
Activation of Cobalt Foil Catalysts for CO Hydrogenation


Abstract
:
1. Introduction
2. Results and Discussion
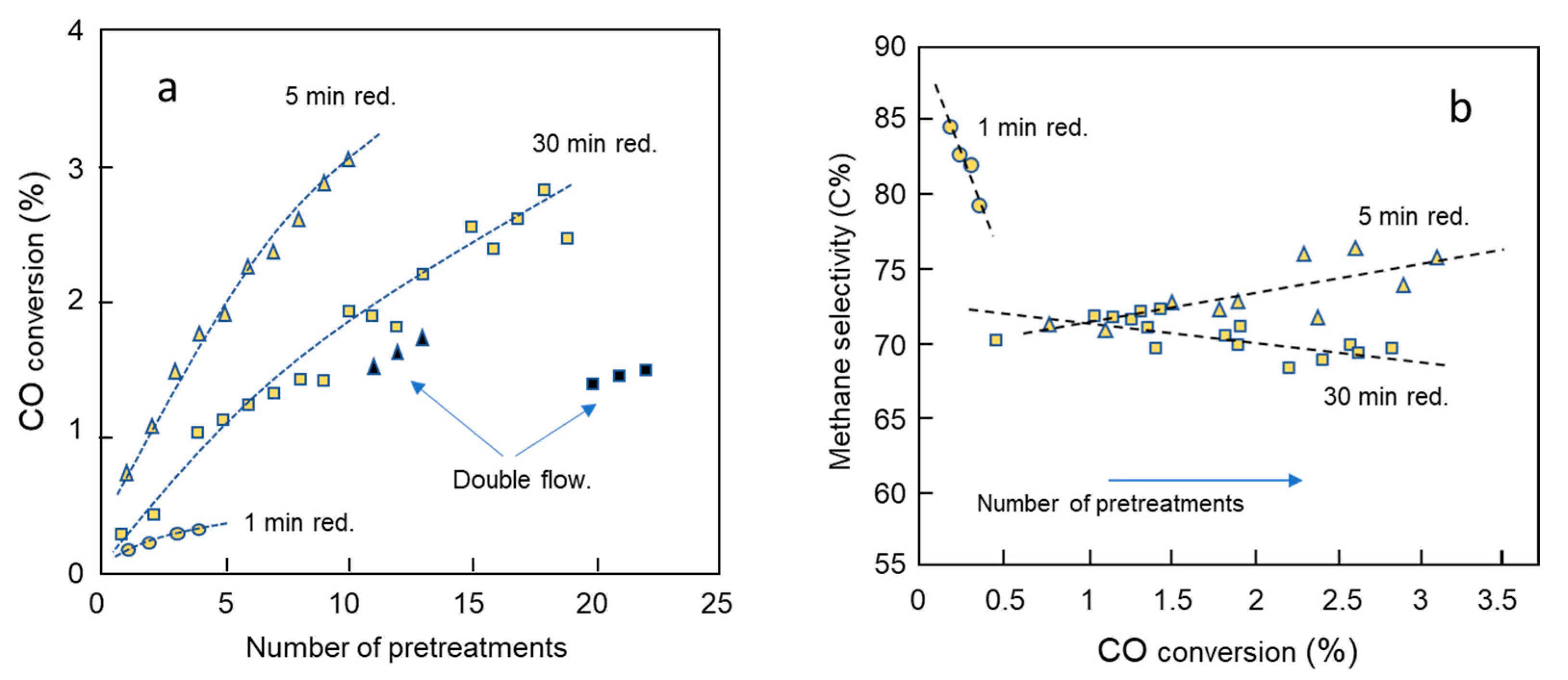
2.1. Series 1: Activity and Selectivity for the Variation in Reduction Time
2.2. Series 1: Paraffin to Olefin Ratio by the Variation of Reduction Time
- Residence time: The gas flow, or space velocity, determining the residence time of the olefins in the reactor often dominates olefin hydrogenation responses. In the present case, however, the flow rate was constant except for a few control experiments (see Figure 2).
- Loading and dispersion of cobalt: The available surface area of cobalt, i.e., the amount and crystallite size of cobalt and the degree of the reduction of the available cobalt oxide, plays a major role. This factor is partly dominating in the present study as the pretreatments constantly changed the amount of available oxide and its reduction.
- Hydrogen coverage: The coverage of hydrogen depends on the total pressure of hydrogen and the H2/CO feed ratio (and conversion), but is also related to the structure sensitivity of the cobalt FT process. The present work was conducted at a high H2/CO ratio of 3, thus favoring hydrogenation.
- Mass transfer: The diffusion of olefins is important for porous catalysts of a size above about 80 μm [3]. The thickness of the cobalt foil is only 50 μm and mass transfer should not be an issue, but actual pore sizes are not known.
- CO conversion (H2O coverage): The CO conversion as, e.g., a result of space velocity alteration, catalyst characteristics, or through a reactor, changes gas composition. In particular, it has been found that produced water inhibits olefin hydrogenation, probably due to the enhanced water coverage on cobalt at high CO conversions displacing hydrogen [7,27,36,37]. Since the conversion in the present study was below 6%, conversion is not considered as a critical parameter.
- Olefin readsorption: The incorporation of olefins during chain growth naturally reduces the amount of primary produced olefins. However, olefin readsorption has been found to be of minor importance for chain growth [17,18]. Nevertheless, it cannot be ruled out that readsorption played a role in the present study due to a light product slate.
- Activation of olefin: Limited information is available on light α-olefin adsorption and double bond activation on cobalt facets under FTS conditions. This factor has been neglected in the following discussion.
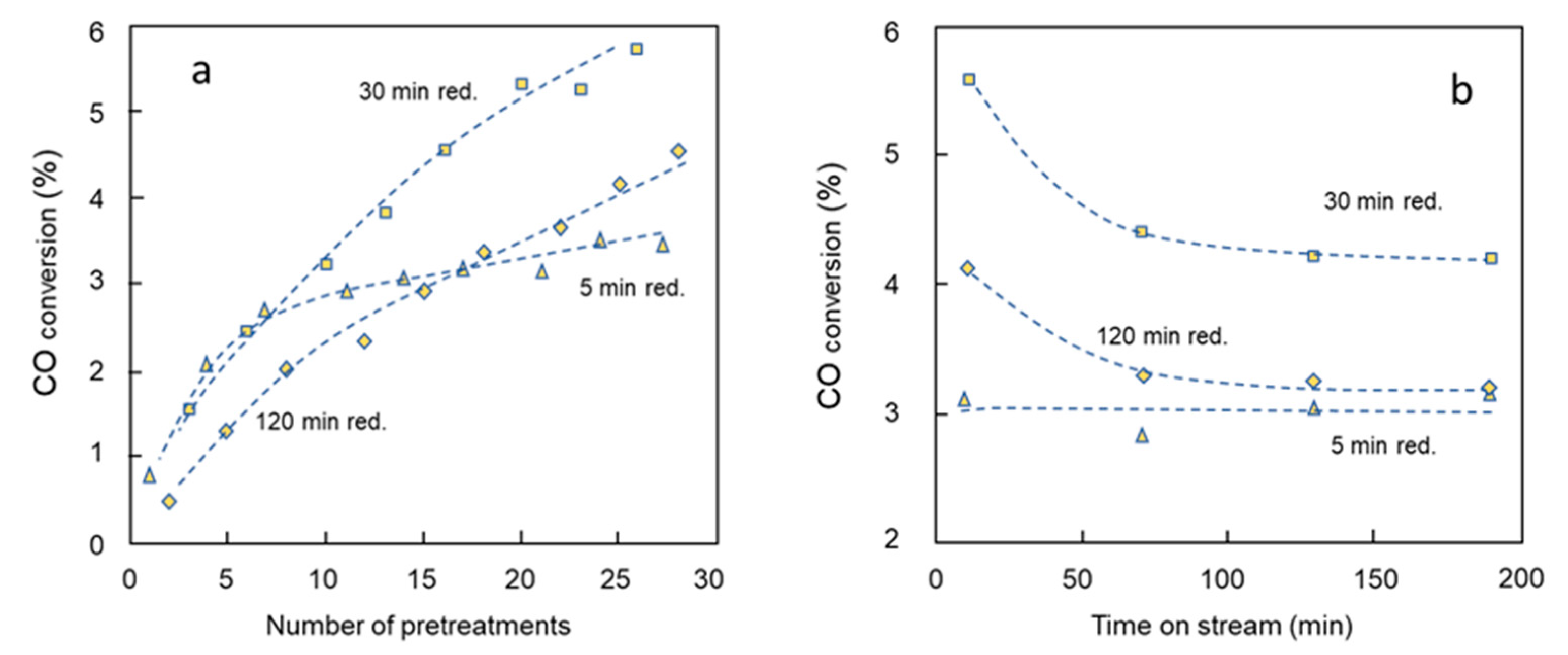
2.3. Series 2: Activity and Selectivity for a Sequential Variation in Reduction Time
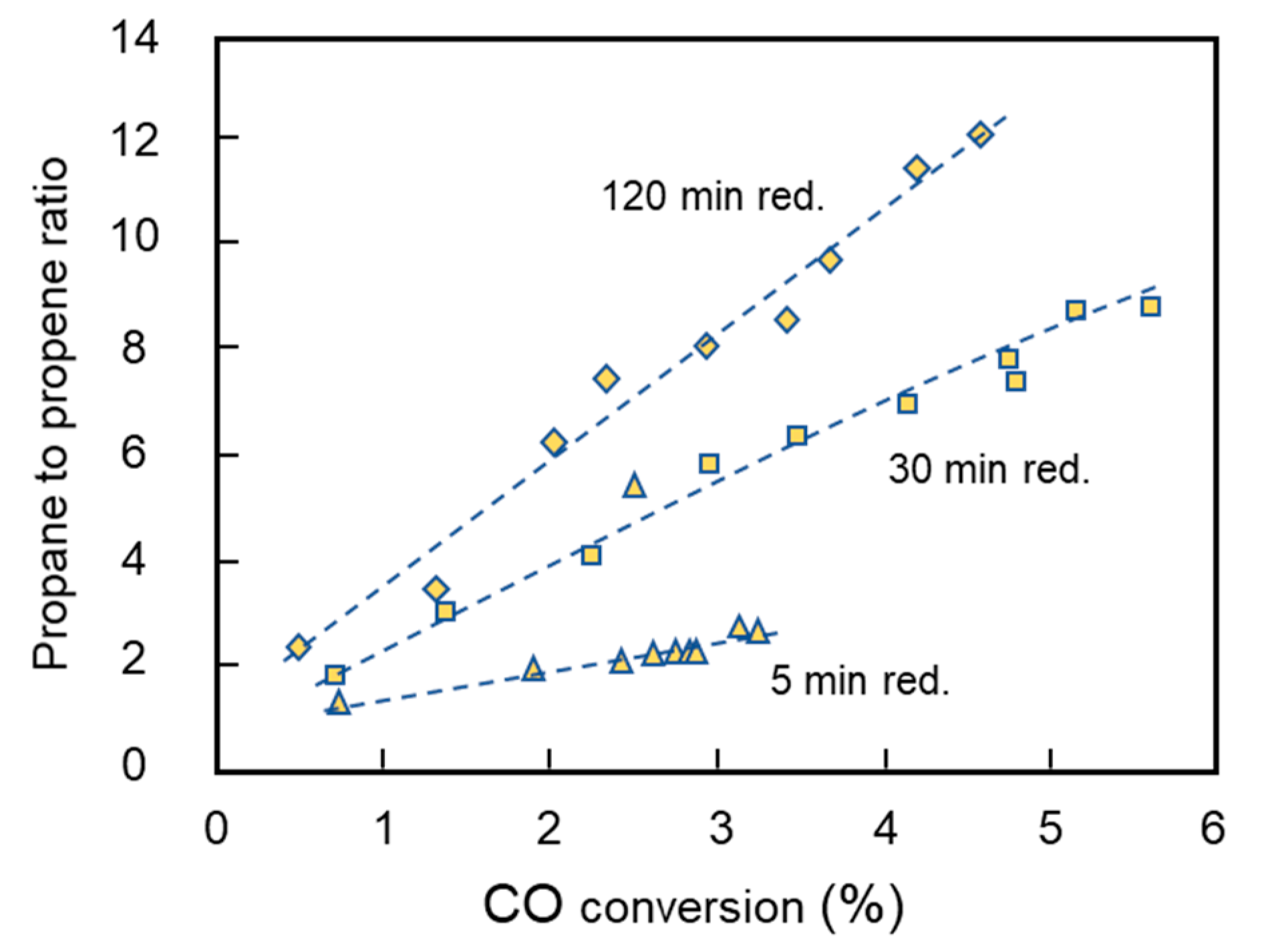
2.4. Series 2: Paraffin to Olefin Ratio by a Sequential Variation in Reduction Time
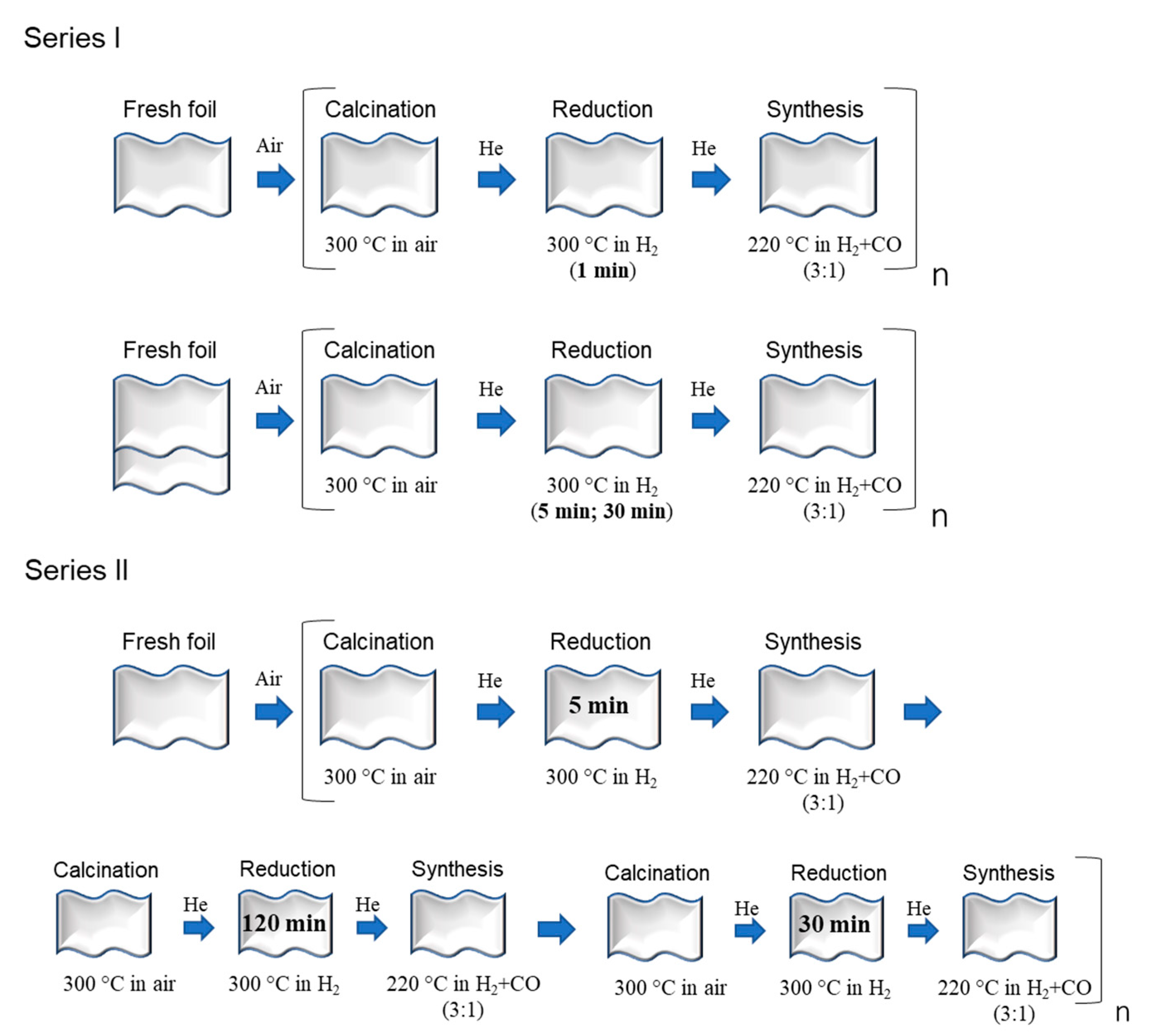
3. Experimental Section
4. Conclusions
- Increased surface area and deeper oxidation is created by repetitive calcination–oxidation–calcination.
- Longer reduction times favor a deepening of the catalytic layer.
- A short reduction time (1 min) gives small cobalt crystallites dispersed on unreduced oxide which are susceptible to low activity by structure sensitivity.
- A 5 min reduction time does not result in appreciable sintering, but does not completely reduce large crystallites formed by a 120 min reduction + calcination.
- A 30 min reduction gives some sintering, but can on the other hand re-disperse large crystallites after following a 120 min reduction + calcination.
- Prolonged reduction causes sintering.
- Small cobalt crystallites promote methane formation due to structure sensitivity.
- The hydrogenation of olefins is favored by repetitive pretreatments and deep oxidation/reduction.
- The hydrogenation of olefins largely follows development in the available cobalt surface area.
- The hydrogenation of olefins is not structure sensitive.
- There are indications of mass transfer limited hydrogenation after redispersion.
- Produced water (CO conversion < 6%) influences neither activity nor selectivity.
Author Contributions
Funding
Conflicts of Interest
References
- Hilmen, A.-M.; Bergene, E.; Lindvåg, O.A.; Schanke, D.; Eri, S.; Holmen, A. Fischer-Tropsch Synthesis Using Monolithic Catalysts. Stud. Surf. Sci. Catal. 2000, 130, 1163–1168. [Google Scholar]
- Iglesia, E. Design, Synthesis, and Use of Cobalt-Based Fischer-Tropsch Synthesis Catalysts. Appl. Catal. A Gen. 1997, 161, 59–78. [Google Scholar] [CrossRef]
- Rytter, E.; Eri, S.; Skagseth, T.H.; Schanke, D.; Bergene, E.; Myrstad, R.; Lindvåg, A. Catalyst Particle Size of Cobalt/Rhenium on Porous Alumina and the Effect on Fischer−Tropsch Catalytic Performance. Ind. Eng. Chem. Res. 2007, 46, 9032–9036. [Google Scholar] [CrossRef]
- Kuipers, E.W.; Scheper, C.; Wilson, J.H.; Vinkenburg, I.H.; Oosterbeek, H. Non-ASF Product Distributions Due to Secondary Reactions during Fischer-Tropsch synthesis. J. Catal. 1996, 158, 288–300. [Google Scholar] [CrossRef]
- Kuipers, E.; Vinkenburg, I.H.; Oosterbeek, H. Chain Length Dependence of α-Olefin Readsorption in Fischer-Tropsch Synthesis. J. Catal. 1995, 152, 137–146. [Google Scholar] [CrossRef]
- Van der Laan, G.P.; Beenackers, A.A.C.M. Kinetics and Selectivity of the Fischer–Tropsch Synthesis: A Literature Review. Catal. Rev. 1999, 41, 255–318. [Google Scholar] [CrossRef]
- Rytter, E.; Yang, J.; Borg, Ø.; Holmen, A. Significance of C3 Olefin to Paraffin Ratio in Cobalt Fischer–Tropsch Synthesis. Catalysts 2020, 10, 967. [Google Scholar] [CrossRef]
- Lojewska, J. Various forms of the Carbonaceous Deposit on the Model Cobalt Catalyst Studied in Hydrogenation of Ethylene. Stud. Surf. Sci. Catal. 2001, 139, 13–20. [Google Scholar]
- Lojewska, J.; Makowski, W.; Tyszewski, T.; Dziembaj, R. Active State of Model Cobalt Foil Catalyst Studied by SEM, TPR/TPO, XPS and TG. Catal. Today 2001, 69, 409–418. [Google Scholar] [CrossRef]
- Palmer, R.L.; Vroom, D.A. Mass-Spectrometric Measurements of Enhanced Methanation Activity over Cobalt and Nickel Foils. J. Catal. 1977, 50, 244–251. [Google Scholar] [CrossRef]
- Frolich, G.; Kestel, U.; Lojewska, J.; Lojewski, T.; Meyer, G.; Vob, M.; Borgmann, D.; Dziembaj, D.; Wedler, G. Activation and Deactivation of Cobalt Catalysts in the Hydrogenation of Carbon Dioxide. Appl. Catal. A 1996, 134, 1–19. [Google Scholar] [CrossRef]
- Jnioui, A.; Eddouasse, M.; Amariglio, A.; Ehrhardt, J.J.; Alnot, M.; Lambert, J.; Amariglio, H. Catalytic Activation of Cobalt Induced by Oxidizing Treatments in the Methanation of Carbon Dioxide. J. Catal. 1987, 106, 144–165. [Google Scholar] [CrossRef]
- Lahtinen, J.; Somorjai, G.A. The Role of Readsorption during CO Hydrogenation over Cobalt Model Catalysts. J. Molec. Catal. A 1994, 91, 387–397. [Google Scholar] [CrossRef]
- Yang, J.; Ma, W.; Chen, D.; Holmen, A.; Davis, B.H. Fischer–Tropsch Synthesis: A Review of the Effect of CO Conversion on Methane Selectivity. Appl. Catal. A Gen. 2014, 470, 250–260. [Google Scholar] [CrossRef]
- Zimmerman, W.; Bukur, D.; Ledakowicz, S. Kinetic Model of Fischer-Tropsch Synthesis Selectivity in the Slurry Phase. Chem. Eng. Sci. 1992, 47, 2707–2712. [Google Scholar] [CrossRef]
- Shi, B.; O’Brien, R.J.; Bao, S.; Davis, B.H. Mechanism of the Isomerization of 1-Alkene during Iron-Catalyzed Fischer-Tropsch Synthesis. J. Catal. 2001, 199, 202–208. [Google Scholar] [CrossRef]
- Shi, B.; Jacobs, G.; Sparks, D.; Davis, B.H. Fischer-Tropsch Synthesis: Accounting for Chain Length Phenomena. Fuel 2005, 84, 1093–1098. [Google Scholar] [CrossRef]
- Yang, J.; Shafer, W.D.; Pendyala, V.R.R.; Jacobs, G.; Chen, D.; Holmen, A.; Davis, B.H. Fischer-Tropsch Synthesis: Using Deuterium as a Tool to Investigate Primary Product Distribution. Catal. Lett. 2014, 144, 524–530. [Google Scholar] [CrossRef]
- Lahtinen, J.; Anraku, T.; Somorjai, G.A. Carbon Monoxide Hydrogenation on Cobalt Foil and on Thin Cobalt Film Model Catalysts. J. Catal. 1993, 142, 206–225. [Google Scholar] [CrossRef]
- Lahtinen, J.; Anraku, T.; Somorjai, G.A. C, CO and CO2 Hydrogenation on Cobalt Foil Model Catalysts: Evidence for the Need of CoO Reduction. Catal. Lett. 1994, 25, 241–255. [Google Scholar] [CrossRef]
- Lahtinen, J.; Somorjai, G.A. The Effects of Promoters in Carbon Monoxide Hydrogenation on Cobalt Foil Model Catalysts. J. Mol. Catal. A 1998, 130, 255–260. [Google Scholar] [CrossRef]
- Wu, C.H.; Eren, B.; Bluhm, H.; Salmeron, M.B. Ambient Pressure Photoelectron Spectroscopy Study of Cobalt Foil Model Catalyst under CO, H2 and Their Mixtures. ACS Catal. 2017, 7, 1150–1157. [Google Scholar] [CrossRef]
- Kobylinski, T.P.; Kibby, C.L.; Pannell, R.B.; Eddy, E.L. Synthesis gas conversion using ROR-activated catalyst. U.S. Patent No. 4605676, 12 August 1986. [Google Scholar]
- Bezemer, G.L.; Bitter, J.H.; Kuipers, H.P.C.E.; Oosterbeek, H.; Holewijn, J.E.; Xu, X.; Kapteijn, F.; van Dillen, A.J.; de Jong, K.P. Cobalt Particle Size Effects in the Fischer-Tropsch Reaction Studied with Carbon Nanofiber Supported Catalysts. J. Am. Chem. Soc. 2006, 128, 3956–3964. [Google Scholar] [CrossRef] [Green Version]
- Borg, Ø.; Dietzel, P.D.C.; Spjelkavik, A.I.; Tveten, E.Z.; Walmsley, J.C.; Eri, S.; Holmen, A.; Rytter, E. Fischer-Tropsch Synthesis: Cobalt Particle Size and Support Effects on Intrinsic Activity and Product Distribution. J. Catal. 2008, 259, 161–164. [Google Scholar] [CrossRef]
- Rane, S.; Borg, Ø.; Rytter, E.; Holmen, A. Relation between Hydrocarbon Selectivity and Cobalt Particle Size for Alumina Supported Cobalt Fischer-Tropsch Catalysts. Appl. Catal. A Gen. 2012, 437–438, 10–17. [Google Scholar] [CrossRef]
- Iglesia, E.; Reyes, S.C.; Madon, R.J.; Soled, S. Selectivity Control and Catalyst Design in the Fischer-Tropsch Synthesis: Sites, Pellets, and Reactors. Adv. Catal. 1993, 39, 221–302. [Google Scholar]
- Borg, Ø.; Eri, S.; Storsæter, S.; Blekkan, E.A.; Wigum, H.; Rytter, E.; Holmen, A. Fischer–Tropsch Synthesis over γ-Alumina-Supported Cobalt Catalysts: Effect of Support Variables. J. Catal. 2007, 248, 89–100. [Google Scholar] [CrossRef]
- Haller, G.L.; Resasco, D.E. Metal-Support Interaction: Group VIII Metals and Reducible Oxides. Adv. Catal. 1989, 36, 173–235. [Google Scholar]
- Ducreux, O.; Rebours, B.; Lynch, J.; Roy-Auberger, M.; Bazin, D. Microstructure of Supported Cobalt Fischer-Tropsch Catalysts. Oil Gas Sci. Technol. Rev. IFP 2009, 64, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Tsakoumis, N.E.; Patanou, E.; Lögdberg, S.; Johnsen, R.E.; Myrstad, R.; van Beek, W.; Rytter, E.; Blekkan, E.A. Structure-Performance Relationships on Co-Based Fischer-Tropsch Synthesis Catalysts: The More Defect-Free, the Better. ACS Catal. 2019, 9, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Keyvanloo, K.; Fischer, M.J.; Hecker, W.C.; Lancee, R.J.; Jacobs, G.; Bartholomew, C.H. Kinetics of Deactivation by Carbon of a Cobalt Fischer-Tropsch Catalyst: Effects of CO and H2 Partial Pressures. J. Catal. 2015, 327, 33–47. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Tu, M.; Ojeda, M.P.; Pinna, D.; Iglesia, E. An Investigation of the Effects of Water on Rate and Selectivity for the Fischer–Tropsch Synthesis on Cobalt-Based Catalysts. J. Catal. 2002, 211, 422–433. [Google Scholar] [CrossRef]
- Bertole, C.J.; Mims, C.A.; Kiss, G. The Effect of Water on the Cobalt-Catalyzed Fischer–Tropsch Synthesis. J. Catal. 2002, 210, 84–96. [Google Scholar] [CrossRef]
- Rytter, E.; Borg, Ø.; Tsakoumis, N.; Holmen, A. Water as Key to Activity and Selectivity in Co Fischer-Tropsch Synthesis: γ-Alumina Based Structure-Performance Relationships. J. Catal. 2018, 365, 334–343. [Google Scholar] [CrossRef]
- Hilmen, A.-M.; Lindvåg, O.A.; Bergene, E.; Schanke, D.; Eri, S.; Holmen, A. Selectivity and Activity Changes Upon Water Addition during Fischer-Tropsch Synthesis. Stud. Surf. Sci. Catal. 2001, 136, 295–300. [Google Scholar]
- Hilmen, A.-M.; Bergene, E.; Lindvag, O.A.; Schanke, D.; Eri, S.; Holmen, A. Fischer–Tropsch Synthesis on Monolithic Catalysts of Different Materials. Catal. Today 2001, 69, 227–232. [Google Scholar] [CrossRef]
- Rytter, E.; Holmen, A. Consorted Vinylene Mechanism for Cobalt Fischer-Tropsch Synthesis Encompassing Water or Hydroxyl Assisted CO-Activation. Top. Catal. 2018, 61, 1024–1034. [Google Scholar] [CrossRef]
- Rytter, E.; Tsakoumis, N.E.; Holmen, A. On the Selectivity to Higher Hydrocarbons in Co-Based Fischer–Tropsch Synthesis. Catal. Today 2016, 261, 3–16. [Google Scholar] [CrossRef]
- Hanssen, K.F.; Blekkan, E.A.; Schanke, D.; Holmen, A. Transient and Steady-State Studies of the Effect of Water on Cobalt Fischer-Tropsch Catalysts. Stud. Surf. Sci. Catal. 1997, 109, 193–202. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reduction Time (min) | Number of Pretreatments | CO Conversion (C%) | C1 (C%) | C2 (C%) | C3 (C%) | C4 (C%) | C5+ (C%) |
|---|---|---|---|---|---|---|---|
| 1 | 4 | 0.3 | 79.6 | 8.9 | 7.3 | 3.7 | 0.4 |
| 5 | 10 | 3.1 | 75.9 | 8.9 | 7.7 | 3.6 | 3.9 |
| 30 | 18 | 2.8 | 70.0 | 11.6 | 9.0 | 4.5 | 5.0 |
| Reduction Time (min) | Number of Pretreatments | CO Conversion (C%) | C1 (C%) | C2 (C%) | C3 (C%) | C4 (C%) | C5+ (C%) |
|---|---|---|---|---|---|---|---|
| 5 | 27 | 3.4 | 75.5 | 10.7 | 7.5 | 3.5 | 2.9 |
| 30 | 29 | 5.6 | 74.1 | 11.3 | 7.8 | 3.9 | 2.9 |
| 120 | 28 | 4.5 | 74.8 | 11.4 | 7.6 | 3.6 | 2.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rytter, E.; Aaserud, C.; Hilmen, A.-M.; Bergene, E.; Holmen, A. Activation of Cobalt Foil Catalysts for CO Hydrogenation. Catalysts 2022, 12, 65. https://doi.org/10.3390/catal12010065
Rytter E, Aaserud C, Hilmen A-M, Bergene E, Holmen A. Activation of Cobalt Foil Catalysts for CO Hydrogenation. Catalysts. 2022; 12(1):65. https://doi.org/10.3390/catal12010065
Chicago/Turabian StyleRytter, Erling, Christian Aaserud, Anne-Mette Hilmen, Edvard Bergene, and Anders Holmen. 2022. "Activation of Cobalt Foil Catalysts for CO Hydrogenation" Catalysts 12, no. 1: 65. https://doi.org/10.3390/catal12010065