | 0.996 |
| PAAD | 65 | 9 | 0.58 | 98.5% | 100% | 0.991 |
| PRAD | 248 | 49 | 0.82 | 90.3% | 91.8% | 0.941 |
| READ | 96 | 7 | 0.50 | 100% | 100% | 1.000 |
| STAD | 260 | 2 | 0.50 | 100% | 100% | 1.000 |
| UCEC | 405 | 46 | 0.79 | 99.0% | 100% | 0.997 |
| Tumor Type 1 | Dataset | #Tumor Samples | #Normal Samples | Sensitivity | Specificity |
|---|---|---|---|---|---|
| BRCA | GSE37754, GSE66695, GSE69914 | 449 | 149 | 79.5% | 94.0% |
| COAD | GSE53051 | 35 | 18 | 94.3% | 83.3% |
| LUAD | GSE53051 | 9 | 11 | 100% | 73% |
| PRAD | GSE112047 | 31 | 16 | 90.3% | 100.0% |
| Tumor Type | Dataset | #Tumor Samples | #Normal Samples | Sensitivity 1 | Specificity 1 |
|---|---|---|---|---|---|
| Lung cancer | [33,34] | 9 | 4 | 100% 2 | 100% 2 |
| Hepatocellular carcinoma | [34,35] | 30 | 36 | 37.3% | 83.3% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Funderburk, K.; Bang-Christensen, S.R.; Miller, B.F.; Tan, H.; Margolin, G.; Petrykowska, H.M.; Baugher, C.; Farney, S.K.; Grimm, S.A.; Jameel, N.; et al. Evaluating Stacked Methylation Markers for Blood-Based Multicancer Detection. Cancers 2023, 15, 4826. https://doi.org/10.3390/cancers15194826
Funderburk K, Bang-Christensen SR, Miller BF, Tan H, Margolin G, Petrykowska HM, Baugher C, Farney SK, Grimm SA, Jameel N, et al. Evaluating Stacked Methylation Markers for Blood-Based Multicancer Detection. Cancers. 2023; 15(19):4826. https://doi.org/10.3390/cancers15194826
Chicago/Turabian StyleFunderburk, Karen, Sara R. Bang-Christensen, Brendan F. Miller, Hua Tan, Gennady Margolin, Hanna M. Petrykowska, Catherine Baugher, S. Katie Farney, Sara A. Grimm, Nader Jameel, and et al. 2023. "Evaluating Stacked Methylation Markers for Blood-Based Multicancer Detection" Cancers 15, no. 19: 4826. https://doi.org/10.3390/cancers15194826
APA StyleFunderburk, K., Bang-Christensen, S. R., Miller, B. F., Tan, H., Margolin, G., Petrykowska, H. M., Baugher, C., Farney, S. K., Grimm, S. A., Jameel, N., Holland, D. O., Altman, N. S., & Elnitski, L. (2023). Evaluating Stacked Methylation Markers for Blood-Based Multicancer Detection. Cancers, 15(19), 4826. https://doi.org/10.3390/cancers15194826