
Oxidation in Flow Using an Ionic Immobilized TEMPO Catalyst on an Ion Exchange Resin

Abstract
:
1. Introduction
2. Results and Discussion
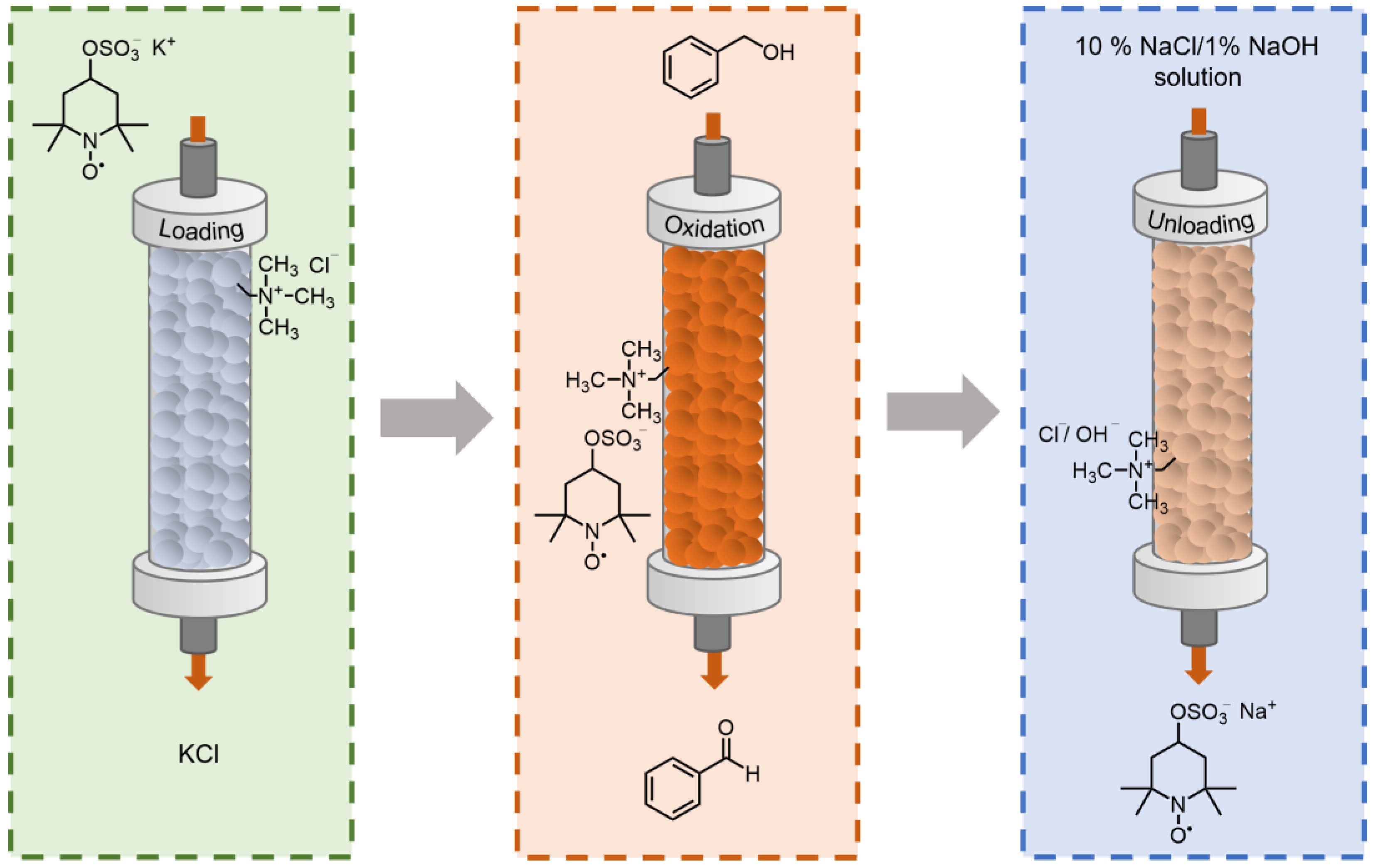
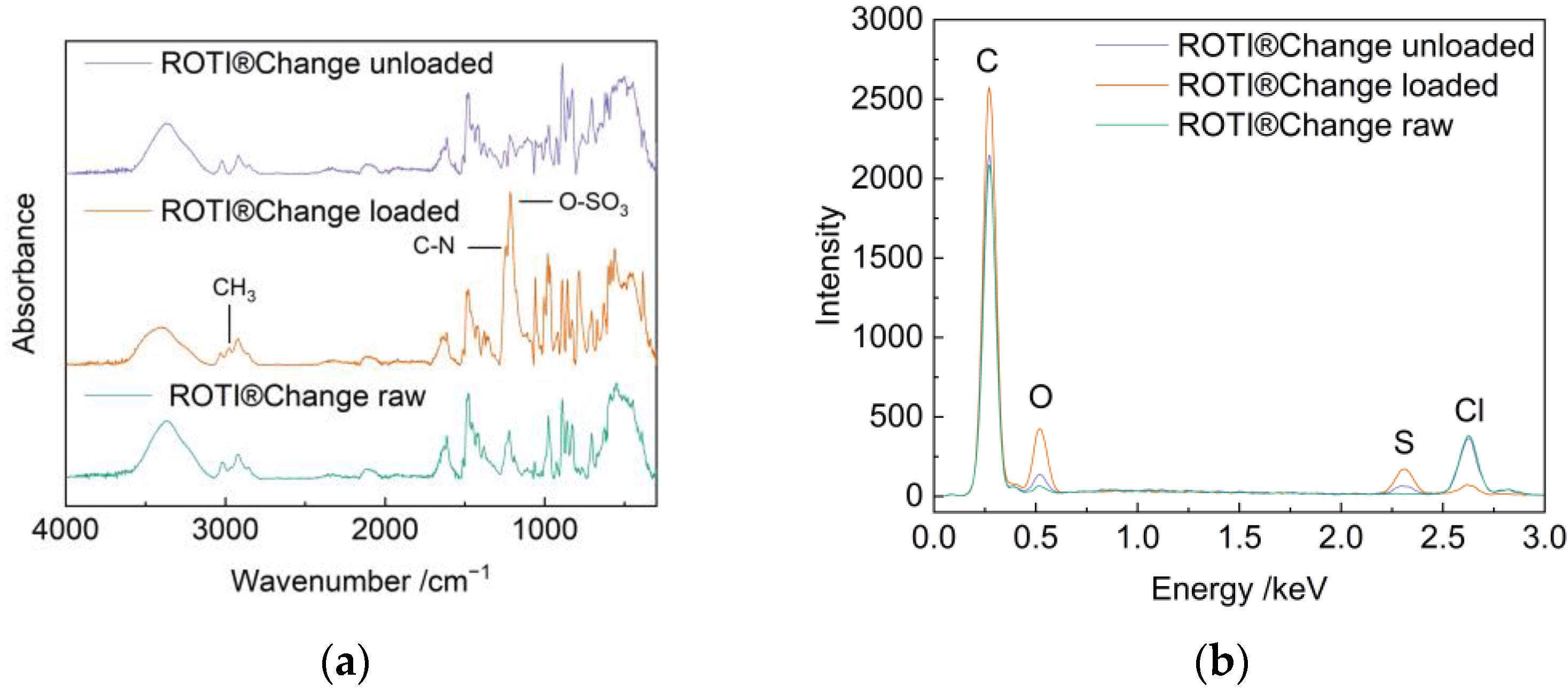
2.1. Preparation and Characterization of the Catalyst
2.2. Batch Oxidation Reactions with Batch-Immobilized TEMPO-4 Sulfate
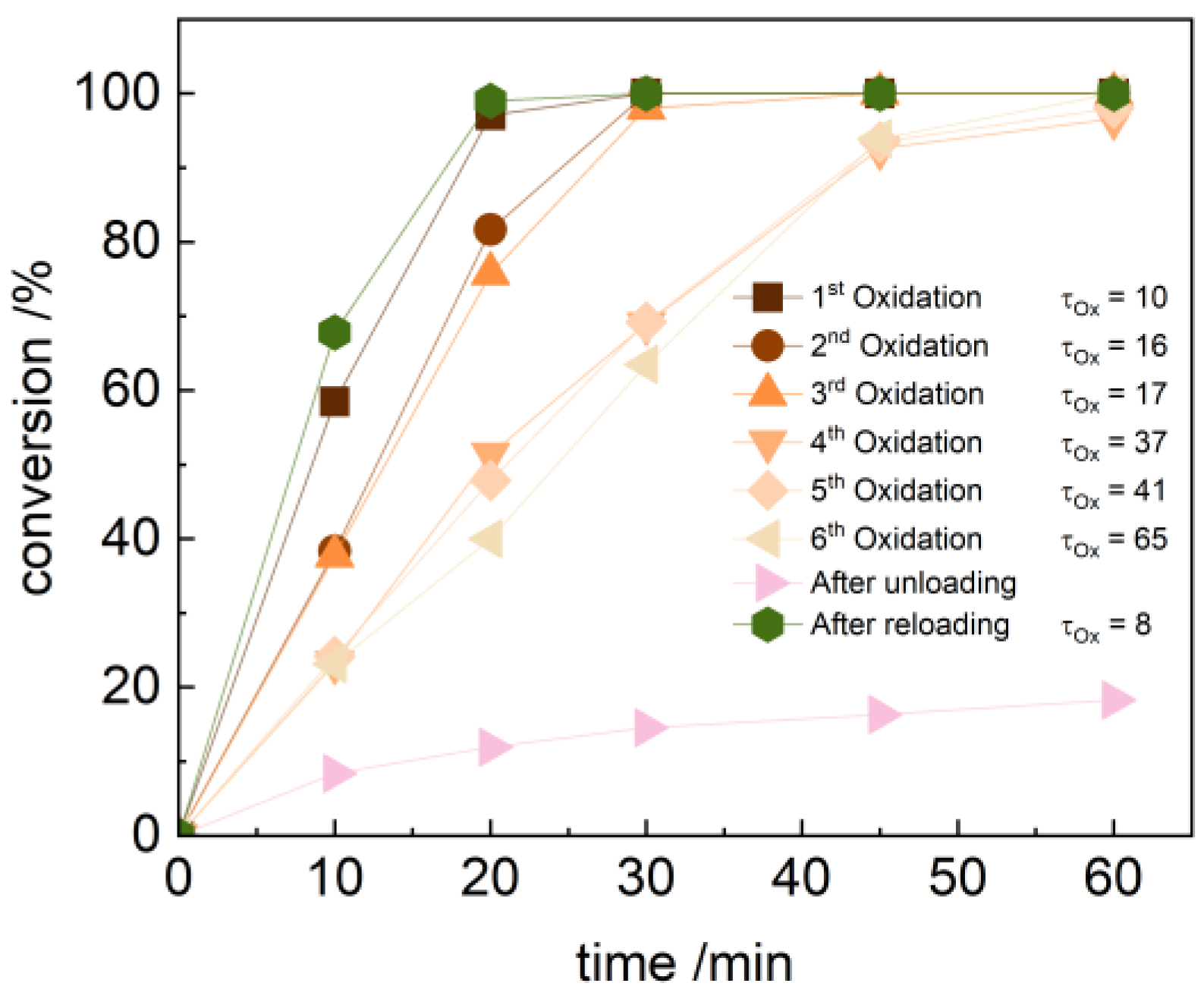
2.3. Catalyst Robustness in Batch Oxidations with Batch-Loaded Catalysts
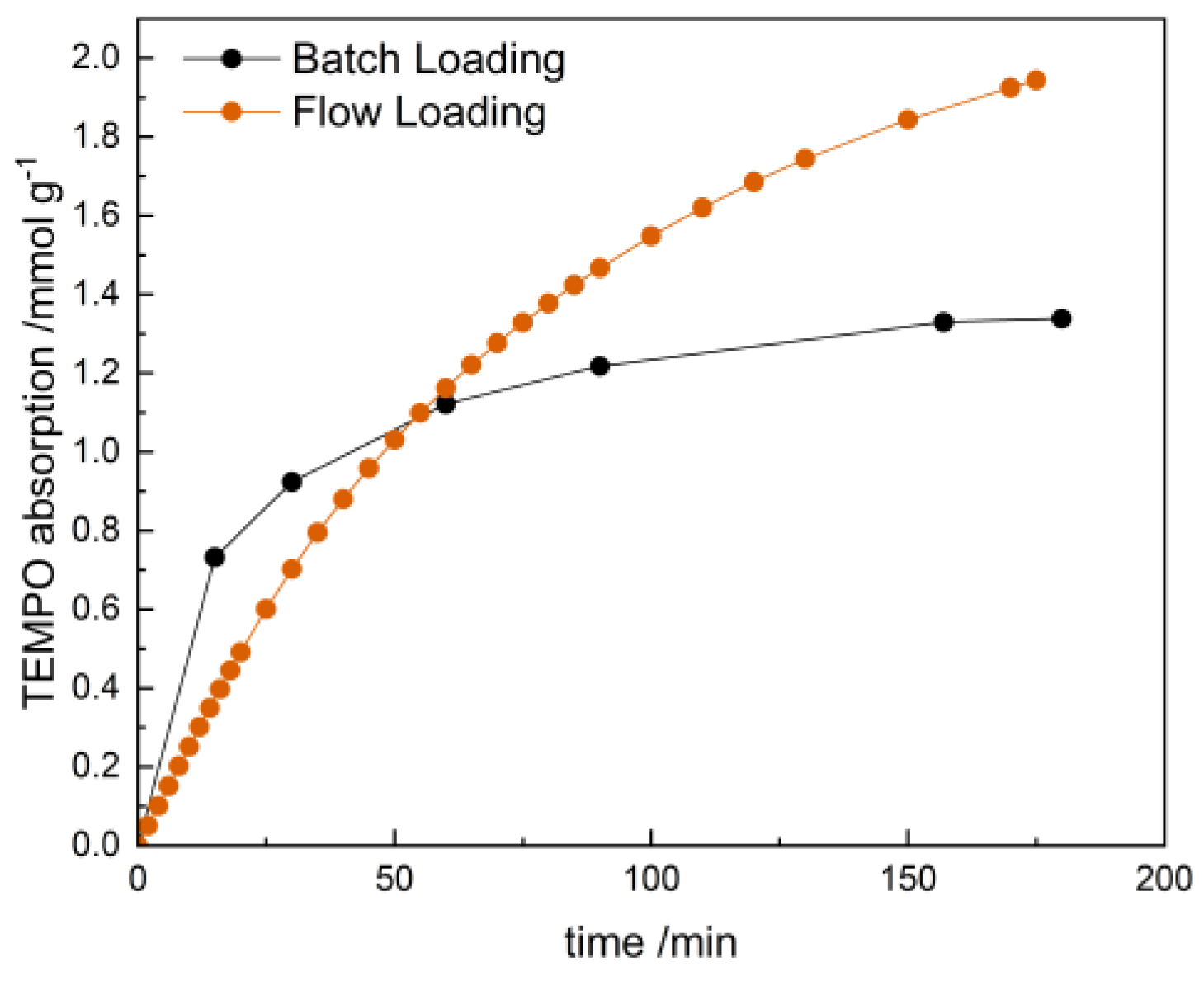
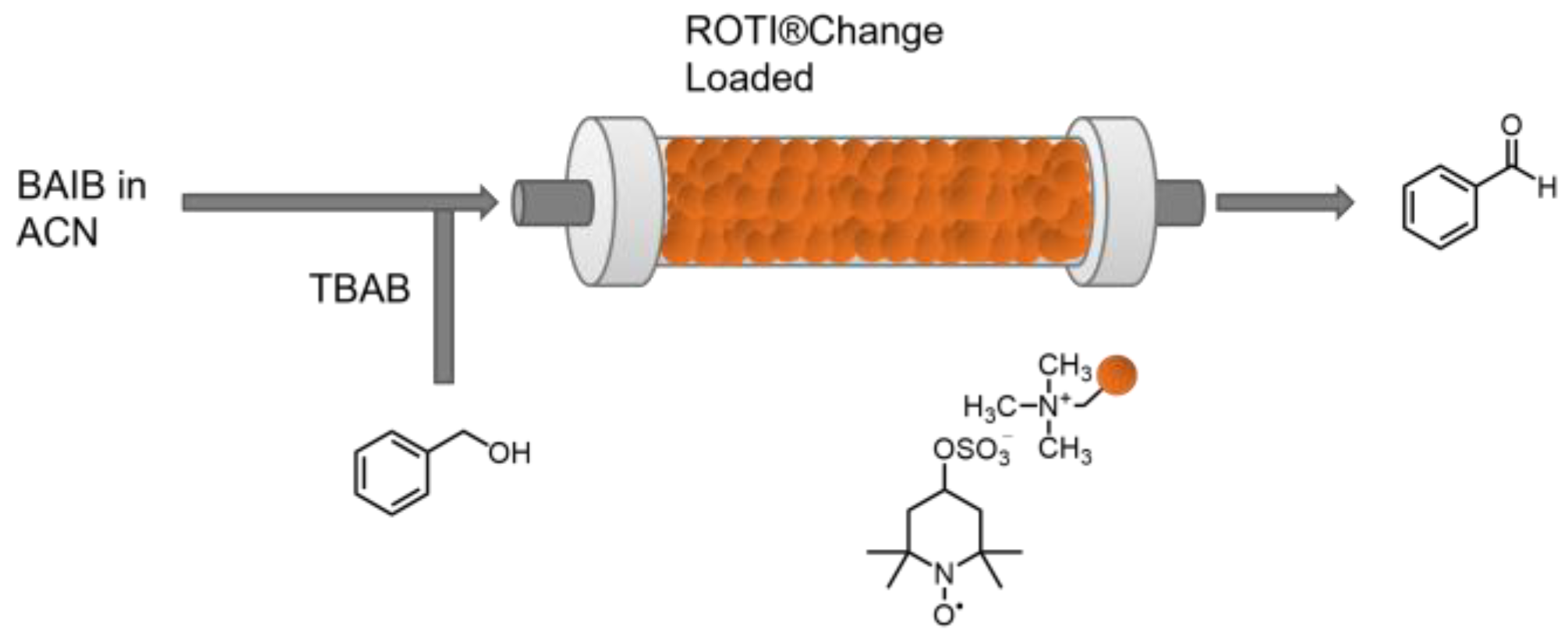
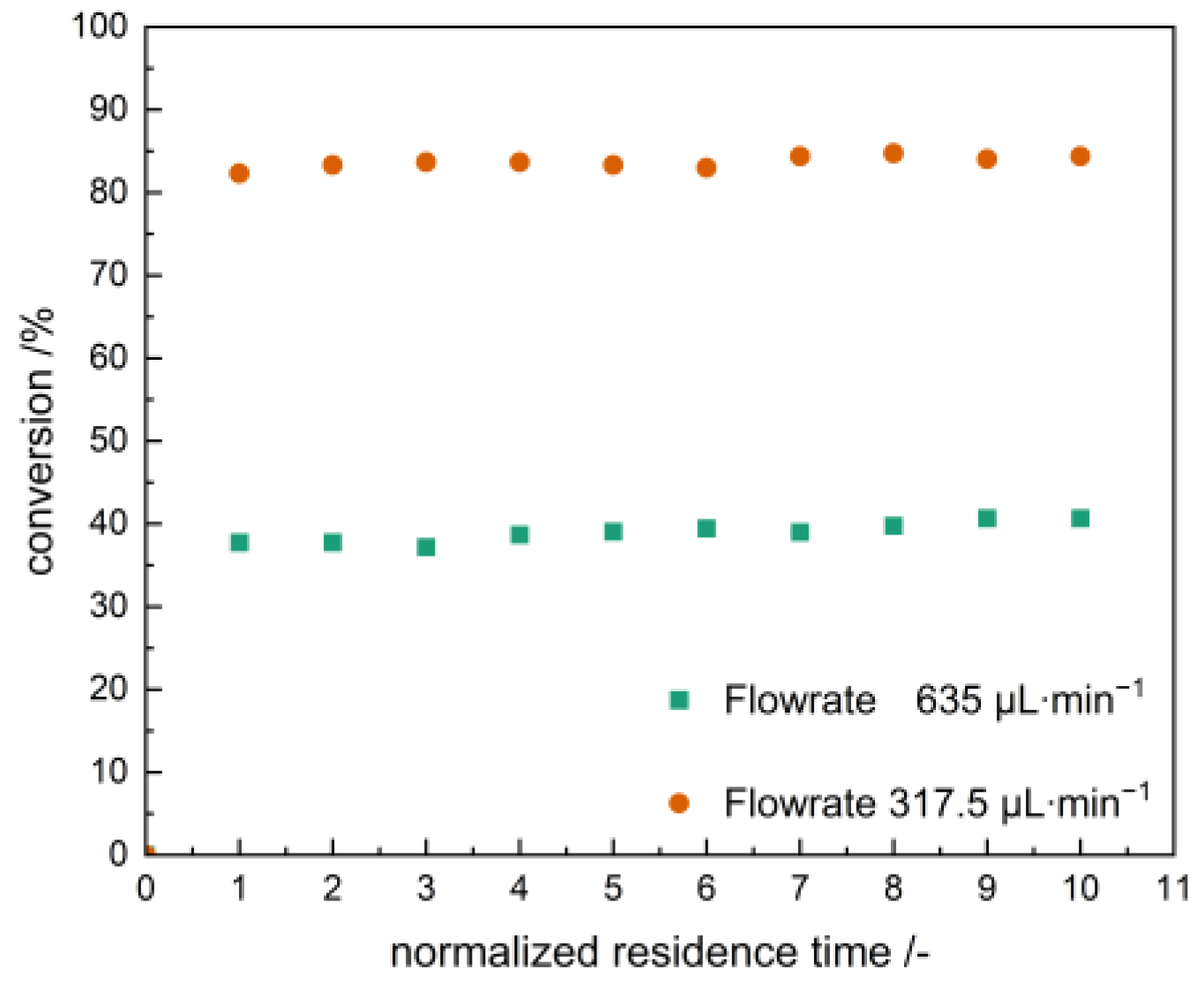
2.4. Oxidation Catalysis in a Flow Reactor with Flow-Loaded Catalyst
3. Materials and Methods
3.1. Materials
3.2. Characterization
3.3. Determination of the Actual Total Exchange Capacity of the Ion Exchange Resin by Chloride Ion Measurement Using Mohr’s Titration
3.4. Preparation of Potassium 4-Sulfonato-Oxy-2,2,6,6-Tetramethylpiperidine-1-Yloxyl (TEMPO-4-Sulfate)
3.5. General Procedure for the Immobilization of TEMPO-4-Sulfate Derivatives on the Ion Exchange Resins
3.6. General Procedure for Regeneration of the Used Catalytic Ion Exchange Resin
3.7. Catalytic Tests
3.7.1. Heterogeneous Catalysis
3.7.2. Immobilization and Heterogeneous Catalysis in Flow
3.7.3. Yield Determination of Aldehydes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shul’pin, G.B.; Süss-Fink, G.; Shul’pina, L.S. Oxidations by the system “hydrogen peroxide–manganese(IV) complex–carboxylic acid”. J. Mol. Catal. A Chem. 2001, 170, 17–34. [Google Scholar] [CrossRef]
- Corey, E.J.; Suggs, J. Pyridinium chlorochromate. An efficient reagent for oxidation of primary and secondary alcohols to carbonyl compounds. Tetrahedron Lett. 1975, 16, 2647–2650. [Google Scholar] [CrossRef]
- Markó, I.E.; Giles, P.R.; Tsukazaki, M.; Chellé-Regnaut, I.; Urch, C.J.; Brown, S.M. Efficient, Aerobic, Ruthenium-Catalyzed Oxidation of Alcohols into Aldehydes and Ketones. J. Am. Chem. Soc. 1997, 119, 12661–12662. [Google Scholar] [CrossRef]
- Musawir, M.; Davey, P.N.; Kelly, G.; Kozhevnikov, I.V. Highly efficient liquid-phase oxidation of primary alcohols to aldehydes with oxygen catalysed by Ru-Co oxide. Chem. Commun. 2003, 12, 1414–1415. [Google Scholar] [CrossRef]
- Tebben, L.; Studer, A. Nitroxides: Applications in synthesis and in polymer chemistry. Angew. Chem. Int. Ed. 2011, 50, 5034–5068. [Google Scholar] [CrossRef]
- Vogler, T.; Studer, A. Applications of TEMPO in Synthesis. Synthesis 2008, 2008, 1979–1993. [Google Scholar] [CrossRef]
- de Luca, L.; Giacomelli, G.; Porcheddu, A. A very mild and chemoselective oxidation of alcohols to carbonyl compounds. Org. Lett. 2001, 3, 3041–3043. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; Arends, I.W.C.E. Organocatalytic Oxidations Mediated by Nitroxyl Radicals. Adv. Synth. Catal. 2004, 346, 1051–1071. [Google Scholar] [CrossRef]
- Fey, T.; Fischer, H.; Bachmann, S.; Albert, K.; Bolm, C. Silica-supported TEMPO catalysts: Synthesis and application in the Anelli oxidation of alcohols. J. Org. Chem. 2001, 66, 8154–8159. [Google Scholar] [CrossRef] [PubMed]
- Kochkar, H.; Lassalle, L.; Morawietz, M.; Hölderich, W.F. Regioselective Oxidation of Hydroxyl Groups of Sugar and Its Derivatives Using Silver Catalysts Mediated by TEMPO and Peroxodisulfate in Water. J. Catal. 2000, 194, 343–351. [Google Scholar] [CrossRef]
- Bolm, C.; Magnus, A.S.; Hildebrand, J.P. Catalytic synthesis of aldehydes and ketones under mild conditions using TEMPO/Oxone. Org. Lett. 2000, 2, 1173–1175. [Google Scholar] [CrossRef]
- Leifert, D.; Studer, A. Organic Synthesis Using Nitroxides. Chem. Rev. 2023, 123, 10302–10380. [Google Scholar] [CrossRef] [PubMed]
- Ciriminna, R.; Pagliaro, M. Industrial Oxidations with Organocatalyst TEMPO and Its Derivatives. Org. Process Res. Dev. 2010, 14, 245–251. [Google Scholar] [CrossRef]
- Sheldon, R.A. E factors, green chemistry and catalysis: An odyssey. Chem. Commun. 2008, 29, 3352–3365. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Why green chemistry and sustainability of resources are essential to our future. J. Environ. Monit. 2008, 10, 406–407. [Google Scholar] [CrossRef]
- Lu, Z.-L.; Lindner, E.; Mayer, H.A. Applications of sol-gel-processed interphase catalysts. Chem. Rev. 2002, 102, 3543–3578. [Google Scholar] [CrossRef] [PubMed]
- Ciriminna, R.; Pandarus, V.; Béland, F.; Xu, Y.-J.; Pagliaro, M. Heterogeneously Catalyzed Alcohol Oxidation for the Fine Chemical Industry. Org. Process Res. Dev. 2015, 19, 1554–1558. [Google Scholar] [CrossRef]
- Ciriminna, R.; Pandarus, V.; Béland, F.; Pagliaro, M. Sol–gel Entrapped Nitroxyl Radicals: Catalysts of Broad Scope. ChemCatChem 2018, 10, 1731–1738. [Google Scholar] [CrossRef]
- Deronzier, A.; Lìmosin, D.; Moutet, J.-C. Electrochemical oxidation of carbinols mediated by nitroxyl radicals in solution or bonded to polypyrrolic coatings on platinum and carbon electrodes. Electrochim. Acta 1987, 32, 1643–1647. [Google Scholar] [CrossRef]
- Dijksman, A.; Arends, I.W.C.E.; Sheldon, R.A. Polymer immobilised TEMPO (PIPO): An efficient catalyst for the chlorinated hydrocarbon solvent-free and bromide-free oxidation of alcohols with hypochlorite. Chem. Commun. 2000, 4, 271–272. [Google Scholar] [CrossRef]
- Gheorghe, A.; Matsuno, A.; Reiser, O. Expedient Immobilization of TEMPO by Copper-Catalyzed Azide-Alkyne [3+2]-Cycloaddition onto Polystyrene Resin. Adv. Synth. Catal. 2006, 348, 1016–1020. [Google Scholar] [CrossRef]
- Miyazawa, T.; Endo, T. Oxidation of benzyl alcohol with Cu(II) mediated by polymeric oxoaminium salt. J. Mol. Catal. 1988, 49, L31–L34. [Google Scholar] [CrossRef]
- Osa, T.; Akiba, U.; Segawa, I.; Bobbitt, J.M. Electrocatalytic Oxidation of Nerol with Nitroxyl Radical Covalently Immobilized to Poly(acrylic acid) Coated on Carbon Electrodes. Chem. Lett. 1988, 17, 1423–1426. [Google Scholar] [CrossRef]
- Schulze, J.S.; Migenda, J.; Becker, M.; Schuler, S.M.M.; Wende, R.C.; Schreiner, P.R.; Smarsly, B.M. TEMPO-functionalized mesoporous silica particles as heterogeneous oxidation catalysts in flow. J. Mater. Chem. A 2020, 8, 4107–4117. [Google Scholar] [CrossRef]
- Vinu, A.; Hossain, K.Z.; Ariga, K. Recent Advances in Functionalization of Mesoporous Silica; American Scientific Publishers: Stevenson Ranch, CA, USA, 2005. [Google Scholar]
- Beejapur, H.A.; Zhang, Q.; Hu, K.; Zhu, L.; Wang, J.; Ye, Z. TEMPO in Chemical Transformations: From Homogeneous to Heterogeneous. ACS Catal. 2019, 9, 2777–2830. [Google Scholar] [CrossRef]
- Megiel, E. Surface modification using TEMPO and its derivatives. Adv. Colloid Interface Sci. 2017, 250, 158–184. [Google Scholar] [CrossRef]
- Hu, K.; Tang, J.; Cao, S.; Zhang, Q.; Wang, J.; Ye, Z. TEMPO-Functionalized Aromatic Polymer as a Highly Active, pH-Responsive Polymeric Interfacial Catalyst for Alcohol Oxidation. J. Phys. Chem. C 2019, 123, 9066–9073. [Google Scholar] [CrossRef]
- Chen, T.; Xu, Z.; Zhou, L.; Hua, L.; Zhang, S.; Wang, J. Temperature responsive polymer-supported TEMPO: An efficient and recoverable catalyst for the selective oxidation of alcohols. Tetrahedron Lett. 2019, 60, 419–422. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, J.; Zhang, M.; Xu, L.; Ji, J. Magnetic Polystyrene Nanosphere Immobilized TEMPO: A Readily Prepared, Highly Reactive and Recyclable Polymer Catalyst in the Selective Oxidation of Alcohols. ChemCatChem 2013, 5, 307–312. [Google Scholar] [CrossRef]
- Mohamad Ali, B.; Yu, Z.; Tao, Z.; Zhang, T.; Wang, L.; He, C.; Zhang, H.; Wang, J. TEMPO-Grafted Polystyrene/Polymethacrylate Organosiloxane Janus Nanohybrids as Efficient Pickering Interfacial Catalyst for Selective Aerobic Oxidation of Cinnamyl Alcohol. ACS Appl. Mater. Interfaces 2024, 16, 20409–20418. [Google Scholar] [CrossRef]
- Zhang, H.; Fu, L.; Zhong, H. Silica gel-supported TEMPO with adsorbed NOx for selective oxidation of alcohols under mild conditions. Chin. J. Catal. 2013, 34, 1848–1854. [Google Scholar] [CrossRef]
- Tsubokawa, N.; Kimoto, T.; Endo, T. Oxidation of alcohols with copper(II) salts mediated by nitroxyl radicals immobilized on ultrafine silica and ferrite surface. J. Mol. Catal. A Chem. 1995, 101, 45–50. [Google Scholar] [CrossRef]
- Kuroboshi, M.; Goto, K.; Tanaka, H. Electrooxidation of Alcohols Using an N-Oxyl-Immobilized Silica Gel. Synfacts 2009, 2009, 694. [Google Scholar] [CrossRef]
- Sharma, S.; Choudhary, A.; Sharma, S.; Shamim, T.; Paul, S. TEMPO supported amine functionalized magnetic titania: A magnetically recyclable catalyst for the aerobic oxidative synthesis of heterocyclic compounds. Monatsh Chem. 2021, 152, 83–94. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, Y.; Song, W.; Zhang, Y.; Zhang, D.; Zhang, J.; Guo, C.; Zhiani, R. Green Synthesis of TEMPO@DFNS-Li2MnO3 Ceramic Nanostructures and Their Application as Photocatalysts under Visible–UV Light for Removal of Organic Dyes in Water. Catal. Lett. 2024, 154, 2513–2526. [Google Scholar] [CrossRef]
- Brunel, D.; Fajula, F.; Nagy, J.; Deroide, B.; Verhoef, M.; Veum, L.; Peters, J.; van Bekkum, H. Comparison of two MCM-41 grafted TEMPO catalysts in selective alcohol oxidation. Appl. Catal. A General. 2001, 213, 73–82. [Google Scholar] [CrossRef]
- Fall, A.; Sene, M.; Gaye, M.; Gómez, G.; Fall, Y. Oxidation of Alcohols with TEMPO Immobilized on an Ionic Liquid. Synfacts 2010, 2010, 1209. [Google Scholar] [CrossRef]
- She, Y.; Chen, X.; Wang, M.; Liu, A.; Wang, X.; Gao, D.; Hu, K.; Hu, M. Heterogeneous solvent-metal-free aerobic oxidation of alcohol under ambient conditions catalyzed by TEMPO-functionalized porous poly(ionic liquid)s. RSC Adv. 2024, 14, 20199–20209. [Google Scholar] [CrossRef] [PubMed]
- Beejapur, H.A.; Campisciano, V.; Giacalone, F.; Gruttadauria, M. Catalytic Synergism in a C 60 IL 10 TEMPO 2 Hybrid in the Efficient Oxidation of Alcohols. Adv. Synth. Catal. 2015, 357, 51–58. [Google Scholar] [CrossRef]
- Chen, T.; Xiao, W.; Wang, Z.; Xie, T.; Yi, C.; Xu, Z. Design and engineering of heterogeneous nitroxide-mediated catalytic systems for selective oxidation: Efficiency and sustainability. Mater. Today Chem. 2022, 24, 100872. [Google Scholar] [CrossRef]
- Hoover, J.M.; Stahl, S.S. Highly practical copper(I)/TEMPO catalyst system for chemoselective aerobic oxidation of primary alcohols. J. Am. Chem. Soc. 2011, 133, 16901–16910. [Google Scholar] [CrossRef] [PubMed]
- Krogul-Sobczak, A.; Pisarek, N.; Cieciórski, P.; Megiel, E. Silver Nanoparticles Densely Grafted with Nitroxides as a Recyclable Green Catalyst in the Selective Oxidation of Alcohols. Nanomaterials 2022, 12, 2542. [Google Scholar] [CrossRef]
- Karimi, B.; Rafiee, M.; Alizadeh, S.; Vali, H. Eco-friendly electrocatalytic oxidation of alcohols on a novel electro generated TEMPO-functionalized MCM-41 modified electrode. Green Chem. 2015, 17, 991–1000. [Google Scholar] [CrossRef]
- Staby, A.; Jensen, I.H. Comparison of chromatographic ion-exchange resins. II. More strong anion-exchange resins. J. Chromatogr. A 2001, 908, 149–161. [Google Scholar] [CrossRef]
- Epp, J.B.; Widlanski, T.S. Facile Preparation of Nucleoside-5′-carboxylic Acids. J. Org. Chem. 1999, 64, 293–295. [Google Scholar] [CrossRef] [PubMed]
- Caneschi, A.; Gatteschi, D.; Sangregorio, C.; Vaz, M.; Costantino, U.; Nocchetti, M.; Vivani, R. Intercalation of a nitronyl nitroxide radical into layered inorganic hosts. Inorganica Chim. Acta 2002, 338, 127–132. [Google Scholar] [CrossRef]
- Grauer, Z.; Waterman, K.C.; Turro, N.J. Reactivity of nitroxide spin probes toward photochemically generated benzyl radicals in clay mineral suspensions. Langmuir 1987, 3, 59–61. [Google Scholar] [CrossRef]
- Hemme, W.L.; Fujita, W.; Awaga, K.; Eckert, H. Solid state NMR strategies for the structural characterization of new hybrid materials based on the intercalation of nitroxide radicals into CdPS3. Solid State Nucl. Magn. Reson. 2011, 39, 106–115. [Google Scholar] [CrossRef]
- Miwa, Y.; Drews, A.R.; Schlick, S. Detection of the Direct Effect of Clay on Polymer Dynamics: The Case of Spin-Labeled Poly(methyl acrylate)/Clay Nanocomposites Studied by ESR, XRD, and DSC. Macromolecules 2006, 39, 3304–3311. [Google Scholar] [CrossRef]
- Röben, C.; Studer, A.; Hemme, W.; Eckert, H. Immobilization of TEMPO Derivatives in Saponite and Use of These Novel Hybrid Materials as Reusable Catalysts. Synlett 2010, 2010, 1110–1114. [Google Scholar] [CrossRef]
- Lucio Anelli, P.; Biffi, C.; Montanari, F.; Quici, S. Fast and selective oxidation of primary alcohols to aldehydes or to carboxylic acids and of secondary alcohols to ketones mediated by oxoammonium salts under two-phase conditions. J. Org. Chem. 1987, 52, 2559–2562. [Google Scholar] [CrossRef]
- Gerken, J.B.; Stahl, S.S. High-Potential Electrocatalytic O2 Reduction with Nitroxyl/NO x Mediators: Implications for Fuel Cells and Aerobic Oxidation Catalysis. ACS Cent. Sci. 2015, 1, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Fontaine, O.; Tarascon, J.-M.; Grimaud, A. Chemical Recognition of Active Oxygen Species on the Surface of Oxygen Evolution Reaction Electrocatalysts. Angew. Chem. Int. Ed. 2017, 56, 8652–8656. [Google Scholar] [CrossRef] [PubMed]
- Żamojć, K.; Zdrowowicz, M.; Wiczk, W.; Jacewicz, D.; Chmurzyński, L. Dihydroxycoumarins as highly selective fluorescent probes for the fast detection of 4-hydroxy-TEMPO in aqueous solution. RSC Adv. 2015, 5, 63807–63812. [Google Scholar] [CrossRef]
- Nakahara, K.; Iwasa, S.; Iriyama, J.; Morioka, Y.; Suguro, M.; Satoh, M.; Cairns, E.J. Electrochemical and spectroscopic measurements for stable nitroxyl radicals. Electrochim. Acta 2006, 52, 921–927. [Google Scholar] [CrossRef]
- Dr. R. Niemand. Specification: Ion Exchange Resin ROTI®Change 1 × 8: 20-50 Mesh, Analytical Grade, Cl—Form. 2023. Available online: https://www.carlroth.com/de/en/anion-exchange-resins/ion-exchange-resin-rotichange-1-x-8/p/6852.1 (accessed on 1 March 2024).
- Liu, S.; Sun, T.; Yang, D.; Cao, M.; Liang, H. Polyacrylic acid supported TEMPO for selective catalytic oxidation of cellulose: Recovered by its pH sensitivity. Cellulose 2018, 25, 5687–5696. [Google Scholar] [CrossRef]
- Tojo, G.; Fernandez, M.I. Oxidation of Primary Alcohols to Carboxylic Acids: A Guide to Current Common Practice; Springer: New York, NY, USA, 2010; ISBN 1441922547. [Google Scholar]
- Sheldon, R.A.; Arends, I.W.C.E.; ten Brink, G.-J.; Dijksman, A. Green, catalytic oxidations of alcohols. Acc. Chem. Res. 2002, 35, 774–781. [Google Scholar] [CrossRef]
- de Luca, L.; Giacomelli, G.; Masala, S.; Porcheddu, A. Trichloroisocyanuric/TEMPO oxidation of alcohols under mild conditions: A close investigation. J. Org. Chem. 2003, 68, 4999–5001. [Google Scholar] [CrossRef]
- de Mico, A.; Margarita, R.; Parlanti, L.; Vescovi, A.; Piancatelli, G. A Versatile and Highly Selective Hypervalent Iodine (III)/2,2,6,6-Tetramethyl-1-piperidinyloxyl-Mediated Oxidation of Alcohols to Carbonyl Compounds. J. Org. Chem. 1997, 62, 6974–6977. [Google Scholar] [CrossRef]
- Salvo, A.; Campisciano, V.; Beejapur, H.; Giacalone, F.; Gruttadauria, M. A Simple Procedure for the Oxidation of Alcohols Using [Bis(acetoxy)iodo]benzene and a Catalytic Amount of Bromide Ions in Ethyl Acetate. Synlett 2015, 26, 1179–1184. [Google Scholar] [CrossRef]
- Ciriminna, R.; Ghahremani, M.; Karimi, B.; Pagliaro, M. Electrochemical Alcohol Oxidation Mediated by TEMPO-like Nitroxyl Radicals. ChemistryOpen 2017, 6, 5–10. [Google Scholar] [CrossRef]
- Rafiee, M.; Karimi, B.; Alizadeh, S. Mechanistic Study of the Electrocatalytic Oxidation of Alcohols by TEMPO and NHPI. ChemElectroChem 2014, 1, 455–462. [Google Scholar] [CrossRef]
- HUANG, J.; DAI, W.; LI, H.; FAN, K. Au/TiO2 as high efficient catalyst for the selective oxidative cyclization of 1,4-butanediol to γ-butyrolactone. J. Catal. 2007, 252, 69–76. [Google Scholar] [CrossRef]
- Aellig, C.; Scholz, D.; Conrad, S.; Hermans, I. Intensification of TEMPO-mediated aerobic alcohol oxidations under three-phase flow conditions. Green Chem. 2013, 15, 1975. [Google Scholar] [CrossRef]
- Winsberg, J.; Stolze, C.; Schwenke, A.; Muench, S.; Hager, M.D.; Schubert, U.S. Aqueous 2,2,6,6-Tetramethylpiperidine- N -oxyl Catholytes for a High-Capacity and High Current Density Oxygen-Insensitive Hybrid-Flow Battery. ACS Energy Lett. 2017, 2, 411–416. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Alcohol | Conv./% | t/min | Products | Selectivity % |
|---|---|---|---|---|---|
| 1 | butane-1-ol | 95 | 30 | butanal | ~100 |
| 2 | benzyl alcohol | 99 | 20 | benzaldehyde | ~100 |
| 3 | 1,4-benzene dimethanol | 100 | 20 | 1,4-benzene dialdehyde 4-(hydroxymethyl) benzaldehyde | 99 1 |
| 4 | 1,4-butanediol | 75 | 90 | γ-butyrolactone | ~100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gmeiner, J.; Luinstra, G.A. Oxidation in Flow Using an Ionic Immobilized TEMPO Catalyst on an Ion Exchange Resin. Catalysts 2024, 14, 542. https://doi.org/10.3390/catal14080542
Gmeiner J, Luinstra GA. Oxidation in Flow Using an Ionic Immobilized TEMPO Catalyst on an Ion Exchange Resin. Catalysts. 2024; 14(8):542. https://doi.org/10.3390/catal14080542
Chicago/Turabian StyleGmeiner, Johannes, and Gerrit A. Luinstra. 2024. "Oxidation in Flow Using an Ionic Immobilized TEMPO Catalyst on an Ion Exchange Resin" Catalysts 14, no. 8: 542. https://doi.org/10.3390/catal14080542