
Advancement and Challenges in Monitoring of CAR-T Cell Therapy: A Comprehensive Review of Parameters and Markers in Hematological Malignancies


Abstract
:Simple Summary
Abstract
1. Introduction
2. CAR-T Cell Monitoring
2.1. Quantitative Monitoring of CAR-T Cells
2.1.1. Polymerase Chain Reaction
2.1.2. Flow Cytometry
2.2. Monitoring of the Activity of CAR-T Cells
2.3. Phenotyping of T-Cell Subsets
3. Biomarkers and Parameters for Monitoring CAR-T Cell Therapy
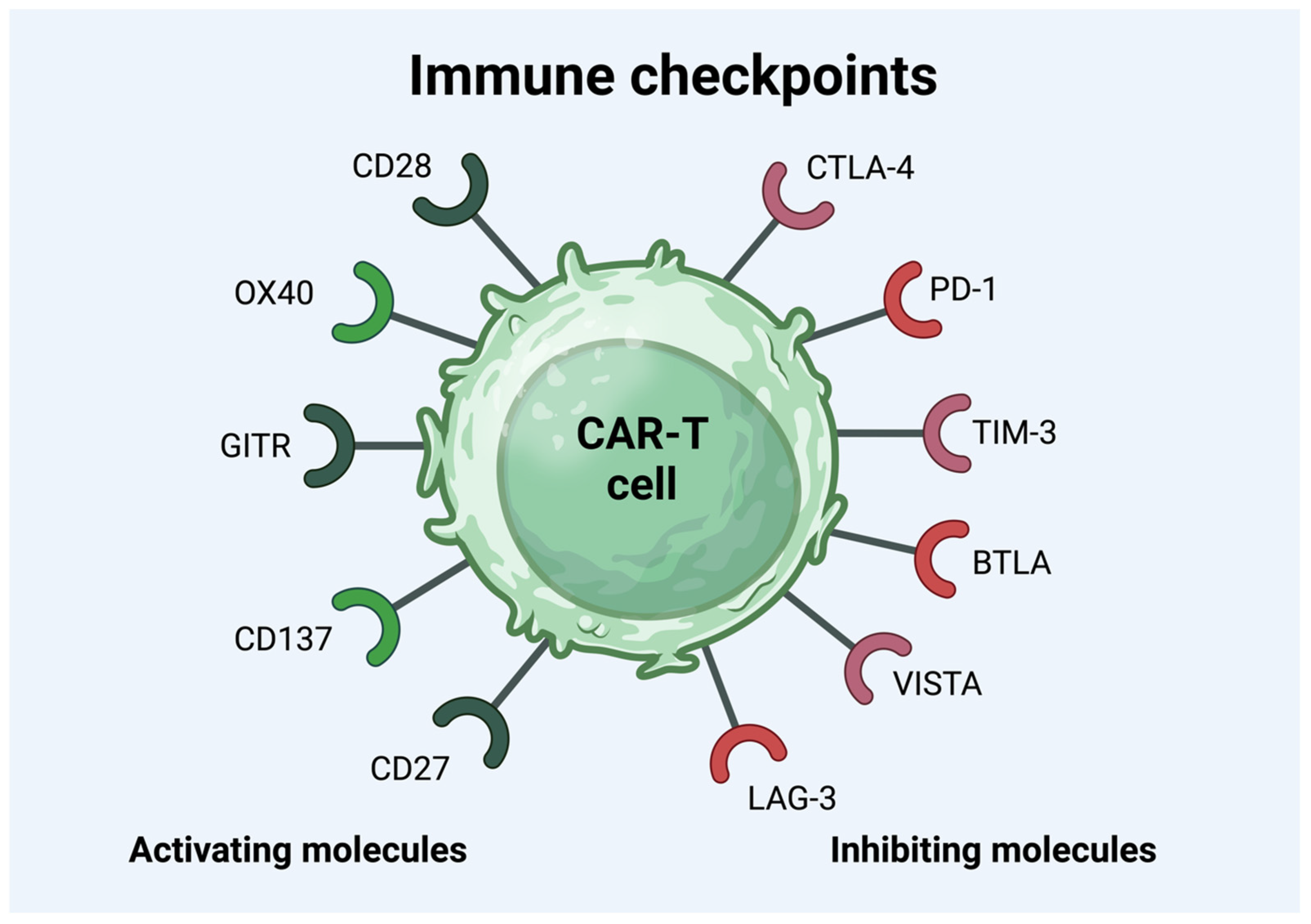
3.1. Immune Checkpoint Molecules
3.2. Cytokines
3.3. ctDNA
4. Monitoring of the Adverse Effects of CAR-T Cell Therapy—Current Practice and Future Options
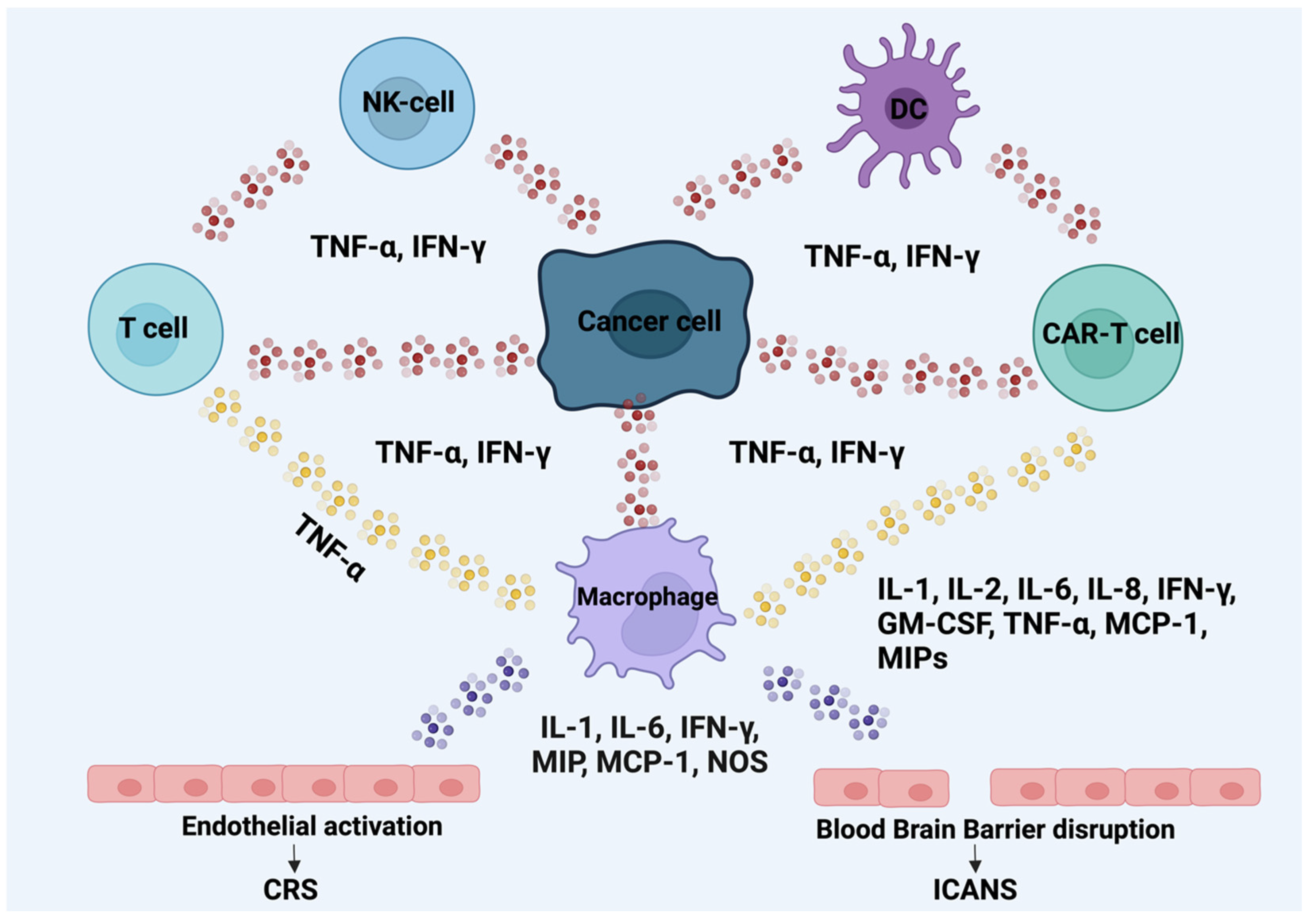
4.1. Cytokine Release Syndrome
4.2. Immune-Effector-Eell-Associated Neurotoxicity Syndrome
4.3. Hemophagocytic Lymphohistiocytosis
4.4. Cytopenias/Marrow Hypoplasia
4.5. B-Cell Aplasia and Hypogammaglobulinemia
5. Conclusions and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Lin, H.; Cheng, J.; Mu, W.; Zhou, J.; Zhu, L. Advances in Universal CAR-T Cell Therapy. Front. Immunol. 2021, 12, 744823. [Google Scholar] [CrossRef] [PubMed]
- Hayden, P.J.; Roddie, C.; Bader, P.; Basak, G.W.; Bonig, H.; Bonini, C.; Chabannon, C.; Ciceri, F.; Corbacioglu, S.; Ellard, R.; et al. Management of adults and children receiving CAR T-cell therapy: 2021 best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE) and the European Haematology Association (EHA). Ann. Oncol. 2022, 33, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Chohan, K.L.; Siegler, E.L.; Kenderian, S.S. CAR-T Cell Therapy: The Efficacy and Toxicity Balance. Curr. Hematol. Malig. Rep. 2023, 18, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Zhu, M.; Huang, H.; Hu, Y. Relapse after CAR-T cell therapy in B-cell malignancies: Challenges and future approaches. J. Zhejiang Univ. Sci. B 2022, 23, 793–811. [Google Scholar] [CrossRef] [PubMed]
- Olejarz, W.; Basak, G. Emerging Therapeutic Targets and Drug Resistance Mechanisms in Immunotherapy of Hematological Malignancies. Cancers 2023, 15, 5765. [Google Scholar] [CrossRef]
- Schanda, N.; Sauer, T.; Kunz, A.; Huckelhoven-Krauss, A.; Neuber, B.; Wang, L.; Hinkelbein, M.; Sedloev, D.; He, B.; Schubert, M.L.; et al. Sensitivity and Specificity of CD19.CAR-T Cell Detection by Flow Cytometry and PCR. Cells 2021, 10, 3208. [Google Scholar] [CrossRef]
- Demaret, J.; Varlet, P.; Trauet, J.; Beauvais, D.; Grossemy, A.; Hego, F.; Yakoub-Agha, I.; Labalette, M. Monitoring CAR T-cells using flow cytometry. Cytom. B Clin. Cytom. 2021, 100, 218–224. [Google Scholar] [CrossRef]
- Garcia-Calderon, C.B.; Sierro-Martinez, B.; Garcia-Guerrero, E.; Sanoja-Flores, L.; Munoz-Garcia, R.; Ruiz-Maldonado, V.; Jimenez-Leon, M.R.; Delgado-Serrano, J.; Molinos-Quintana, A.; Guijarro-Albaladejo, B.; et al. Monitoring of kinetics and exhaustion markers of circulating CAR-T cells as early predictive factors in patients with B-cell malignancies. Front. Immunol. 2023, 14, 1152498. [Google Scholar] [CrossRef]
- Sadowski, K.; Olejarz, W.; Basak, G. Modern Advances in CARs Therapy and Creating a New Approach to Future Treatment. Int. J. Mol. Sci. 2022, 23, 15006. [Google Scholar] [CrossRef]
- de la Iglesia-San Sebastián, I.; Carbonell, D.; Bastos-Oreiro, M.; Pérez-Corral, A.; Bailén, R.; Chicano, M.; Muñiz, P.; Monsalvo, S.; Escudero-Fernández, A.; Oarbeascoa, G.; et al. Digital PCR Improves Sensitivity and Quantification in Monitoring CAR-T Cells in B Cell Lymphoma Patients. Transplant. Cell Ther. 2024, 30, e301–e306. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.L.; Berger, C.; Kunz, A.; Schmitt, A.; Badbaran, A.; Neuber, B.; Zeschke, S.; Wang, L.; Riecken, K.; Huckelhoven-Krauss, A.; et al. Comparison of single copy gene-based duplex quantitative PCR and digital droplet PCR for monitoring of expansion of CD19-directed CAR T cells in treated patients. Int. J. Oncol. 2022, 60, 48. [Google Scholar] [CrossRef] [PubMed]
- Selim, A.G.; Minson, A.; Blombery, P.; Dickinson, M.; Harrison, S.J.; Anderson, M.A. CAR-T cell therapy: Practical guide to routine laboratory monitoring. Pathology 2021, 53, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Turicek, D.P.; Giordani, V.M.; Moraly, J.; Taylor, N.; Shah, N.N. CAR T-cell detection scoping review: An essential biomarker in critical need of standardization. J. Immunother. Cancer 2023, 11, e006596. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Martin, T.; Usmani, S.Z.; Berdeja, J.G.; Agha, M.; Cohen, A.D.; Hari, P.; Avigan, D.; Deol, A.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, an Anti-B-cell Maturation Antigen Chimeric Antigen Receptor T-Cell Therapy, for Relapsed/Refractory Multiple Myeloma: CARTITUDE-1 2-Year Follow-Up. J. Clin. Oncol. 2023, 41, 1265–1274. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Dickinson, M.; Munoz, J.; Ulrickson, M.L.; Thieblemont, C.; Oluwole, O.O.; Herrera, A.F.; Ujjani, C.S.; Lin, Y.; Riedell, P.A.; et al. Axicabtagene ciloleucel as first-line therapy in high-risk large B-cell lymphoma: The phase 2 ZUMA-12 trial. Nat. Med. 2022, 28, 735–742. [Google Scholar] [CrossRef]
- He, J.; Xiong, X.; Yang, H.; Li, D.; Liu, X.; Li, S.; Liao, S.; Chen, S.; Wen, X.; Yu, K.; et al. Defined tumor antigen-specific T cells potentiate personalized TCR-T cell therapy and prediction of immunotherapy response. Cell Res. 2022, 32, 530–542. [Google Scholar] [CrossRef]
- Gardner, C.M.; Tan, H.; Hull, E.L.; Lisauskas, J.B.; Sum, S.T.; Meese, T.M.; Jiang, C.; Madden, S.P.; Caplan, J.D.; Burke, A.P.; et al. Detection of lipid core coronary plaques in autopsy specimens with a novel catheter-based near-infrared spectroscopy system. JACC Cardiovasc. Imaging 2008, 1, 638–648. [Google Scholar] [CrossRef]
- Myers, R.M.; Li, Y.; Barz Leahy, A.; Barrett, D.M.; Teachey, D.T.; Callahan, C.; Fasano, C.C.; Rheingold, S.R.; DiNofia, A.; Wray, L.; et al. Humanized CD19-Targeted Chimeric Antigen Receptor (CAR) T Cells in CAR-Naive and CAR-Exposed Children and Young Adults With Relapsed or Refractory Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2021, 39, 3044–3055. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Milgrom, S.A.; Rechner, L.; Berthelsen, A. The optimal use of PET/CT in the management of lymphoma patients. Br. J. Radiol. 2021, 94, 20210470. [Google Scholar] [CrossRef] [PubMed]
- Georgi, T.W.; Kurch, L.; Franke, G.N.; Jentzsch, M.; Schwind, S.; Perez-Fernandez, C.; Petermann, N.; Merz, M.; Metzeler, K.; Borte, G.; et al. Prognostic value of baseline and early response FDG-PET/CT in patients with refractory and relapsed aggressive B-cell lymphoma undergoing CAR-T cell therapy. J. Cancer Res. Clin. Oncol. 2023, 149, 6131–6138. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.C.; Fehse, B.; Akyuz, N.; Geffken, M.; Wolschke, C.; Janson, D.; Gagelmann, N.; Luther, M.; Wichmann, D.; Frenzel, C.; et al. Molecular monitoring of T-cell kinetics and migration in severe neurotoxicity after real-world CD19-specific chimeric antigen receptor T cell therapy. Haematologica 2023, 108, 444–456. [Google Scholar] [CrossRef]
- Cheson, B.D.; Fisher, R.I.; Barrington, S.F.; Cavalli, F.; Schwartz, L.H.; Zucca, E.; Lister, T.A.; Alliance, A.L.; Lymphoma, G.; Eastern Cooperative Oncology, G.; et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: The Lugano classification. J. Clin. Oncol. 2014, 32, 3059–3068. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Gökbuget, N.; Dombret, H.; Bassan, R.; Wadleigh, M.; Doubek, M.; Ribera, J. Inclusion and response criteria for clinical trials in relapsed/refractory acute lymphoblastic leukemia and usefulness of historical control trials. Haematologica 2017, 102, e118–e119. [Google Scholar] [CrossRef]
- Lin, Y.; Qiu, L.; Usmani, S.; Joo, C.W.; Costa, L.; Derman, B.; Du, J.; Einsele, H.; Fernandez de Larrea, C.; Hajek, R.; et al. Consensus guidelines and recommendations for the management and response assessment of chimeric antigen receptor T-cell therapy in clinical practice for relapsed and refractory multiple myeloma: A report from the International Myeloma Working Group Immunotherapy Committee. Lancet Oncol. 2024, 25, e374–e387. [Google Scholar] [CrossRef]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.-V.; et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef] [PubMed]
- Kiesgen, S.; Messinger, J.C.; Chintala, N.K.; Tano, Z.; Adusumilli, P.S. Comparative analysis of assays to measure CAR T-cell-mediated cytotoxicity. Nat. Protoc. 2021, 16, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A.; Frigault, M.J.; Ansari, A.; Gliner, B.; Heery, C.; Shah, B. Dose-response correlation for CAR-T cells: A systematic review of clinical studies. J. Immunother. Cancer 2022, 10, e005678. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Rossi, J.M.; Neelapu, S.S.; Jacobson, C.A.; Miklos, D.B.; Ghobadi, A.; Oluwole, O.O.; Reagan, P.M.; Lekakis, L.J.; Lin, Y.; et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 2020, 4, 4898–4911. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.C.; Boissel, N.; Cassaday, R.D.; Leguay, T.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: Phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 2021, 398, 491–502. [Google Scholar] [CrossRef]
- Gupta, A.; Gill, S. CAR-T cell persistence in the treatment of leukemia and lymphoma. Leuk. Lymphoma 2021, 62, 2587–2599. [Google Scholar] [CrossRef]
- Schubert, M.L.; Kunz, A.; Schmitt, A.; Neuber, B.; Wang, L.; Hückelhoven-Krauss, A.; Langner, S.; Michels, B.; Wick, A.; Daniel, V.; et al. Assessment of CAR T Cell Frequencies in Axicabtagene Ciloleucel and Tisagenlecleucel Patients Using Duplex Quantitative PCR. Cancers 2020, 12, 2820. [Google Scholar] [CrossRef]
- Wang, Y.; Chan, L.L.; Grimaud, M.; Fayed, A.; Zhu, Q.; Marasco, W.A. High-Throughput Image Cytometry Detection Method for CAR-T Transduction, Cell Proliferation, and Cytotoxicity Assays. Cytom. A 2021, 99, 689–697. [Google Scholar] [CrossRef]
- Reichman, A.; Kunz, A.; Joedicke, J.J.; Höpken, U.E.; Keib, A.; Neuber, B.; Sedloev, D.; Wang, L.; Jiang, G.; Hückelhoven-Krauss, A.; et al. Comparison of FACS and PCR for Detection of BCMA-CAR-T Cells. Int. J. Mol. Sci. 2022, 23, 903. [Google Scholar] [CrossRef]
- Masilamani, M.; Jawa, V.; Dai, Y.; Das, R.; Park, A.; Lamba, M.; Wu, F.; Zheng, X.; Lu, E.; Gleason, C.; et al. Bioanalytical Methods for Characterization of CAR-T Cellular Kinetics: Comparison of PCR Assays and Matrices. Clin. Pharmacol. Ther. 2023, 114, 664–672. [Google Scholar] [CrossRef]
- Ginzinger, D.G. Gene quantification using real-time quantitative PCR: An emerging technology hits the mainstream. Exp. Hematol. 2002, 30, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [PubMed]
- Stahl, T.; Böhme, M.U.; Kröger, N.; Fehse, B. Digital PCR to assess hematopoietic chimerism after allogeneic stem cell transplantation. Exp. Hematol. 2015, 43, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Fehse, B.; Badbaran, A.; Berger, C.; Sonntag, T.; Riecken, K.; Geffken, M.; Kröger, N.; Ayuk, F.A. Digital PCR Assays for Precise Quantification of CD19-CAR-T Cells after Treatment with Axicabtagene Ciloleucel. Mol. Ther. Methods Clin. Dev. 2020, 16, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Mock, U.; Hauber, I.; Fehse, B. Digital PCR to assess gene-editing frequencies (GEF-dPCR) mediated by designer nucleases. Nat. Protoc. 2016, 11, 598–615. [Google Scholar] [CrossRef]
- Cheng, J.; Mao, X.; Chen, C.; Long, X.; Chen, L.; Zhou, J.; Zhu, L. Monitoring anti-CD19 chimeric antigen receptor T cell population by flow cytometry and its consistency with digital droplet polymerase chain reaction. Cytom. A 2023, 103, 16–26. [Google Scholar] [CrossRef]
- Hu, Y.; Huang, J. The Chimeric Antigen Receptor Detection Toolkit. Front. Immunol. 2020, 11, 1770. [Google Scholar] [CrossRef]
- Blache, U.; Weiss, R.; Boldt, A.; Kapinsky, M.; Blaudszun, A.R.; Quaiser, A.; Pohl, A.; Miloud, T.; Burgaud, M.; Vucinic, V.; et al. Advanced Flow Cytometry Assays for Immune Monitoring of CAR-T Cell Applications. Front. Immunol. 2021, 12, 658314. [Google Scholar] [CrossRef]
- Zaninelli, S.; Meli, C.; Borleri, G.; Quaroni, M.; Pavoni, C.; Gaipa, G.; Biondi, A.; Introna, M.; Golay, J.; Rambaldi, A.; et al. Optimization and validation of in vivo flow cytometry chimeric antigen receptor T cell detection method using CD19his indirect staining. Cytom. A 2024, 105, 112–123. [Google Scholar] [CrossRef]
- Sarikonda, G.; Mathieu, M.; Natalia, M.; Pahuja, A.; Xue, Q.; Pierog, P.L.; Trampont, P.C.; Decman, V.; Reynolds, S.; Hanafi, L.A.; et al. Best practices for the development, analytical validation and clinical implementation of flow cytometric methods for chimeric antigen receptor T cell analyses. Cytom. B Clin. Cytom. 2021, 100, 79–91. [Google Scholar] [CrossRef]
- Brunner, K.T.; Mauel, J.; Cerottini, J.C.; Chapuis, B. Quantitative assay of the lytic action of immune lymphoid cells on 51-Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology 1968, 14, 181–196. [Google Scholar] [PubMed]
- Karimi, M.A.; Lee, E.; Bachmann, M.H.; Salicioni, A.M.; Behrens, E.M.; Kambayashi, T.; Baldwin, C.L. Measuring cytotoxicity by bioluminescence imaging outperforms the standard chromium-51 release assay. PLoS ONE 2014, 9, e89357. [Google Scholar] [CrossRef] [PubMed]
- Contag, C.H.; Bachmann, M.H. Advances in in vivo bioluminescence imaging of gene expression. Annu. Rev. Biomed. Eng. 2002, 4, 235–260. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, X.; Xu, X.; Abassi, Y.A. Dynamic and label-free monitoring of natural killer cell cytotoxic activity using electronic cell sensor arrays. J. Immunol. Methods 2006, 309, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.Z.; Zhu, L.; Jackson, J.A.; Gabos, S.; Sun, X.J.; Wang, X.B.; Xu, X. Dynamic monitoring of cytotoxicity on microelectronic sensors. Chem. Res. Toxicol. 2005, 18, 154–161. [Google Scholar] [CrossRef]
- Solly, K.; Wang, X.; Xu, X.; Strulovici, B.; Zheng, W. Application of real-time cell electronic sensing (RT-CES) technology to cell-based assays. Assay. Drug Dev. Technol. 2004, 2, 363–372. [Google Scholar] [CrossRef]
- Erskine, C.L.; Henle, A.M.; Knutson, K.L. Determining optimal cytotoxic activity of human Her2neu specific CD8 T cells by comparing the Cr51 release assay to the xCELLigence system. J. Vis. Exp. 2012, 66, 3683. [Google Scholar] [CrossRef]
- Peper, J.K.; Schuster, H.; Löffler, M.W.; Schmid-Horch, B.; Rammensee, H.G.; Stevanović, S. An impedance-based cytotoxicity assay for real-time and label-free assessment of T-cell-mediated killing of adherent cells. J. Immunol. Methods 2014, 405, 192–198. [Google Scholar] [CrossRef]
- Eugene-Norbert, M.; Cuffel, A.; Riou, G.; Jean, L.; Blondel, C.; Dehayes, J.; Bisson, A.; Giverne, C.; Brotin, E.; Denoyelle, C.; et al. Development of optimized cytotoxicity assays for assessing the antitumor potential of CAR-T cells. J. Immunol. Methods 2024, 525, 113603. [Google Scholar] [CrossRef]
- Xi, B.; Berahovich, R.; Zhou, H.; Xu, S.; Wei, Y.; Guan, J.; Harto, H.; Guan, J.; Wu, L.; Santa Ana, D.; et al. A Real-time Potency Assay for Chimeric Antigen Receptor T Cells Targeting Solid and Hematological Cancer Cells. J. Vis. Exp. 2019, e59033. [Google Scholar] [CrossRef]
- Lisby, A.N.; Carlson, R.D.; Baybutt, T.R.; Weindorfer, M.; Snook, A.E. Evaluation of CAR-T cell cytotoxicity: Real-time impedance-based analysis. Methods Cell Biol. 2022, 167, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chahroudi, A.; Silvestri, G.; Wernett, M.E.; Kaiser, W.J.; Safrit, J.T.; Komoriya, A.; Altman, J.D.; Packard, B.Z.; Feinberg, M.B. Visualization and quantification of T cell-mediated cytotoxicity using cell-permeable fluorogenic caspase substrates. Nat. Med. 2002, 8, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Packard, B.Z.; Komoriya, A. Intracellular protease activation in apoptosis and cell-mediated cytotoxicity characterized by cell-permeable fluorogenic protease substrates. Cell Res. 2008, 18, 238–247. [Google Scholar] [CrossRef]
- Jedema, I.; van der Werff, N.M.; Barge, R.M.; Willemze, R.; Falkenburg, J.H. New CFSE-based assay to determine susceptibility to lysis by cytotoxic T cells of leukemic precursor cells within a heterogeneous target cell population. Blood 2004, 103, 2677–2682. [Google Scholar] [CrossRef]
- Mousset, C.M.; Hobo, W.; Woestenenk, R.; Preijers, F.; Dolstra, H.; van der Waart, A.B. Comprehensive Phenotyping of T Cells Using Flow Cytometry. Cytom. A 2019, 95, 647–654. [Google Scholar] [CrossRef]
- Lopez-Cantillo, G.; Uruena, C.; Camacho, B.A.; Ramirez-Segura, C. CAR-T Cell Performance: How to Improve Their Persistence? Front. Immunol. 2022, 13, 878209. [Google Scholar] [CrossRef]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef]
- Rossi, J.; Paczkowski, P.; Shen, Y.W.; Morse, K.; Flynn, B.; Kaiser, A.; Ng, C.; Gallatin, K.; Cain, T.; Fan, R.; et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 2018, 132, 804–814. [Google Scholar] [CrossRef]
- Deng, Q.; Han, G.; Puebla-Osorio, N.; Ma, M.C.J.; Strati, P.; Chasen, B.; Dai, E.; Dang, M.; Jain, N.; Yang, H.; et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med. 2020, 26, 1878–1887. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, X.Q.; Zhang, R.L.; Liu, F.; Wang, Y.; Yan, Z.L.; Song, Y.P.; Yang, T.; Li, P.; Wang, Z.; et al. Donor-derived CD19 CAR-T cell therapy of relapse of CD19-positive B-ALL post allotransplant. Leukemia 2021, 35, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Kochenderfer, J.N. Long-term outcomes following CAR T cell therapy: What we know so far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Baur, K.; Buser, A.; Jeker, L.T.; Khanna, N.; Laubli, H.; Heim, D.; Dirks, J.C.; Widmer, C.C.; Volken, T.; Passweg, J.R.; et al. CD4+ CAR T-cell expansion is associated with response and therapy related toxicities in patients with B-cell lymphomas. Bone Marrow Transplant. 2023, 58, 1048–1050. [Google Scholar] [CrossRef] [PubMed]
- Attardi, E.; Di Cesare, S.; Amodio, D.; Giancotta, C.; Cotugno, N.; Cifaldi, C.; Chiriaco, M.; Palma, P.; Finocchi, A.; Di Matteo, G.; et al. Phenotypical T Cell Differentiation Analysis: A Diagnostic and Predictive Tool in the Study of Primary Immunodeficiencies. Front. Immunol. 2019, 10, 2735. [Google Scholar] [CrossRef]
- Gattinoni, L.; Klebanoff, C.A.; Palmer, D.C.; Wrzesinski, C.; Kerstann, K.; Yu, Z.; Finkelstein, S.E.; Theoret, M.R.; Rosenberg, S.A.; Restifo, N.P. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Investig. 2005, 115, 1616–1626. [Google Scholar] [CrossRef]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef]
- Gattinoni, L.; Klebanoff, C.A.; Restifo, N.P. Paths to stemness: Building the ultimate antitumour T cell. Nat. Rev. Cancer 2012, 12, 671–684. [Google Scholar] [CrossRef]
- Rathod, S. Phenotyping of CAR T cells. Methods Cell Biol. 2022, 167, 71–80. [Google Scholar] [CrossRef]
- Peinelt, A.; Bremm, M.; Kreyenberg, H.; Cappel, C.; Banisharif-Dehkordi, J.; Erben, S.; Rettinger, E.; Jarisch, A.; Meisel, R.; Schlegel, P.G.; et al. Monitoring of Circulating CAR T Cells: Validation of a Flow Cytometric Assay, Cellular Kinetics, and Phenotype Analysis Following Tisagenlecleucel. Front. Immunol. 2022, 13, 830773. [Google Scholar] [CrossRef] [PubMed]
- Popa, D.C.; Sandu, H.M.; Suciu, R.; Ţica, V.G.; Şerbănică, A.; Şerbănică, I.; Jercan, C.; Coriu, D.; Tanase, A.; Coliţă, A. Monitoring CAR T cells in peripheral blood by flow cytometry following Tisagenlecleucel in Fundeni Clinical Institute, Bucharest. Rev. Romana Med. Lab. 2023, 31, 175–184. [Google Scholar] [CrossRef]
- Ahmed, R.; Bevan, M.J.; Reiner, S.L.; Fearon, D.T. The precursors of memory: Models and controversies. Nat. Rev. Immunol. 2009, 9, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Lecuroux, C.; Girault, I.; Urrutia, A.; Doisne, J.M.; Deveau, C.; Goujard, C.; Meyer, L.; Sinet, M.; Venet, A. Identification of a particular HIV-specific CD8+ T-cell subset with a CD27+ CD45RO-/RA+ phenotype and memory characteristics after initiation of HAART during acute primary HIV infection. Blood 2009, 113, 3209–3217. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Teichgraber, V.; Becker, T.C.; Masopust, D.; Kaech, S.M.; Antia, R.; von Andrian, U.H.; Ahmed, R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 2003, 4, 225–234. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Gattinoni, L.; Torabi-Parizi, P.; Kerstann, K.; Cardones, A.R.; Finkelstein, S.E.; Palmer, D.C.; Antony, P.A.; Hwang, S.T.; Rosenberg, S.A.; et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9571–9576. [Google Scholar] [CrossRef]
- Sabatino, M.; Hu, J.; Sommariva, M.; Gautam, S.; Fellowes, V.; Hocker, J.D.; Dougherty, S.; Qin, H.; Klebanoff, C.A.; Fry, T.J.; et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood 2016, 128, 519–528. [Google Scholar] [CrossRef]
- Biasco, L.; Izotova, N.; Rivat, C.; Ghorashian, S.; Richardson, R.; Guvenel, A.; Hough, R.; Wynn, R.; Popova, B.; Lopes, A.; et al. Clonal expansion of T memory stem cells determines early anti-leukemic responses and long-term CAR T cell persistence in patients. Nat. Cancer 2021, 2, 629–642. [Google Scholar] [CrossRef]
- Wang, X.; Popplewell, L.L.; Wagner, J.R.; Naranjo, A.; Blanchard, M.S.; Mott, M.R.; Norris, A.P.; Wong, C.W.; Urak, R.Z.; Chang, W.C.; et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood 2016, 127, 2980–2990. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef]
- Kaartinen, T.; Luostarinen, A.; Maliniemi, P.; Keto, J.; Arvas, M.; Belt, H.; Koponen, J.; Makinen, P.I.; Loskog, A.; Mustjoki, S.; et al. Low interleukin-2 concentration favors generation of early memory T cells over effector phenotypes during chimeric antigen receptor T-cell expansion. Cytotherapy 2017, 19, 689–702. [Google Scholar] [CrossRef] [PubMed]
- McLellan, A.D.; Ali Hosseini Rad, S.M. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol. Cell Biol. 2019, 97, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Blaeschke, F.; Stenger, D.; Kaeuferle, T.; Willier, S.; Lotfi, R.; Kaiser, A.D.; Assenmacher, M.; Doring, M.; Feucht, J.; Feuchtinger, T. Induction of a central memory and stem cell memory phenotype in functionally active CD4(+) and CD8(+) CAR T cells produced in an automated good manufacturing practice system for the treatment of CD19(+) acute lymphoblastic leukemia. Cancer Immunol. Immunother. 2018, 67, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Busch, D.H.; Frassle, S.P.; Sommermeyer, D.; Buchholz, V.R.; Riddell, S.R. Role of memory T cell subsets for adoptive immunotherapy. Semin. Immunol. 2016, 28, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Riddell, S.R.; Sommermeyer, D.; Berger, C.; Liu, L.S.; Balakrishnan, A.; Salter, A.; Hudecek, M.; Maloney, D.G.; Turtle, C.J. Adoptive therapy with chimeric antigen receptor-modified T cells of defined subset composition. Cancer J. 2014, 20, 141–144. [Google Scholar] [CrossRef]
- Hinrichs, C.S.; Borman, Z.A.; Cassard, L.; Gattinoni, L.; Spolski, R.; Yu, Z.; Sanchez-Perez, L.; Muranski, P.; Kern, S.J.; Logun, C.; et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 17469–17474. [Google Scholar] [CrossRef]
- Brummelman, J.; Pilipow, K.; Lugli, E. The Single-Cell Phenotypic Identity of Human CD8(+) and CD4(+) T Cells. Int. Rev. Cell Mol. Biol. 2018, 341, 63–124. [Google Scholar] [CrossRef]
- van den Broek, T.; Borghans, J.A.M.; van Wijk, F. The full spectrum of human naive T cells. Nat. Rev. Immunol. 2018, 18, 363–373. [Google Scholar] [CrossRef]
- Benoit-Lizon, I.; Jacquin, E.; Rivera Vargas, T.; Richard, C.; Roussey, A.; Dal Zuffo, L.; Martin, T.; Melis, A.; Vinokurova, D.; Shahoei, S.H.; et al. CD4 T cell-intrinsic STING signaling controls the differentiation and effector functions of T(H)1 and T(H)9 cells. J. Immunother. Cancer 2022, 10, e003459. [Google Scholar] [CrossRef]
- Xu, N.; Palmer, D.C.; Robeson, A.C.; Shou, P.; Bommiasamy, H.; Laurie, S.J.; Willis, C.; Dotti, G.; Vincent, B.G.; Restifo, N.P.; et al. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J. Exp. Med. 2021, 218, e20200844. [Google Scholar] [CrossRef]
- Chan, J.D.; Lai, J.; Slaney, C.Y.; Kallies, A.; Beavis, P.A.; Darcy, P.K. Cellular networks controlling T cell persistence in adoptive cell therapy. Nat. Rev. Immunol. 2021, 21, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2015, 112, 15408–15413. [Google Scholar] [CrossRef] [PubMed]
- Samji, T.; Khanna, K.M. Understanding memory CD8(+) T cells. Immunol. Lett. 2017, 185, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Koyama, S. Mechanisms of regulatory T cell infiltration in tumors: Implications for innovative immune precision therapies. J. Immunother. Cancer 2021, 9, e002591. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Nishikawa, H. Roles of regulatory T cells in cancer immunity. Int. Immunol. 2016, 28, 401–409. [Google Scholar] [CrossRef]
- Kang, J.H.; Zappasodi, R. Modulating Treg stability to improve cancer immunotherapy. Trends Cancer 2023, 9, 911–927. [Google Scholar] [CrossRef]
- Frank, M.J.; Hossain, N.M.; Bukhari, A.; Dean, E.; Spiegel, J.Y.; Claire, G.K.; Kirsch, I.; Jacob, A.P.; Mullins, C.D.; Lee, L.W.; et al. Monitoring of Circulating Tumor DNA Improves Early Relapse Detection After Axicabtagene Ciloleucel Infusion in Large B-Cell Lymphoma: Results of a Prospective Multi-Institutional Trial. J. Clin. Oncol. 2021, 39, 3034–3043. [Google Scholar] [CrossRef]
- Caligaris-Cappio, F.; Bertilaccio, M.T.; Scielzo, C. How the microenvironment wires the natural history of chronic lymphocytic leukemia. Semin. Cancer Biol. 2014, 24, 43–48. [Google Scholar] [CrossRef]
- Olejarz, W.; Sadowski, K.; Szulczyk, D.; Basak, G. Advancements in Personalized CAR-T Therapy: Comprehensive Overview of Biomarkers and Therapeutic Targets in Hematological Malignancies. Int. J. Mol. Sci. 2024, 25, 7743. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Rabinovich, E.; Pradhan, K.; Sica, R.A.; Bachier-Rodriguez, L.; Mantzaris, I.; Kornblum, N.; Shastri, A.; Gritsman, K.; Goldfinger, M.; Verma, A.; et al. Elevated LDH greater than 400 U/L portends poorer overall survival in diffuse large B-cell lymphoma patients treated with CD19 CAR-T cell therapy in a real world multi-ethnic cohort. Exp. Hematol. Oncol. 2021, 10, 55. [Google Scholar] [CrossRef] [PubMed]
- Vercellino, L.; Di Blasi, R.; Kanoun, S.; Tessoulin, B.; Rossi, C.; D’Aveni-Piney, M.; Oberic, L.; Bodet-Milin, C.; Bories, P.; Olivier, P.; et al. Predictive factors of early progression after CAR T-cell therapy in relapsed/refractory diffuse large B-cell lymphoma. Blood Adv. 2020, 4, 5607–5615. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, A.V.; Gauthier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Gooley, T.; Li, D.; Cherian, S.; Chen, X.; Pender, B.S.; et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood 2019, 133, 1876–1887. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wu, Z.; Luo, Y.; Shi, J.; Yu, J.; Pu, C.; Liang, Z.; Wei, G.; Cui, Q.; Sun, J.; et al. Potent Anti-leukemia Activities of Chimeric Antigen Receptor-Modified T Cells against CD19 in Chinese Patients with Relapsed/Refractory Acute Lymphocytic Leukemia. Clin. Cancer Res. 2017, 23, 3297–3306. [Google Scholar] [CrossRef] [PubMed]
- Odorizzi, P.M.; Wherry, E.J. Inhibitory receptors on lymphocytes: Insights from infections. J. Immunol. 2012, 188, 2957–2965. [Google Scholar] [CrossRef]
- Zolov, S.N.; Rietberg, S.P.; Bonifant, C.L. Programmed cell death protein 1 activation preferentially inhibits CD28.CAR-T cells. Cytotherapy 2018, 20, 1259–1266. [Google Scholar] [CrossRef]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef]
- Armand, P. Immune checkpoint blockade in hematologic malignancies. Blood 2015, 125, 3393–3400. [Google Scholar] [CrossRef]
- Tunger, A.; Sommer, U.; Wehner, R.; Kubasch, A.S.; Grimm, M.O.; Bachmann, M.P.; Platzbecker, U.; Bornhauser, M.; Baretton, G.; Schmitz, M. The Evolving Landscape of Biomarkers for Anti-PD-1 or Anti-PD-L1 Therapy. J. Clin. Med. 2019, 8, 1534. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Finney, O.C.; Brakke, H.M.; Rawlings-Rhea, S.; Hicks, R.; Doolittle, D.; Lopez, M.; Futrell, R.B.; Orentas, R.J.; Li, D.; Gardner, R.A.; et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J. Clin. Investig. 2019, 129, 2123–2132. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- Berraondo, P.; Etxeberria, I.; Ponz-Sarvise, M.; Melero, I. Revisiting Interleukin-12 as a Cancer Immunotherapy Agent. Clin. Cancer Res. 2018, 24, 2716–2718. [Google Scholar] [CrossRef]
- Liu, J.; Cao, S.; Kim, S.; Chung, E.Y.; Homma, Y.; Guan, X.; Jimenez, V.; Ma, X. Interleukin-12: An update on its immunological activities, signaling and regulation of gene expression. Curr. Immunol. Rev. 2005, 1, 119–137. [Google Scholar] [CrossRef]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef]
- Kueberuwa, G.; Kalaitsidou, M.; Cheadle, E.; Hawkins, R.E.; Gilham, D.E. CD19 CAR T Cells Expressing IL-12 Eradicate Lymphoma in Fully Lymphoreplete Mice through Induction of Host Immunity. Mol. Ther. Oncolytics 2018, 8, 41–51. [Google Scholar] [CrossRef]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.; Yeh, R.; Bernal, Y.; Riviere, I.; Sadelain, M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: Case report of an unforeseen adverse event in a phase I clinical trial. Mol. Ther. 2010, 18, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Mirlekar, B. Tumor promoting roles of IL-10, TGF-beta, IL-4, and IL-35: Its implications in cancer immunotherapy. SAGE Open Med. 2022, 10, 20503121211069012. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [PubMed]
- Klaver, Y.; van Steenbergen, S.C.L.; Sleijfer, S.; Debets, R.; Lamers, C.H.J. Plasma IFN-gamma and IL-6 levels correlate with peripheral T-cell numbers but not toxicity in RCC patients treated with CAR T-cells. Clin. Immunol. 2016, 169, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Underhill, H.R.; Kitzman, J.O.; Hellwig, S.; Welker, N.C.; Daza, R.; Baker, D.N.; Gligorich, K.M.; Rostomily, R.C.; Bronner, M.P.; Shendure, J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016, 12, e1006162. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef]
- Acharya, U.H.; Dhawale, T.; Yun, S.; Jacobson, C.A.; Chavez, J.C.; Ramos, J.D.; Appelbaum, J.; Maloney, D.G. Management of cytokine release syndrome and neurotoxicity in chimeric antigen receptor (CAR) T cell therapy. Expert Rev. Hematol. 2019, 12, 195–205. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Turtle, C.J. Toxicities of CD19 CAR-T cell immunotherapy. Am. J. Hematol. 2019, 94, S42–S49. [Google Scholar] [CrossRef]
- Chou, C.K.; Turtle, C.J. Insight into mechanisms associated with cytokine release syndrome and neurotoxicity after CD19 CAR-T cell immunotherapy. Bone Marrow Transplant. 2019, 54, 780–784. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.L.; Schmitt, M.; Wang, L.; Ramos, C.A.; Jordan, K.; Muller-Tidow, C.; Dreger, P. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann. Oncol. 2021, 32, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Rasche, L.; Kortüm, K.M.; Danhof, S.; Hudecek, M.; Einsele, H. Toxicities of Chimeric Antigen Receptor T Cell Therapy in Multiple Myeloma: An Overview of Experience From Clinical Trials, Pathophysiology, and Management Strategies. Front. Immunol. 2020, 11, 620312. [Google Scholar] [CrossRef] [PubMed]
- Markouli, M.; Ullah, F.; Unlu, S.; Omar, N.; Lopetegui-Lia, N.; Duco, M.; Anwer, F.; Raza, S.; Dima, D. Toxicity Profile of Chimeric Antigen Receptor T-Cell and Bispecific Antibody Therapies in Multiple Myeloma: Pathogenesis, Prevention and Management. Curr. Oncol. 2023, 30, 6330–6352. [Google Scholar] [CrossRef] [PubMed]
- Cosenza, M.; Sacchi, S.; Pozzi, S. Cytokine Release Syndrome Associated with T-Cell-Based Therapies for Hematological Malignancies: Pathophysiology, Clinical Presentation, and Treatment. Int. J. Mol. Sci. 2021, 22, 7652. [Google Scholar] [CrossRef]
- Abramson, J.S.; Solomon, S.R.; Arnason, J.; Johnston, P.B.; Glass, B.; Bachanova, V.; Ibrahimi, S.; Mielke, S.; Mutsaers, P.; Hernandez-Ilizaliturri, F.; et al. Lisocabtagene maraleucel as second-line therapy for large B-cell lymphoma: Primary analysis of the phase 3 TRANSFORM study. Blood 2023, 141, 1675–1684. [Google Scholar] [CrossRef]
- National Cancer Institute. Common Terminology Criteria for Adverse Events Version 6.0; National Cancer Institute: Bethesda, MD, USA, 2020. [Google Scholar]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Maus, M.V.; Alexander, S.; Bishop, M.R.; Brudno, J.N.; Callahan, C.; Davila, M.L.; Diamonte, C.; Dietrich, J.; Fitzgerald, J.C.; Frigault, M.J.; et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune effector cell-related adverse events. J. Immunother. Cancer 2020, 8, e001511. [Google Scholar] [CrossRef]
- Frey, N.; Porter, D. Cytokine release syndrome with chimeric antigen receptor T cell therapy. Biol. Blood Marrow Transplant. 2018, 25, e123–e127. [Google Scholar] [CrossRef]
- Xiao, X.; Huang, S.; Chen, S.; Wang, Y.; Sun, Q.; Xu, X.; Li, Y. Mechanisms of cytokine release syndrome and neurotoxicity of CAR T-cell therapy and associated prevention and management strategies. J. Exp. Clin. Cancer Res. 2021, 40, 367. [Google Scholar] [CrossRef]
- Chou, C.K.; Turtle, C.J. Assessment and management of cytokine release syndrome and neurotoxicity following CD19 CAR-T cell therapy. Expert Opin. Biol. Ther. 2020, 20, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.; Frey, N.; Wood, P.A.; Weng, Y.; Grupp, S.A. Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J. Hematol. Oncol. 2018, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, M.; Jain, T.; Santomasso, B.D.; Mead, E.; Wudhikarn, K.; Silverberg, M.L.; Batlevi, Y.; Shouval, R.; Devlin, S.M.; Batlevi, C.; et al. Comparing CAR T-cell toxicity grading systems: Application of the ASTCT grading system and implications for management. Blood Adv. 2020, 4, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Yanez, L.; Sanchez-Escamilla, M.; Perales, M.A. CAR T Cell Toxicity: Current Management and Future Directions. Hemasphere 2019, 3, e186. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra225. [Google Scholar] [CrossRef]
- Hay, K.A.; Hanafi, L.A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; Lopez, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef]
- Fishman, J.A.; Hogan, J.I.; Maus, M.V. Inflammatory and Infectious Syndromes Associated With Cancer Immunotherapies. Clin. Infect. Dis. 2019, 69, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Westin, J.R.; Neelapu, S.S.; Green, M.R.; Shpall, E.J.; Nair, R.; Rodriguez, M.A.; Do, B.; Brown, A.R.T.; Horowitz, S.B.; Watson, G.; et al. Clinical efficacy of anakinra to mitigate CAR T-cell therapy–associated toxicity in large B-cell lymphoma. Blood Adv. 2020, 4, 3123–3127. [Google Scholar] [CrossRef]
- Holtzman, N.G.; Xie, H.; Bentzen, S.; Kesari, V.; Bukhari, A.; El Chaer, F.; Lutfi, F.; Siglin, J.; Hutnick, E.; Gahres, N.; et al. Immune effector cell-associated neurotoxicity syndrome after chimeric antigen receptor T-cell therapy for lymphoma: Predictive biomarkers and clinical outcomes. Neuro Oncol. 2021, 23, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Karschnia, P.; Jordan, J.T.; Forst, D.A.; Arrillaga-Romany, I.C.; Batchelor, T.T.; Baehring, J.M.; Clement, N.F.; Gonzalez Castro, L.N.; Herlopian, A.; Maus, M.V.; et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood 2019, 133, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef]
- Brown, B.D.; Tambaro, F.P.; Kohorst, M.; Chi, L.; Mahadeo, K.M.; Tewari, P.; Petropoulos, D.; Slopis, J.M.; Sadighi, Z.; Khazal, S. Immune Effector Cell Associated Neurotoxicity (ICANS) in Pediatric and Young Adult Patients Following Chimeric Antigen Receptor (CAR) T-Cell Therapy: Can We Optimize Early Diagnosis? Front. Oncol. 2021, 11, 634445. [Google Scholar] [CrossRef]
- Rubin, D.B.; Al Jarrah, A.; Li, K.; LaRose, S.; Monk, A.D.; Ali, A.B.; Spendley, L.N.; Nikiforow, S.; Jacobson, C.; Vaitkevicius, H. Clinical Predictors of Neurotoxicity After Chimeric Antigen Receptor T-Cell Therapy. JAMA Neurol. 2020, 77, 1536–1542. [Google Scholar] [CrossRef]
- Maziarz, R.T.; Schuster, S.J.; Romanov, V.V.; Rusch, E.S.; Li, J.; Signorovitch, J.E.; Maloney, D.G.; Locke, F.L. Grading of neurological toxicity in patients treated with tisagenlecleucel in the JULIET trial. Blood Adv. 2020, 4, 1440–1447. [Google Scholar] [CrossRef]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Chavez, J.C.; Sehgal, A.R.; William, B.M.; Munoz, J.; Salles, G.; Munshi, P.N.; Casulo, C.; Maloney, D.G.; de Vos, S.; et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): A single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022, 23, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Somerville, R.P.T.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J. Clin. Oncol. 2017, 35, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Santomasso, B.; Riviere, I.; Senechal, B.; Wang, X.; Purdon, T.; Wang, Y.; Halton, E.; Diamonte, C.; Li, D.; et al. Baseline and early post-treatment clinical and laboratory factors associated with severe neurotoxicity following 19-28z CAR T cells in adult patients with relapsed B-ALL. J. Clin. Oncol. 2017, 35, 7024. [Google Scholar] [CrossRef]
- Gofshteyn, J.S.; Shaw, P.A.; Teachey, D.T.; Grupp, S.A.; Maude, S.; Banwell, B.; Chen, F.; Lacey, S.F.; Melenhorst, J.J.; Edmonson, M.J.; et al. Neurotoxicity after CTL019 in a pediatric and young adult cohort. Ann. Neurol. 2018, 84, 537–546. [Google Scholar] [CrossRef]
- Shalabi, H.; Wolters, P.L.; Martin, S.; Toledo-Tamula, M.A.; Roderick, M.C.; Struemph, K.; Kane, E.; Yates, B.; Delbrook, C.; Mackall, C.L.; et al. Systematic Evaluation of Neurotoxicity in Children and Young Adults Undergoing CD22 Chimeric Antigen Receptor T-Cell Therapy. J. Immunother. 2018, 41, 350–358. [Google Scholar] [CrossRef]
- Singh, N.; Hofmann, T.J.; Gershenson, Z.; Levine, B.L.; Grupp, S.A.; Teachey, D.T.; Barrett, D.M. Monocyte lineage-derived IL-6 does not affect chimeric antigen receptor T-cell function. Cytotherapy 2017, 19, 867–880. [Google Scholar] [CrossRef]
- Faramand, R.; Jain, M.; Staedtke, V.; Kotani, H.; Bai, R.; Reid, K.; Lee, S.B.; Spitler, K.; Wang, X.; Cao, B.; et al. Tumor Microenvironment Composition and Severe Cytokine Release Syndrome (CRS) Influence Toxicity in Patients with Large B-Cell Lymphoma Treated with Axicabtagene Ciloleucel. Clin. Cancer Res. 2020, 26, 4823–4831. [Google Scholar] [CrossRef]
- Butt, O.H.; Zhou, A.Y.; Caimi, P.F.; Luckett, P.H.; Wisch, J.K.; Derenoncourt, P.R.; Lee, K.; Wu, G.F.; de Lima, M.J.G.; Campian, J.L.; et al. Assessment of Pretreatment and Posttreatment Evolution of Neurofilament Light Chain Levels in Patients Who Develop Immune Effector Cell-Associated Neurotoxicity Syndrome. JAMA Oncol. 2022, 8, 1652–1657. [Google Scholar] [CrossRef]
- Amidi, Y.; Eckhardt, C.A.; Quadri, S.A.; Malik, P.; Firme, M.S.; Jones, D.K.; Jain, A.; Danish, H.H.; Rubin, D.B.; Jacobson, C.A.; et al. Forecasting immune effector cell-associated neurotoxicity syndrome after chimeric antigen receptor t-cell therapy. J. Immunother. Cancer 2022, 10, e005459. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood J. Am. Soc. Hematol. 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Stone, J.B.; DeAngelis, L.M. Cancer-treatment-induced neurotoxicity--focus on newer treatments. Nat. Rev. Clin. Oncol. 2016, 13, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Danish, H.; Santomasso, B.D. Neurotoxicity Biology and Management. Cancer J. 2021, 27, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Sandler, R.D.; Tattersall, R.S.; Schoemans, H.; Greco, R.; Badoglio, M.; Labopin, M.; Alexander, T.; Kirgizov, K.; Rovira, M.; Saif, M.; et al. Diagnosis and management of secondary HLH/MAS following HSCT and CAR-T cell therapy in adults; A review of the literature and a survey of practice within EBMT centres on behalf of the Autoimmune Diseases Working Party (ADWP) and Transplant Complications Working Party (TCWP). Front. Immunol. 2020, 11, 524. [Google Scholar] [CrossRef]
- Hashmi, H.; Bachmeier, C.; Chavez, J.C.; Song, J.; Hussaini, M.; Krivenko, G.; Nishihori, T.; Kotani, H.; Davila, M.L.; Locke, F.L.; et al. Haemophagocytic lymphohistiocytosis has variable time to onset following CD19 chimeric antigen receptor T cell therapy. Br. J. Haematol. 2019, 187, e35–e38. [Google Scholar] [CrossRef] [PubMed]
- Penack, O.; Peczynski, C.; Koenecke, C.; Polge, E.; Kuhnl, A.; Fegueux, N.; Daskalakis, M.; Kroger, N.; Dreger, P.; Besley, C.; et al. Severe cytopenia after CD19 CAR T-cell therapy: A retrospective study from the EBMT Transplant Complications Working Party. J. Immunother. Cancer 2023, 11, e006406. [Google Scholar] [CrossRef]
- Logue, J.M.; Peres, L.C.; Hashmi, H.; Colin-Leitzinger, C.M.; Shrewsbury, A.M.; Hosoya, H.; Gonzalez, R.M.; Copponex, C.; Kottra, K.H.; Hovanky, V.; et al. Early cytopenias and infections after standard of care idecabtagene vicleucel in relapsed or refractory multiple myeloma. Blood Adv. 2022, 6, 6109–6119. [Google Scholar] [CrossRef]
- Santomasso, B.D.; Nastoupil, L.J.; Adkins, S.; Lacchetti, C.; Schneider, B.J.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated With Chimeric Antigen Receptor T-Cell Therapy: ASCO Guideline. J. Clin. Oncol. 2021, 39, 3978–3992. [Google Scholar] [CrossRef]
- Sharma, N.; Reagan, P.M.; Liesveld, J.L. Cytopenia after CAR-T Cell Therapy-A Brief Review of a Complex Problem. Cancers 2022, 14, 1501. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, J.; Li, J.; Zhang, L.; Li, J.; Fan, L.; Chen, L. Cytopenias following anti-CD19 chimeric antigen receptor (CAR) T cell therapy: A systematic analysis for contributing factors. Ann. Med. 2022, 54, 2951–2965. [Google Scholar] [CrossRef]
- Jain, T.; Olson, T.S.; Locke, F.L. How I treat cytopenias after CAR T-cell therapy. Blood 2023, 141, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Nahas, G.R.; Komanduri, K.V.; Pereira, D.; Goodman, M.; Jimenez, A.M.; Beitinjaneh, A.; Wang, T.P.; Lekakis, L.J. Incidence and risk factors associated with a syndrome of persistent cytopenias after CAR-T cell therapy (PCTT). Leuk. Lymphoma 2020, 61, 940–943. [Google Scholar] [CrossRef]
- Schaefer, A.; Saygin, C.; Maakaron, J.; Hoelscher, T.; Purdin, Z.; Robinson, J.; Lamprecht, M.; Penza, S.; Brammer, J.E.; Efebera, Y.A.; et al. Cytopenias after Chimeric Antigen Receptor T-Cells (CAR-T) Infusion; Patterns and Outcomes. Biol. Blood Marrow Transplant. 2019, 25, S171. [Google Scholar] [CrossRef]
- Gabelli, M.; Oporto-Espuelas, M.; Bonney, D.K.; Burridge, S.; Farish, S.; Mullanfiroze, K.; Lazareva, A.; Samarasinghe, S.; Ancliff, P.; Vora, A.; et al. Maintenance therapy for early loss of B-cell aplasia after CD19 CAR T-cell therapy. Blood Adv. 2023, 8, 1959–1963. [Google Scholar] [CrossRef] [PubMed]
- Wat, J.; Barmettler, S. Hypogammaglobulinemia After Chimeric Antigen Receptor (CAR) T-Cell Therapy: Characteristics, Management, and Future Directions. J. Allergy Clin. Immunol. Pract. 2022, 10, 460–466. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Arnold, D.E.; Maude, S.L.; Callahan, C.A.; DiNofia, A.M.; Grupp, S.A.; Heimall, J.R. Subcutaneous immunoglobulin replacement following CD19-specific chimeric antigen receptor T-cell therapy for B-cell acute lymphoblastic leukemia in pediatric patients. Pediatr. Blood Cancer 2020, 67, e28092. [Google Scholar] [CrossRef]
- Hill, J.A.; Giralt, S.; Torgerson, T.R.; Lazarus, H.M. CAR-T—And a side order of IgG, to go?—Immunoglobulin replacement in patients receiving CAR-T cell therapy. Blood Rev. 2019, 38, 100596. [Google Scholar] [CrossRef]
- Hernani, R.; Benzaquen, A.; Solano, C. Toxicities following CAR-T therapy for hematological malignancies. Cancer Treat. Rev. 2022, 111, 102479. [Google Scholar] [CrossRef] [PubMed]
- Kampouri, E.; Walti, C.S.; Gauthier, J.; Hill, J.A. Managing hypogammaglobulinemia in patients treated with CAR-T-cell therapy: Key points for clinicians. Expert Rev. Hematol. 2022, 15, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Buechner, J.; Grupp, S.A.; Hiramatsu, H.; Teachey, D.T.; Rives, S.; Laetsch, T.W.; Yanik, G.A.; Wood, P.; Awasthi, R.; Yi, L.; et al. Practical guidelines for monitoring and management of coagulopathy following tisagenlecleucel CAR T-cell therapy. Blood Adv. 2021, 5, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, C.; Xia, J.; Li, P.; Cao, J.; Pan, B.; Tan, X.; Li, H.; Qi, K.; Wang, X.; et al. Humoral immune reconstitution after anti-BCMA CAR T-cell therapy in relapsed/refractory multiple myeloma. Blood Adv. 2021, 5, 5290–5299. [Google Scholar] [CrossRef]
- Los-Arcos, I.; Iacoboni, G.; Aguilar-Guisado, M.; Alsina-Manrique, L.; Diaz de Heredia, C.; Fortuny-Guasch, C.; Garcia-Cadenas, I.; Garcia-Vidal, C.; Gonzalez-Vicent, M.; Hernani, R.; et al. Recommendations for screening, monitoring, prevention, and prophylaxis of infections in adult and pediatric patients receiving CAR T-cell therapy: A position paper. Infection 2021, 49, 215–231. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| T-Cell Subset | Surface Marker Expression | References | |
|---|---|---|---|
| Naive T Cells (TN) | CD45RO-, CCR7+, CD45RA+, CD62L+, CD95-, | [22,72,96,97,98,99] |  |
| Stem-cell-memory T cells (TSCM) | CD45RO+, CCR7+, CD45RA+, CD62L+, CD95+ | [22,72,96,97,99] | |
| Central memory T cells (TCM) | CD45RO+, CD45RA-, CCR7+, CD62L+, CD95+ | [22,72,96,97,99,100] | |
| Effector memory T cells (TEM) | CD45RO+, CD45RA-, CCR7-, CD62L-, CD95+ | [22,72,96,97,99,100] | |
| Effector T cells (TEFF) | CD45RO-, CD45RA+, CCR7-, CD62L-, CD95+ | [72,97,103] |
| CRS Parameter | Grade 1 | Grade 2 | Grade 3 | Grade 4 |
|---|---|---|---|---|
| Fever | Temperature ≥ 38 °C | Temperature ≥ 38 °C | Temperature ≥ 38 °C | Temperature ≥ 38 °C |
| Hypotension | None | Not requiring vasopressors | Requiring a vasopressor with or without vasopressin | Requiring multiple vasopressors (excluding vasopressin) |
| Hypoxia | None | Requiring low-flow nasal cannula‡ or blow-by | Requiring high-flow nasal cannula‡, facemask, nonrebreather mask, or Venturi mask | Requiring positive pressure (e.g., CPAP, BiPAP, intubation and mechanical ventilation) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ploch, W.; Sadowski, K.; Olejarz, W.; Basak, G.W. Advancement and Challenges in Monitoring of CAR-T Cell Therapy: A Comprehensive Review of Parameters and Markers in Hematological Malignancies. Cancers 2024, 16, 3339. https://doi.org/10.3390/cancers16193339
Ploch W, Sadowski K, Olejarz W, Basak GW. Advancement and Challenges in Monitoring of CAR-T Cell Therapy: A Comprehensive Review of Parameters and Markers in Hematological Malignancies. Cancers. 2024; 16(19):3339. https://doi.org/10.3390/cancers16193339
Chicago/Turabian StylePloch, Weronika, Karol Sadowski, Wioletta Olejarz, and Grzegorz W. Basak. 2024. "Advancement and Challenges in Monitoring of CAR-T Cell Therapy: A Comprehensive Review of Parameters and Markers in Hematological Malignancies" Cancers 16, no. 19: 3339. https://doi.org/10.3390/cancers16193339