1. Introduction
Enzymes are macromolecular biocatalysts, displaying high levels of stereo-, chemo- and regioselectivity in a variety of chemical or biochemical reactions under mild conditions. Enzymes are routinely leveraged to enable unique applications throughout research, industrial production and molecular diagnostics as they are known to accelerate reactions by >10
17 fold [
1]. Enzyme immobilization has been an area of great interest since it was first reported in 1916 [
2], with many reviews available [
3,
4,
5,
6,
7,
8,
9,
10,
11]. Immobilization is achieved by anchoring enzymes to or within solid supports in order to facilitate efficient separation from the reaction media in heterogeneous systems and increase the reusability of enzymes. In addition, immobilization of enzymes on the support materials usually leads to better enzymatic stability under both storage and operational conditions [
7]. Generation of an immobilized enzyme requires a thorough understanding of the biochemical/physical properties of the enzyme (i.e., homogeneity, availability, stability, conformational flexibility, molecular weight, size, surface charge, pH and temperature profile, and specific activity) and the support materials (available functional groups, chemical and physical properties) so that an appropriate immobilization process can be tailored to maximize the specific enzyme activity and stability throughout the intended application.
Generating a standard methodology that is universally amenable to the immobilization of any enzyme is not possible; and finding the optimal strategy often requires iterative improvements through trial and error. The most frequently employed immobilization techniques can be described by one of the following strategies: non-covalent adsorption and deposition, immobilization via ionic interactions, covalent attachment, cross-linking of an enzyme, or entrapment within a solid support via a polymer gel lattice [
3,
7,
12,
13,
14,
15]. For simplicity, this article will broadly categorize methodologies as either covalent or non-covalent immobilization; the focus herein is on covalent immobilization. Covalently immobilized enzymes are typically considered to be superior to non-covalently immobilized enzymes as the potential for enzyme leaching from the surface is minimized, thereby avoiding protein contamination of the product.
The reactions employed to effectively immobilize the enzyme depend on the chemical and physical properties of the support, as well as the environment in which it is applied [
16]. In this regard, we believe it is advantageous to utilize stable, pre-existing solids as core support materials, such as agarose, chitin, silicates and synthetic (co-)polymer encapsulated magnetic beads because of their known physical properties, chemical functionality and compatibility within molecular biology workflows (i.e., immunoprecipitation, affinity purification, or pull-down reactions).
Advancements in chemical/molecular biology and material science have provided the means to easily produce large amounts of recombinant protein and enable unique strategies for enzyme immobilization. Researchers can easily manipulate the gene of interest, clone it in optimized expression vectors, transform them into the host of choice, induce protein expression and then, the protein is ready for purification, characterization and immobilization.
Traditionally, covalent immobilization was performed through amino acids containing an ionizable side chain (aspartic acid, glutamic acid, lysine, cysteine, histidine, tyrosine and arginine) or the N-terminal α-amine. Such covalent immobilization strategies require tedious experimentation and analysis for selection of the appropriate reaction conditions, as loss of enzymatic activity can result from sub-optimal orientation of the enzyme active site (steric hindrance/blocking of substrate accessibility/product release), reduced conformational flexibility of the enzyme, or the resulting chemical modifications imparted on the amino acid constituents, prosthetic groups/cofactors of the enzyme [
17,
18]. Recently, advancements in site-specific protein for characterization and immobilization of enzymes have been driven by the development of bio-orthogonal reactions that proceed under physiological conditions between chemical groups that are absent in, and do not cross-react with, endogenous functionalities in proteins [
19,
20].
These new site-specific protein immobilization approaches by taking advantages of bio-orthogonal reactions, enable better control of both the site and level of enzyme modification (i.e., one can append bio-orthogonal reactive handles to the enzyme as opposed to leveraging the native amino acid sequence) as well as direct the enzyme immobilization to preserve the optimal orientation of the active site, although some disadvantages such as alteration of enzyme 3D structure and complicated preparation process were also reported [
21]. Introducing these bio-orthogonal groups into proteins typically relies on the cellular incorporation of unnatural amino acids for which the site-specific modification can be performed [
22]. An alternative approach for site specific enzyme immobilization relies on the expression of recombinant enzyme as a fusion protein with the SNAP-tag. The SNAP-tag is a small protein (20 kDa) based on human O
6-alkylguanine-DNA-alkyltransferase (hAGT). During the self-labelling reaction of the SNAP-tag with its substrate, O
6-benzylguanine (BG), a stable thioether bond is formed between the reactive cysteine of the tag and the label/support with high specificity and defined stoichiometry [
23,
24]. Therefore, use of a fusion partner immobilization strategy results in each enzyme molecule carrying only a single conjugation site. In this regard, we have succeeded in immobilizing several DNA modifying enzymes on BG-functionalized magnetic beads by taking advantage of self-labeling SNAP-tag as a fusion partner of the relevant enzymes [
25]. These immobilized enzymes have been employed in construction of human genomic libraries by performing sequential manipulation of DNA fragments, resulting in improved sequence coverage and reduced GC-bias in Illumina next-generation sequencing [
26]. Employing immobilized enzymes throughout library construction removes the requirement for high temperature treatment and permits easy separation of the end-polished library DNA from the enzymes linked to magnetic beads at ambient temperature.
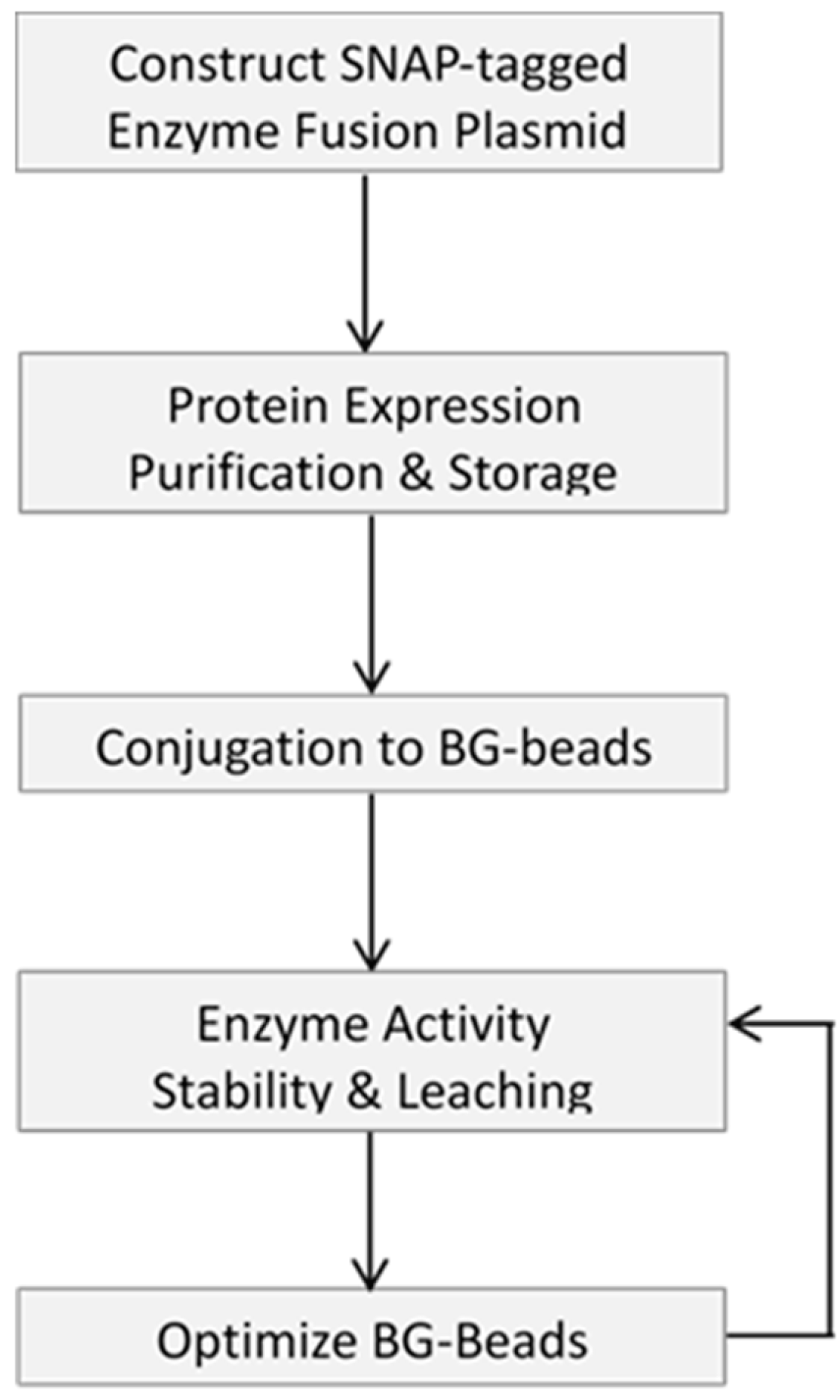
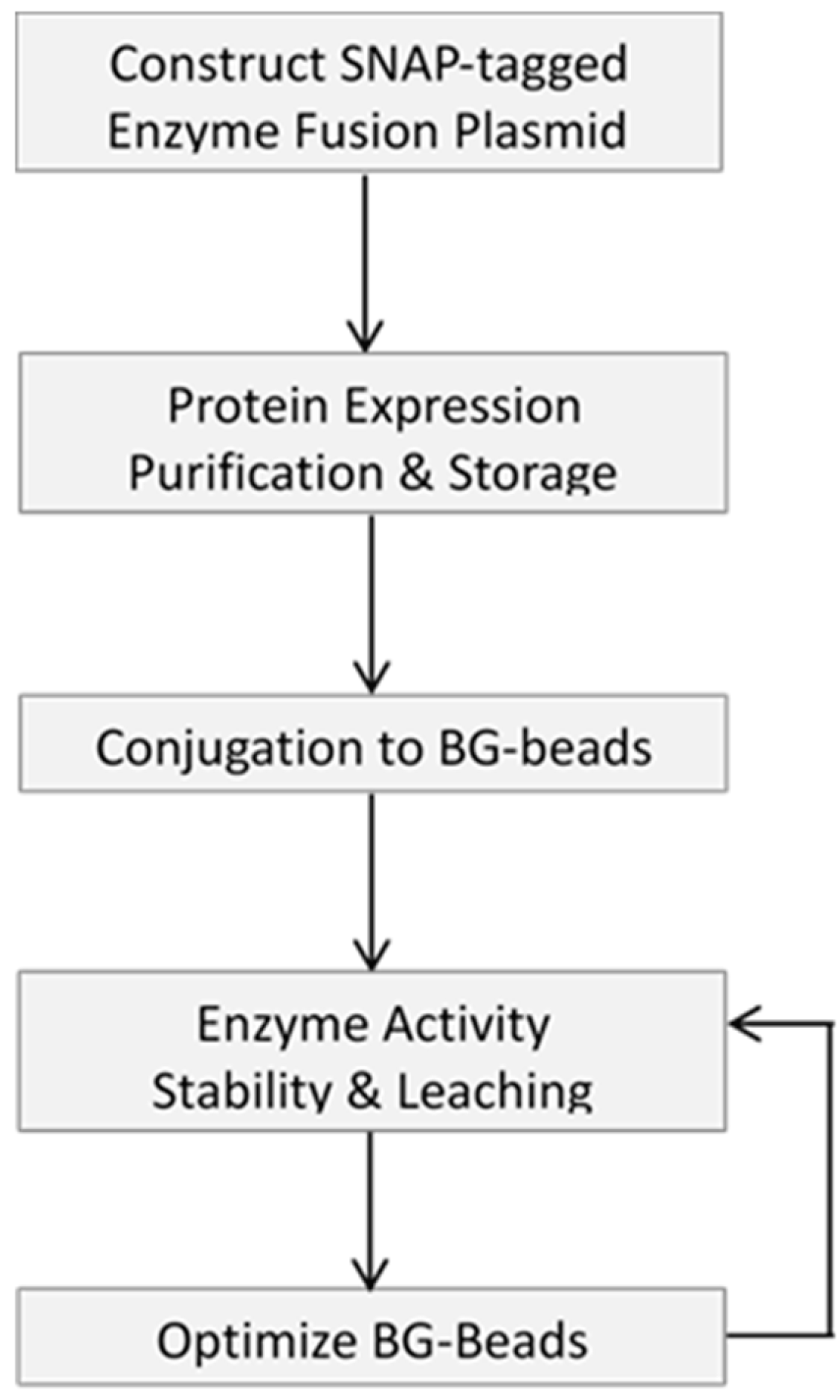
This article describes a methodology to express, purify and site-specifically immobilize (through bio-orthogonal reactivity) a fusion protein of interest onto various microbead supports. Attempts to optimize the support surfaces, to enable the application of the immobilized enzymes for next-generation sequencing are also reported. We dedicate the first part of this article to outlining the basic steps to generate and validate site-specific immobilized SNAP-tagged fusion proteins, using recombinant SNAP-tagged T4 DNA ligase as a model. Then, we show that the support material (i.e., agarose, chitin, magnetic and Si-magnetic chitin beads) is of tremendous influence on the performance of an immobilized enzyme by performing gel electrophoresis for its DNA ligation evaluation. Different strategies to modify the bead surface such as using different concentrations of PEG spacers and coatings are shown to impact the performance of the immobilized enzyme. This is followed by highlighting the utility of immobilized enzymes for construction of a genomic DNA library of AT-rich Clostridium acetobutylicum for the Illumina sequencing platform. The goal of this research article is to provide the researcher with a broad picture of the merits and challenges of covalent enzyme immobilization and its potential applications in molecular workflows.
3. Materials and Methods
3.1. Materials
All DNA modifying enzymes, including T4 DNA ligase, T4 DNA Polymerase, T4 Polynucleotide Kinase, and Taq DNA Polymerase were from New England Biolabs (NEB, Ipswich, MA, USA). Carboxylate-modified magnetic beads (average diameter 1 µm) were from GE Healthcare Bio-Sciences (Pittsburgh, PA, USA). HPLC-purified synthetic single stranded oligonucleotides were obtained from Integrated DNA Technologies (IDT, Coralville, IA, USA) as lyophilized solids. Oligonucleotides were dissolved in nuclease-free water prior to use. T4 DNA Ligase Reaction Buffer, Quick Ligation Buffer, NEBNext
® End Repair Reaction Buffer and NEB Buffer 2 were from NEB. pSNAP
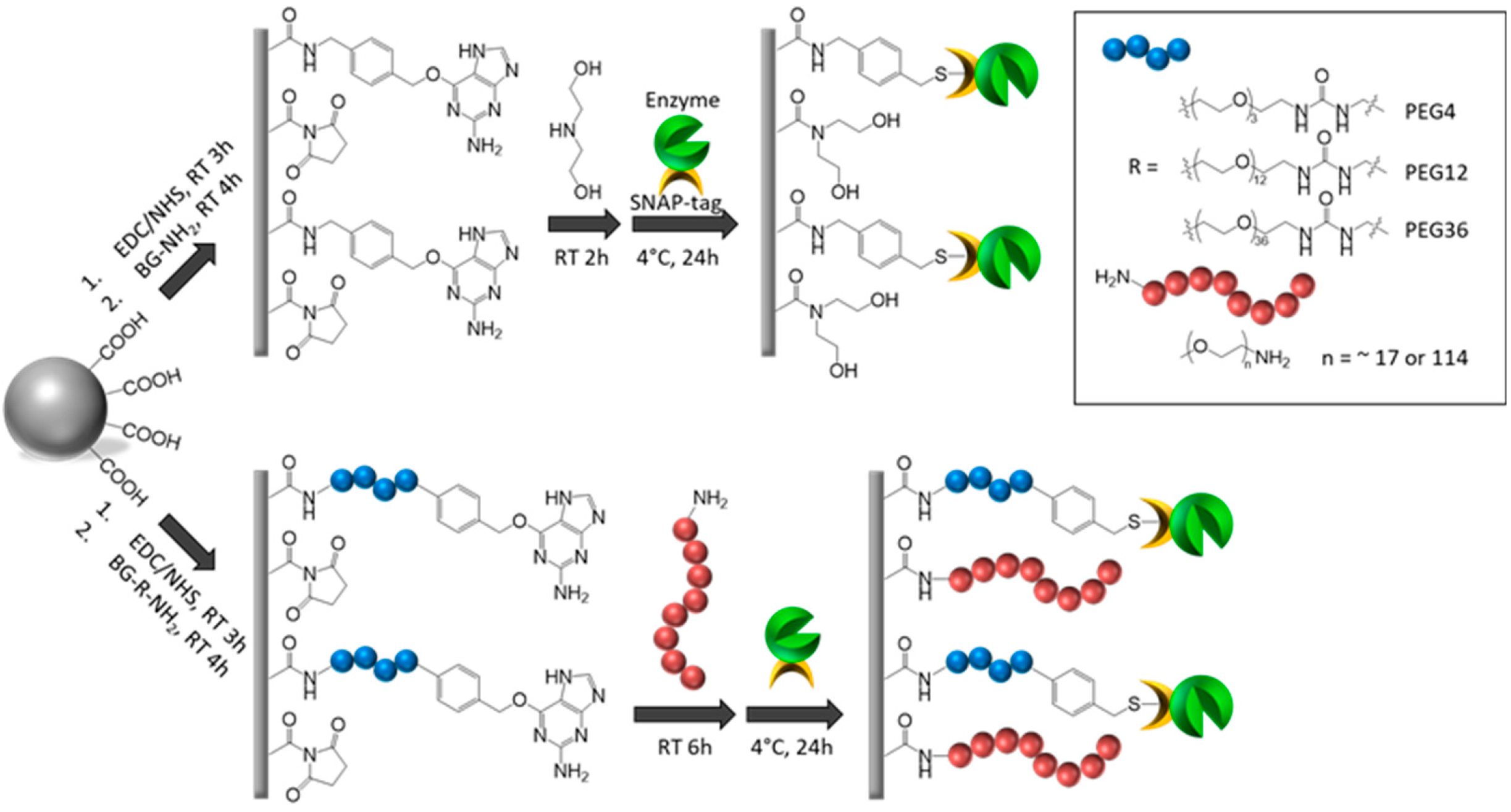
f-tag(T7) vector was used for construction of recombinant plasmids expressing SNAP-tagged enzymes and is commercially available (NEB). In this report four microbeads were functionalized with BG ligand and used for immobilization of T4 DNA ligase. The sources and average bead size are described below. Carboxylate-modified magnetic beads (average diameter 1 µm), obtained from GE Healthcare Bio-Sciences (Pittsburgh, PA, USA), were used to generate BG-Mag beads. BG-SiMag (1 µm, SiM in
Figure 2) was derived from SiMag-Chitosan (Product no. 14095, Chemicell, Berlin, Germany). BG-Agarose (100–200 µm) was commercially available (SNAP-capture, NEB); BG-Chitin (100–200 µm) was made by BG-modification of Chitin Beads (NEB). 1 µm beads can typically stay in suspension during reaction without agitation. BG-NH
2 and BG-PEG
4-NH
2 (sold under the name BG-PEG-NH
2) were from NEB. Methoxypolyethylene glycol amines with average molecular weight 750 Da, referred to as PEG
750, was purchased from Sigma-Aldrich (St Louis, MO, USA) and used for coating BG-Mag beads.
3.2. Beads Surface Modifications
Surface modifications of beads were previously reported [
25]. 2 mL of a 50 mg/mL suspension of carboxylate magnetic beads (average diameter 1 µm) in water were separated with aid of an external magnet and were washed three times with 10 mL of water followed with 3 washes with 10 mL of a 50 mM 2-(
N-morpholino) ethanesulfonic acid (MES) buffer pH 6.0. The supernatant was removed and a mixture containing 50 mM
N-(3-Dimethylaminopropyl)-
N′-ethylcarbodiimide (EDC) and 50 mM NHS in 10 mL of 50 mM MES buffer pH 6.0 was added to the magnetic beads. The suspension mixture was gently shaken at room temperature for 3 h. The supernatant was removed, and the beads were washed three times with ice-cold 50 mM MES. After bead washing, a solution of 3.7 mM BG-NH
2 or BG-PEG
n-NH
2 (wherein n = 4) in 10 mL of isopropanol/water 1:1 was added. The reaction mixture was shaken for 4 h. The supernatant was removed and a solution of 0.2 M ethanolamine in isopropanol/water 1:1 was added. The reaction mixture was shaken at room temperature for 2 h. For the PEG-coated beads, instead of diethanolamine, a solution of 20 mM methoxypolyethylene glycol amines PEG
750 in isopropanol/water 1:1 solution was added to the beads and shaken at room temperature for 24 h. The supernatant was then removed, and the beads were washed with water (3 × 10 mL) and isopropanol/water 1:1 (3 × 10 mL). The beads were stored in isopropanol/water 1:1 at 10 mg/mL concentration.
3.3. Protein Immobilization
SNAP-tagged enzymes were conjugated to benzylguanine-functionalized magnetic beads by mixing 18.8 nmol of protein with 100 mg beads resuspended in 500 µL of a 10 mM phosphate-buffered saline (PBS) pH 7.4 (10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, and 137 mM NaCl) with 1 mM dithiothreitol (DTT). The mixture was shaken at 4 °C for 24 h. Beads were washed 8 times with 1 mL of 10 mM PBS buffer pH 7.4. The immobilized enzymes were stored at −20 °C in a storage buffer containing 1 mM DTT, 0.1% Triton® X-100, 10 mM Tris-HCl, 0.1 mM EDTA, 100 mM KCl, 50% glycerol, pH 7.4.
3.4. Plasmids and Expression of SNAP-Tagged Enzymes
Construction of SNAP-tagged T4 DNA polymerase, T4 polynucleotide kinase and Taq DNA polymerase were previously described using pSNAP
f-tag (T7) [
25]. pSNAP
f-tag(T7) vector was used to subclone the genes encoding T4 DNA ligase, resulting in a construct termed T4 DNA ligase-SNAP-6× His expresses a fusion protein consisting of an N-terminal T4 DNA ligase, SNAP-tag and C-terminal six histidine residues.
E. coli NEB T7 Express strain was used for inducible expression of SNAP-tagged enzymes, typically at 16 °C for 16 h. Soluble fusion proteins were purified from clarified cell lysate by affinity chromatography using Ni-NTA agarose (Qiagen, Hilden, Germany), after extensive wash with 800 mL of buffer containing 10 mM Tris-HCl, pH 8.0, 20 mM Imidazole, 1 mM DTT and 0.3 M NaCl, per 10 mL of Ni-NTA agarose resin. Purified protein was dialyzed into a storage buffer (1 mM DTT, 0.1% Triton
® X-100, 10 mM Tris-HCl, 0.1 mM EDTA, 100 mM KCl, pH 7.4) containing 50% glycerol for long-term storage at −80 °C.
3.5. Enzyme Immobilization
SNAP-tagged enzymes were conjugated to BG-functionalized magnetic beads by mixing approximately 18.8 nmol of protein with 10 mg of beads resuspended in 500 µL of a 10 mM phosphate-buffered saline (PBS) pH 7.4 (10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, and 135 mM NaCl) with 1 mM dithiothreitol (DTT). The mixture was shaken at 4 °C for 24 h. Beads were washed 8 times with 1 mL of 10 mM PBS buffer (pH 7.4). The immobilized enzymes were stored at −20°C in a storage buffer containing 1 mM DTT, 0.1% Triton® X-100, 10 mM Tris-HCl, 0.1 mM EDTA, 100 mM KCl, 50% glycerol, pH 7.4.
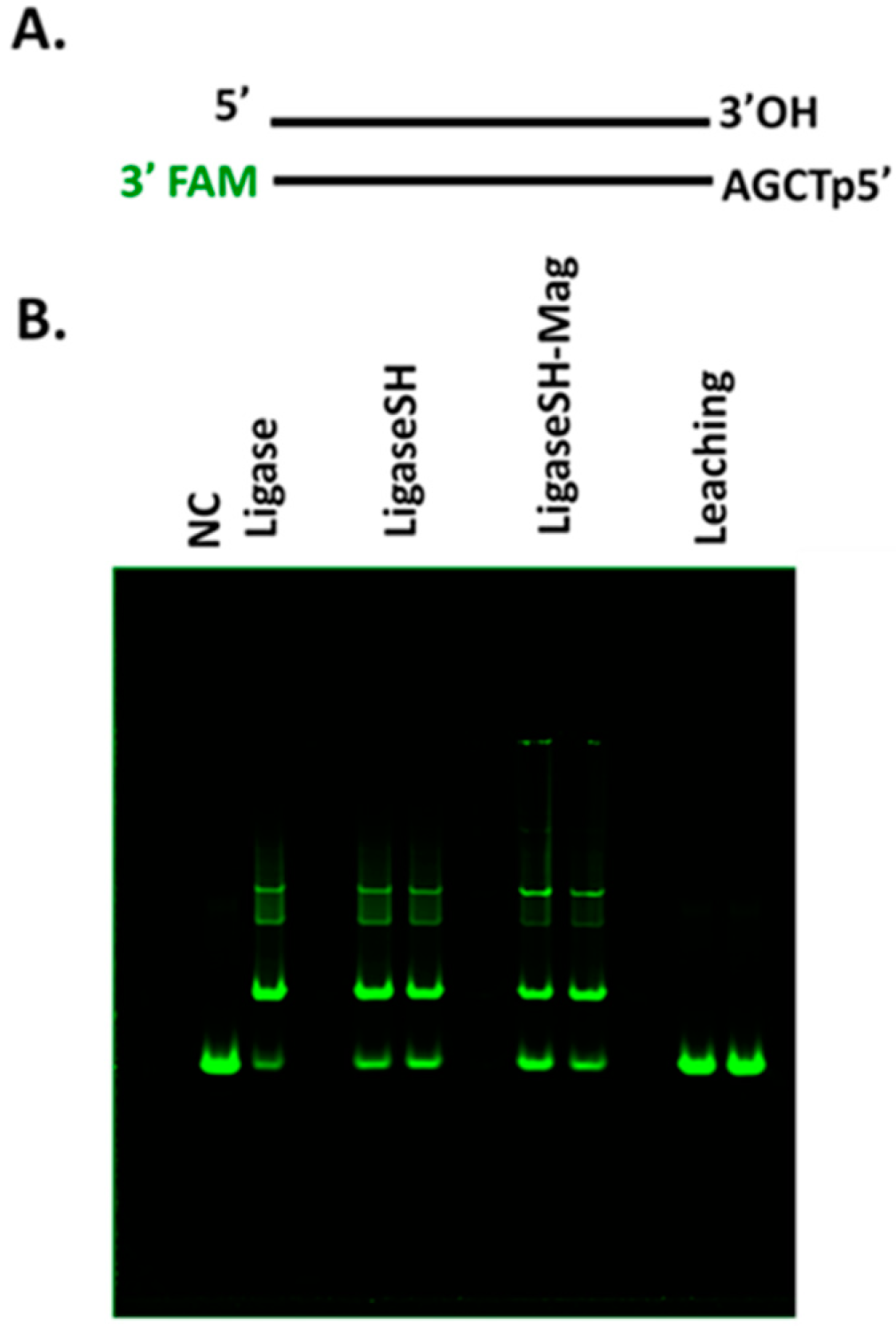
3.6. Ligase Activity Assays
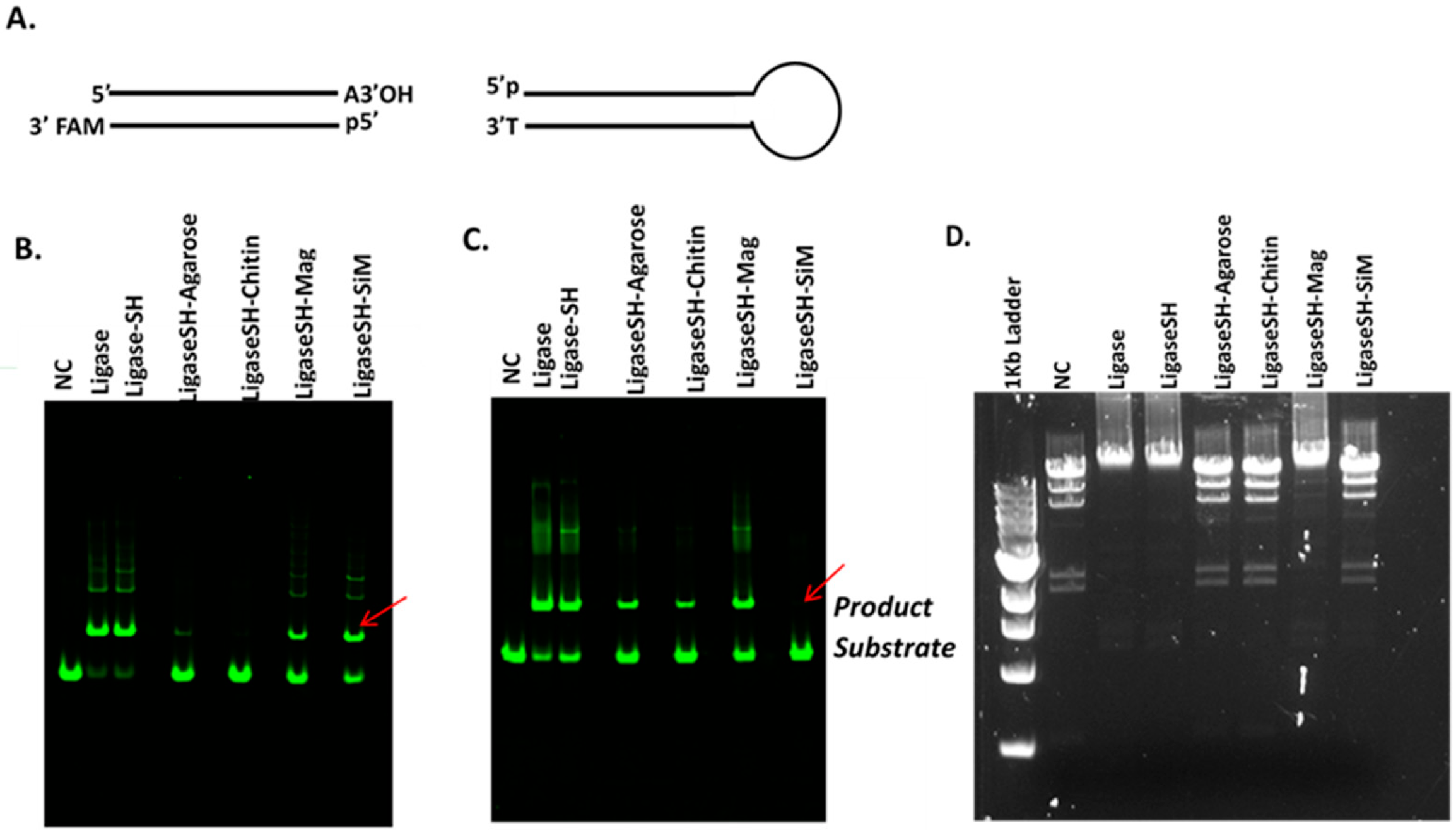
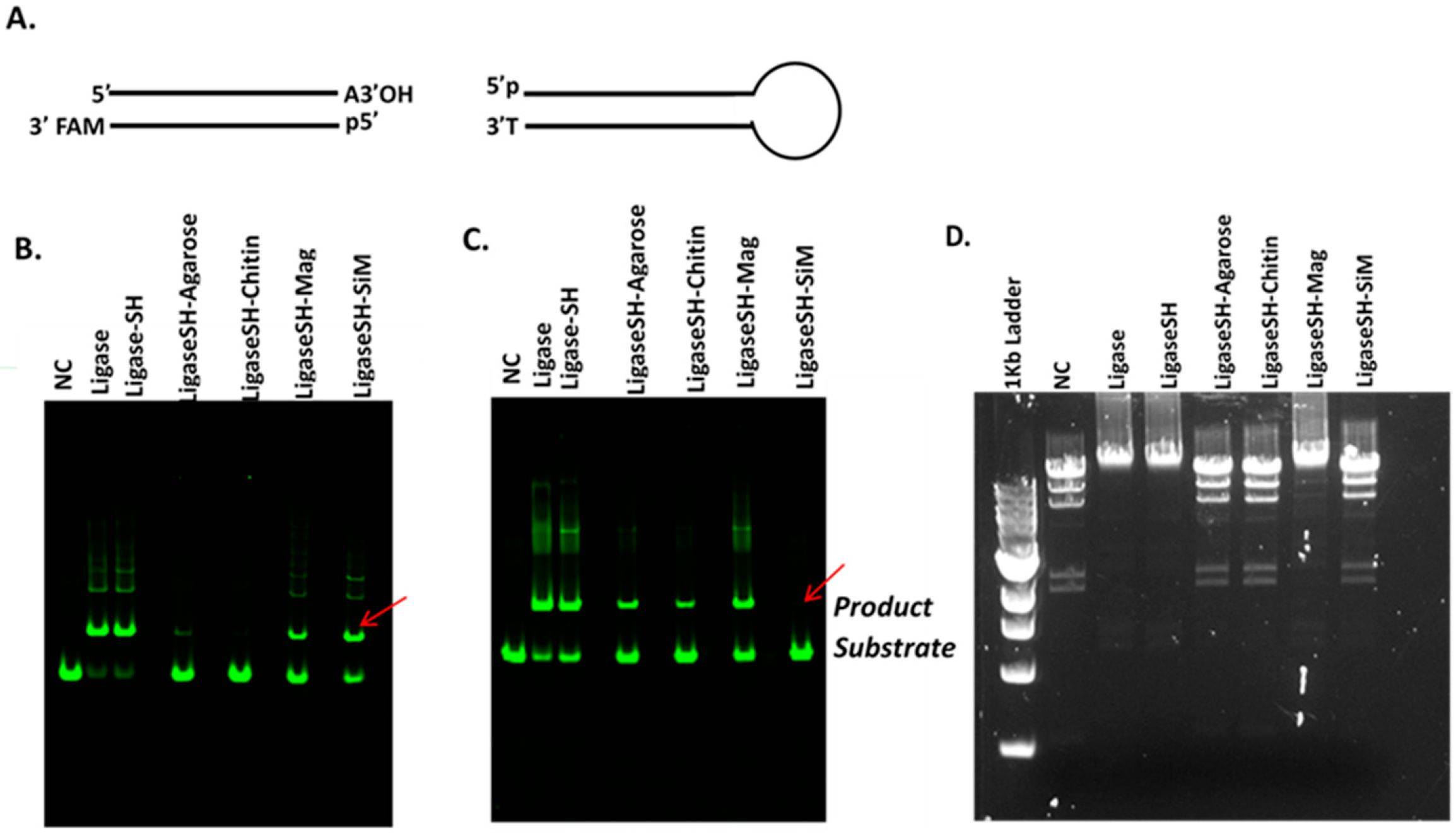
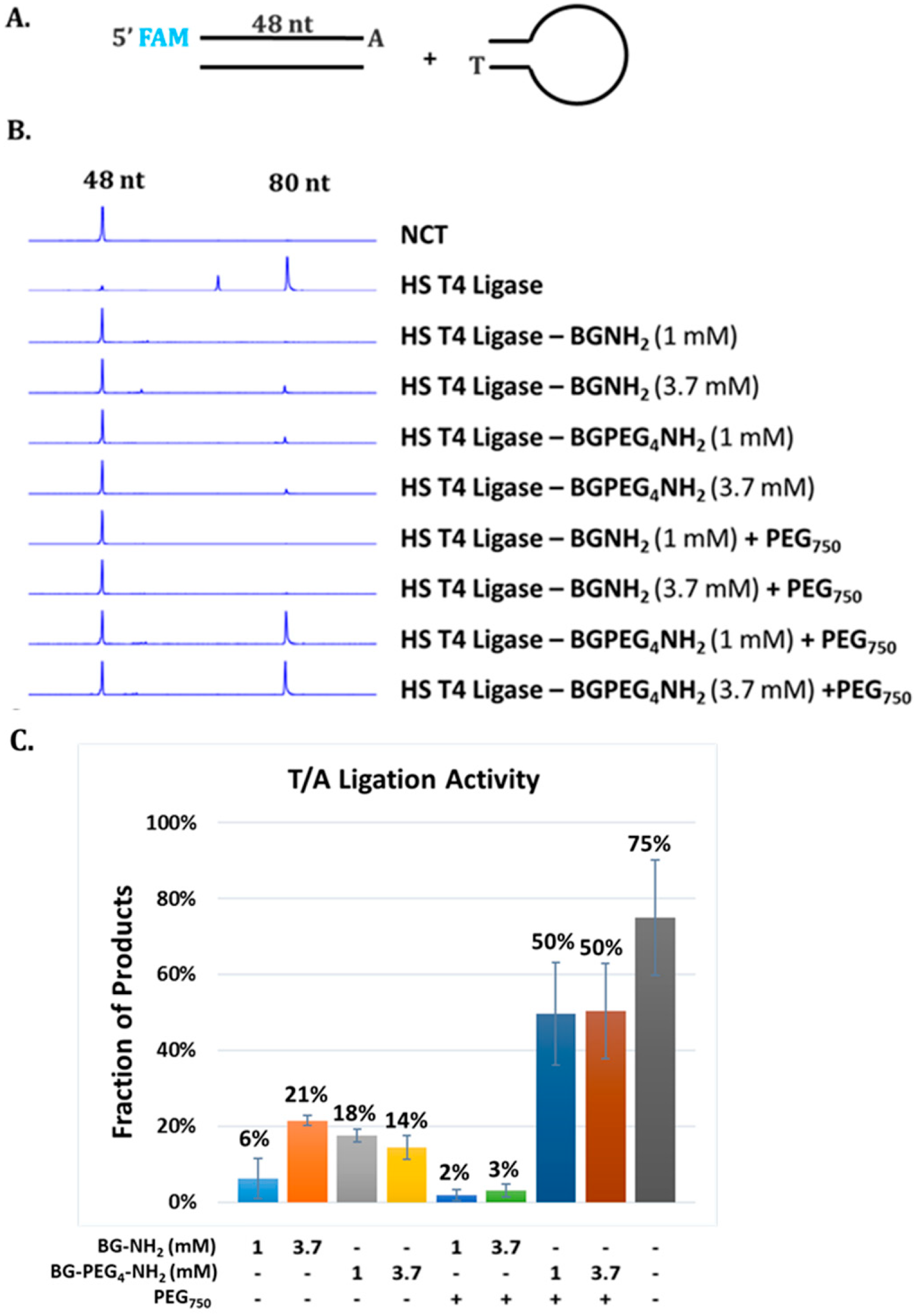
Synthetic DNA duplexes possessing a fluorescent 5′-FAM probe were used to monitor ligation efficiency via capillary gel electrophoresis (CE) analysis. DNA duplex was prepared by mixing each pair of complementary oligos at 10 µM final concentration in 1× NEB Buffer 2. The mixture was heated to 80 °C and cooled to room temperature slowly. 5′ FAM-54mer/50mer and 5′ FAM-66mer/70mer substrates, with complementary four nucleotide overhangs, were used for standard activity assays using 1× T4 DNA Ligase Reaction Buffer (NEB) to determine the unit in comparison to that of soluble T4 DNA ligase. A 5′ FAM-labeled 48-bp synthetic DNA containing a single 3′A and an Illumina adaptor (NEB) containing a 3′T were ligated using Blunt/TA Ligation Buffer provided by NEB. Lambda DNA digested with restriction enzyme HindIII (NEB) was used to assess ligation of large DNA fragments via agarose gel electrophoresis. All DNA fragments are 5′ phosphorylated. Ligation reactions were typically performed at 25 °C for 2 h in 1× T4 DNA Ligation Buffer or at 25 °C for 15 min in T/A Ligation Buffer or Quick Ligation Buffer prior to CE analysis as described below.
3.7. Capillary Gel Electrophoresis Assays
T4 DNA ligase activity was determined using DNA duplex end-labeled with a fluorescent FAM probe. Briefly, 100 nM DNA substrate and 1.5 nM soluble or immobilized T4 DNA ligase were incubated in 10 µL of 1× T4 DNA ligation Buffer, T/A Ligation Buffer or Quick Ligation Buffet at 20 °C for 30 min. The reaction was terminated by addition of 40 µL of 12.5 mM EDTA solution (10 mM final concentration). The resulting mixture was diluted to a 5 nM final concentration of FAM-labeled substrate. Capillary gel electrophoresis (CE) analysis was performed on an Applied Biosystems 3730 xl Genetic Analyzer to measure substrate depletion and product formation. DNA fragment analysis and quantification were performed using Peak Scanner software v1.0 (Applied Biosystems, Foster City, CA, USA).
3.8. DNA Library Preparation and Sequencing
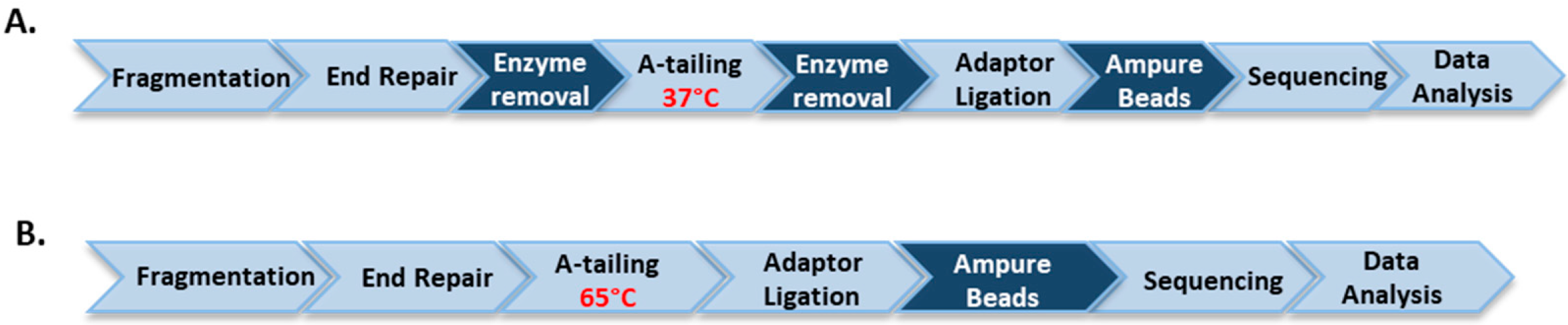
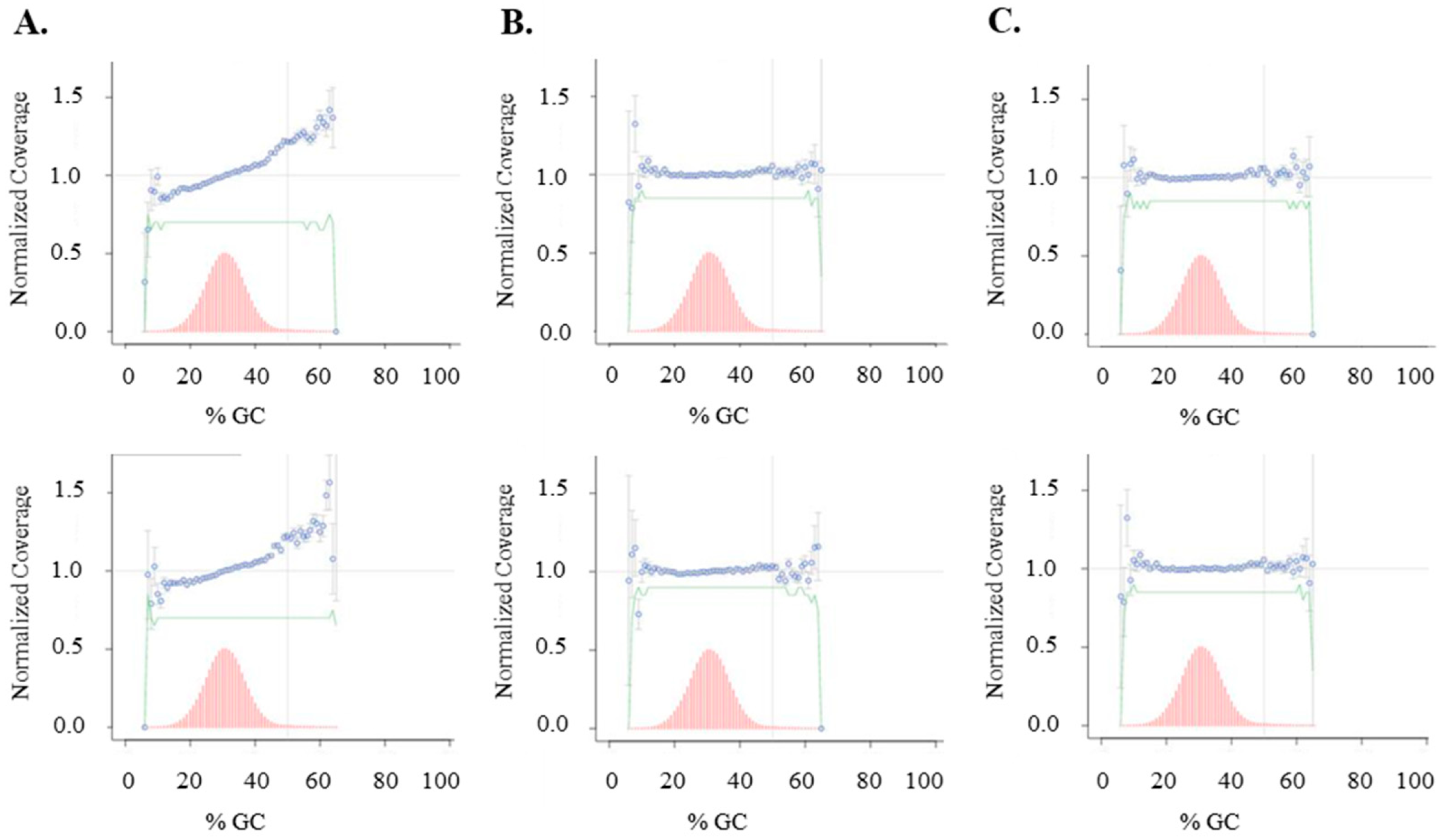
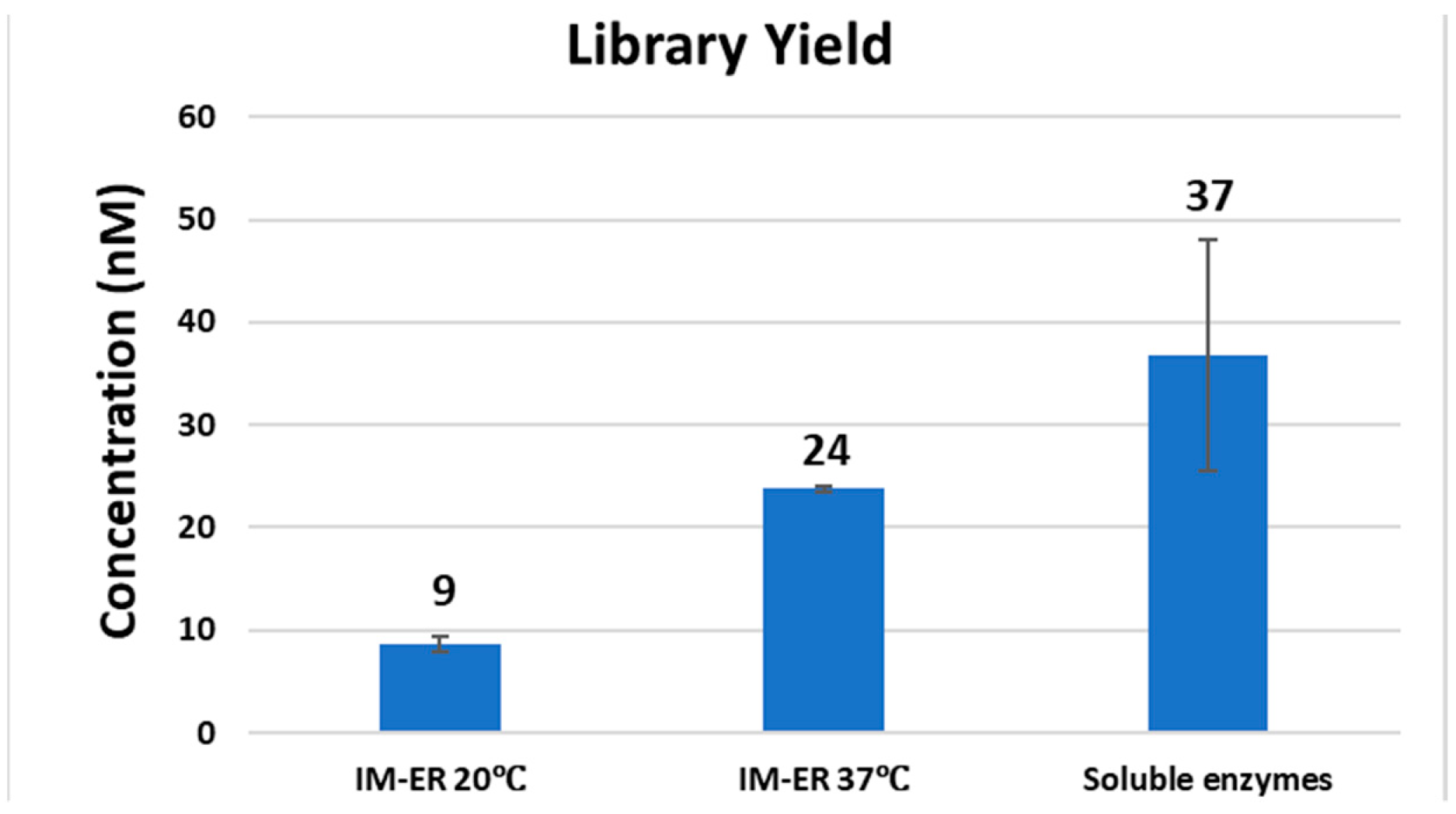
Clostridium acetobutylicum genomic DNA, purchased from ATCC (Manassas, VA, USA), was diluted to a final concentration of 100 µg/mL in 10 mM Tris-HCl, 1 mM EDTA, pH 7.5 and processed for ultrasonication shearing using Covaris AFA S2 system (Covaris, Woburn, MA, USA) with the settings of 5% duty cycle, intensity 10 and 200 cycles per burst for 6 min. For library construction, 1 µg of genomic DNA fragments was treated with either soluble untagged T4 DNA pol, T4 PNK and Taq DNA pol or the corresponding immobilized enzymes according to the workflow of the NEBNext Ultra DNA Library Prep Kit. DNA was first treated at 20 °C for 30 min with soluble end-repair enzymes or at 20 °C or 37 °C for 30 min with immobilized enzymes. For the workflow of soluble enzymes, end-repaired DNA was treated for A-tailing at 65 °C for 30 min. For the workflow of immobilized enzymes, after the removal of immobilized T4 DNA pol and T4 PNK, an aliquot of Taq DNA polymerase conjugated to magnetic beads was added to the end-repaired library and A-tailing was performed at 37 °C for 30 min. Each library was ligated to pre-annealed full-length paired-end Illumina adaptors according to the protocol of TruSeq DNA PCR-free LT Library Preparation Kit (Illumina, San Diego, CA, USA). DNA libraries were size-selected and analyzed to determine the size distribution using an Agilent High Sensitivity DNA Kit on a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA) and by qPCR using the NEBNext Library Quant Kit for Illumina.
The libraries were sequenced on an Illumina MiSeq in paired-end mode (2 × 75 bp). Reads were adapter trimmed (SeqPrep, v1.1
https://github.com/jstjohn/SeqPrep, GitHub, San Francisco, CA, USA) before alignment to the GRCh37 reference genome (bowtie 2.3.2, end-to-end, -X 1000, GitHub, San Francisco, CA, USA) [
35]. GC Bias was assessed using Picard’s Collect GC Bias Metrics (Picard 2.7.1, GitHub, San Francisco, CA, USA). Relevant low-GC regions were identified by intersecting 100 bp windows (Bedtools v2.25.0, Quinlan Lab, University of Utah, Salt Lake City, UT, USA) having GC fractions <0.2 with 80% overlap and with features in the Gencode v26 basic genes. Coverage of low-GC regions was assessed using Bedtools genomecov, Quinlan Lab, University of Utah, Salt Lake City, UT, USA.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}