
On the Binding Energy of Atoms in Crystals of Noble Gases and Metals and the Speed of Sound


Framatome, 1 Place Jean Millier, Paris la Défense CEDEX, 92084 Paris, France
†
Retired.
Crystals 2024, 14(10), 852; https://doi.org/10.3390/cryst14100852 (registering DOI)
Submission received: 12 August 2024
/
Revised: 24 September 2024
/
Accepted: 26 September 2024
/
Published: 29 September 2024
(This article belongs to the Section Crystal Engineering)
Abstract
:The speed of sound depends on the structure and material properties of the crystal, such as density and Young’s modulus. On the other hand, from atomistic arguments it is possible to associate Young’s modulus with other material properties. These observations lead to a relationship between binding energy of atoms in a crystal (which is one of the parameters appearing in Mie-Lennard-Jones potential), speed of sound in the longitudinal direction and mass of one atom in the lattice. This subject was addressed by several authors, providing different implementations of this relation. A literature review on this topic is made and the mathematical derivation of the relation is carried out. Applications of this relationship to rare gases, some metals and some rare earths are presented and the results compared to others taken from literature.
1. Introduction
Many interatomic potentials were developed to describe the interaction between atoms or molecules in condensed matter [1]. Among them, parametric two-body potentials, expressed by analytical relationships, allow the description of the intra-particle potential energy as a function of position, with the minimum value being the interatomic binding energy. Within this framework, potentials such as Morse, Mie and Lennard-Jones were successfully used to describe the bonding of atoms in crystal structures. Variations of these potentials have been proposed. For example, for the Lennard-Jones potential a variation has been implemented with truncation and shifting in order to mitigate performance issues and increase accuracy [2,3]. In solids, the binding energy, which appears as a parameter in the above mentioned potentials, can be evaluated from experiment via measurements of certain properties such as sublimation energy [4] or from simulation via molecular dynamics associated with ab initio calculations such as density functional theory (DFT) with periodic boundary conditions for crystalline structure optimisation [2,5,6,7]. Interatomic binding energy can be associated with physical phenomena in condensed matter; for example: a binding energy relationship can describe cohesion and other properties of metals [8]; a periodic binding energy dependence on atomic mass can be observed within the table of elements [4]; in nuclear reactor physics, the inter-nuclei binding energy can be associated with the neutron-nucleus interaction, expressed by the reaction’s cross sections [9]; and the binding energy of a crystal can be related to its melting temperature: the smaller the binding energy, the lower the melting temperature [10]. The aim of the present work is to explore the relationship between binding energy and sound velocity in crystals. This topic was previously addressed by several authors. Trachenko et al. [11] and Erokhin and Kalashnikov [12] proposed a proportionality between the speed of sound and the square root of the ratio between binding energy and the mass of the atom. Vos et al. [13] reported the square of the speed of sound as proportional to the ratio between the second derivative of the potential and the mass of the atom. Khrapak [14] related the product of the mass of the atom multiplied by the square of the speed of sound to an expression involving the first and second derivatives of the Lennard-Jones potential, making therefore appear the binding energy. The above kind of relationships is addressed in this work.
2. Theory
2.1. Preliminary Observations
The speed of sound in solids depends on the structure and on the material properties of the crystal. When the medium is a bar, the square of the speed of sound , in the longitudinal direction, which is the constant appearing in the one-dimensional wave equation, can be expressed in terms of the ratio between the Young’s modulus E and the density [15]:
In the theoretical derivation we consider a simple cubic crystal with Poisson’s ratio equal to zero, in order to prevent elastic deformation in the longitudinal direction from causing transverse deformation. Under this assumption, the Young’s modulus and the elastic constant coincide. This behavior can be transposed to fcc but not to bcc crystals. For symmetry reasons (cf. Appendix A) any of the axes of the referential perpendicular to the sides of the cubic structure can be chosen as direction of the sound wave. The chosen direction is perpendicular to the 100 face. The Young’s modulus, defined as the ratio between the strain and the stress, can be derived from atomistic arguments [16,17] based on the Mie-Lennard-Jones potential [18,19]. Combining the two expressions it is possible to obtain a relationship between the binding energy and the speed of sound in the crystal, in the longitudinal direction. Its derivation is presented in Section 2.2 and Section 2.3. Applications are presented in Section 3.
2.2. Young’s Modulus from Mie and Lennard-Jones Potential
The Mie potential is a simple particle-pair potential composed of two terms representing the repulsive and attractive forces between particles, such as the atoms in the crystal. Following the approach proposed in refs. [16,17], it is used here to represent the forces acting between the atoms in the crystal. Its formulation is as follows [20,21,22]
where is the binding energy of the interacting particles (the energy required to separate them), r is the distance between the particles, and is their equilibrium distance. The exponents depend on the material.
A most used form, with and , is the 12-6 Lennard-Jones potential [23]:
As shown in Figure 1, which plots function , parameters and are the depth of the potential and its position, respectively.
The Young’s modulus can be obtained from the definition of the Mie potential, knowing that it is the curvature of the potential with regard to the distance between atoms, i.e., the second derivative [16]. More in details, taking the derivative of the potential provides the force between the particles:
From Equation (4), it can be seen that , which proves that for the potential has a minimum. In the framework of our approach, , which is the equilibrium distance, is also the size a of the crystal cell as illustrated by the simple cube (sc) lattice in Figure 2, representative of the internal structure of the bar.
Let us apply a force F to the ends of the bar. The distance between the atoms will change from a to r. Young’s modulus E is defined as the ratio between stress (force per unit surface, ) and strain (relative deformation, ) in the limit as the strain goes to 0. Therefore, in mathematical terms, Young’s modulus can be written as:
Taking into account that vanishes for , Equation (5) can be written as:
which, in virtue of the derivative definition, is equivalent to:
Computing the derivative of at the point from Equation (4), after some manipulations, the relationship between Young’s modulus and binding energy is obtained:
2.3. Relationship between Binding Energy and Speed of Sound
The term at denominator, , i.e., the density multiplied by the atom-cell volume, is the mass M of a single atom-cell in a cubic lattice, where a simple cubic structure was assumed, which allows to write:
Parameters and a and therefore the density , are assumed to be at room temperature and atmospheric pressure, consistently with the conditions related to the sound velocity. The combination of Equation (10) and Equation (9) provides the relationship between binding energy and speed of sound in the longitudinal direction:
Even if it was assumed a simple cubic structure, this relationship is valid for face centered cubic (fcc) structure (see Figure 2) as well (cf. Appendix B). Erokhin and Kalashnikov [12] proposed the above relation with another proportionality constant, because their work was based on another interatomic potential. A similar relationship was proposed by Trachenko et al. [11] who pointed out that elastic constants are governed by the density of electromagnetic energy in condensed matter phases. As a consequence a relation between the bulk modulus and the binding energy was provided, resulting in a proportionality relation between the binding energy, the square of the sound velocity and the mass of the atom.
2.4. Binding Energy in a Crystal and the Associated Mass Defect
An observation from a relativistic point of view can be made about the crystal binding energy. If we want to separate an atom from the crystal structure, the amount of energy that binds the atom to its neighbors must be supplied on the form of latent heat of sublimation. Inversely, if a crystal is formed from these free atoms, the same amount of energy is released and a mass defect appears. For each atom the amount of energy is the value expressed by Equation (11) multiplied by a factor Z/2, where Z is the number of neighbors a given atom interacts with, which differs between sc, bcc and fcc crystals. According to the mass-energy equivalence, the associated mass defect responds to the relationship:
where c is the speed of light. Substituting Equation (12) into Equation (11) after some algebra we obtain:
Equation (13) shows that the relative mass defect associated with the crystal binding energy (i.e., the ratio between this mass defect and the mass of the atom) is proportional to the square of the ratio between speed of sound and speed of light.
3. Values of Binding Energy for Several Elements
Equation (11) was used to compute the binding energy for several elements having cubic structure, where the mass of the atom M is defined as the ratio between atomic weight A and Avogadro number :
The elements studied are: 4 rare gases, Ne, Ar, Kr and Xe, which at solid state have fcc crystal structure [25], 7 metals with fcc, 3 metals with bcc and 3 rare earth metals with fcc structure (Ce, Yb, Th). Both the 12-6 Lennard-Jones and Mie potentials were used. The data used in the formula expressed by Equation (11), taken from references [26,27,28,29,30,31], is presented in Table 1. It is pointed out that certain metals have more than one crystalline structure in nature. For example for iron, we have Fe which is bcc and Fe which is fcc. Within our approach it is assumed that the different forms have the same behavior with respect to atom-pair binding energy.
3.1. Values of Binding Energy Using the 12-6 Lennard-Jones Potential
For this formulation Equation (11) was used with the values and . The results for 4 rare gases are presented in Table 2 and compared to the ones obtained by Horton [21] solving a two-equation system set by fit of two crystal properties: the sublimation energy and the 0 °K lattice size. The results for 8 metals are presented in Table 3 and compared to the ones obtained by: Heinz et al. [23] and Kanhaiya et al. [32], who employed an isothermal–isobaric (NPT) molecular dynamics simulation with the Parrinello-Rahman method using a cubic supercell; Jacobson and Thompson [33] who determined the values of by calculating the total energy that binds an atom to the others in the crystal, without limiting the sum to the nearest neighbors, and imposing that the result be equal to the cohesion energy. The results for molybdenum are also provided but no comparison is available. Table 4 presents the results obtained for 3 fcc rare earth metals, which are compared to the ones obtained by Kanhaiya et al. [32].
3.2. Values of Binding Energy Using the Mie Potential
For this formulation Equation (11) was used with the values of n and m taken from the works of Magomedov [20] and Fürth [22]. The results for 4 rare gases, 10 metals and 3 fcc rare earth metals are presented in Table 5, Table 6 and Table 7, respectively, and compared to the ones obtained by Magomedov [20], who used an approach based on the preservation of measured quantities such as sublimation energy and thermal expansion coefficient. Table 8 shows the comparison with Fürth’s results for 10 metals [22]. The approximation of interaction of only nearest-neighbor atoms was adopted by both authors.
4. Conclusions
A relationship between the binding energy of atoms in crystals and the speed of sound was derived on the basis of the Mie and Lennard-Jones potentials. Comparing with other results taken from literature, its application to the 12-6 Lennard-Jones potential shows a quite good agreement in the case of rare gases. As far as metals are concerned, the deviations are higher but in the case of fcc structure they are consistent with the uncertainties inherent in the methods used by their authors to compute them [23]. However, results for rare earth metals show higher differences. Consistence is also observed in the case of the Mie potential, even though a difference of about 40% is observed for platinum. In both cases of Lennard-Jones and Mie potential, the comparison related to bcc metals shows higher differences. This is because the theory was derived for sc crystals and the behavior can be extended to fcc structures, as shown in Appendix B, but for bcc metals the extension is not justified. It is emphasized that this relationship, expressed by Equation (11), was obtained with a theoretical approach, while the models that were used to compute the binding energy values we compared with belong to force field classes that provide deeper insight into the physics of crystals. They use more complex algorithms to perform detailed calculations of interatomic interactions in crystals. These force fields are applicable to all types of crystals and other forms of condensed matter, while our approach is limited to cubic crystals of pure elements. Extension of this work to hpc crystals and salt crystals is ongoing.
The values of the binding energy computed for various elements depend on the form of the potential. Values associated with the 12-6 Lennard-Jones potential cannot be compared with the ones associated with the Mie potential. This is due to the fact that the values of the binding energy , the exponents n and m, the distance, have to be seen as part of an inseparable whole.
Funding
No funding was received for this study.
Data Availability Statement
The data used for this study can be found inside the article.
Acknowledgments
The author thanks Hendrik Heinz (University of Colorado-Boulder, Boulder, USA) and Mahach N. Magomedov (Russian Academy of Sciences, Makhachkala, Russia) for their help in the interpretation of the results. Special thanks to Joe Wolfe (School of Physics, UNSW Sydney, Bidjigal Country, Australia), who inspired this work.
Conflicts of Interest
The author declares no conflict of interest.
Appendix A. About Anisotropy in Crystals
Although metal crystals are anisotropic solids [34,35], their elastic properties are the same in the directions perpendicular to the sides of the elementary cube (100), (010), (001). This means that the elastic properties are fully represented by only the set of elastic constants, , , and . However, some metals behave as they were isotropic. On the basis of the elastic anisotropy, defined as:
Ziegenhain et al. [36] showed that Al, which has X = 1.20, is nearly isotropic while Cu, with X = 3.22, is quite anisotropic. Quantitatively, deviations from the isotropic value were only 1% for Al, while thy were up to 10% for Cu.
Appendix B. Case of Face Centered Cubic Structure
Equation (11), expressing the relationship between the binding energy and the square of speed of sound, was obtained in case of simple cubic structure. We show here that the case of face centered cubic structure (fcc) respond to the same relationship. In case of fcc structure (Figure 2), the lattice can be seen as 4 embedded lattices with cells of size a, where one of the lattices is related to the vertices and 3 others to the center of the faces. Therefore the surface is affected by 4 pairs of interacting particles and the force appearing in Equation (5) has to be multiplied by 4, which conducts to a Young modulus 4 times higher than in the case of simple cubic structure:
This means that the square of the speed of sound, which is proportional to E according to Equation (1), is 4 times higher too and Equation (9) becomes:
On the other hand, in a fcc lattice cell there is more than one atom. This number is 4, which leads to replacing Equation (10) by the following one:
Substituting Equation (A4) into Equation (A3) and solving with respect to we obtain the relationship between binding energy and speed of sound, which is the same as Equation (11):
Appendix C. Determination of Fürth’s Mie Potential Parameters
Fürth’s article [22] does not report the values of the Mie potential parameter , but the values of the sublimation energy and the equation relating them:
where q is a constant depending on the lattice type, with its value being 6 for fcc and 4 for bcc crystals. The values of are reported in Table A1 and were used to compute inverting Equation (A6) (see Table 8).

{kind=link}
{kind=link}
Table A1.
Sublimation energy for 10 metals from Fürth’s work.
| Element | (kcal/mol) |
|---|---|
| Al | 67.60 |
| Au | 90.70 |
| Pb | 47.00 |
| Ni | 98.00 |
| Pt | 124.7 |
| Ag | 69.40 |
| Cu | 81.70 |
| Fe | 96.50 |
| Mo | 156.0 |
| W | 203.9 |
References
- Müsera, M.H.; Sukhomlinov, S.V.; Pastewka, L. Interatomic potentials: Achievements and challenges. Adv. Phys. X 2023, 8, 2093129. [Google Scholar] [CrossRef]
- Kshirsagar, S.; Mandadapu, K.K.; Papadopoulos, P. Classical molecular dynamics simulations of crystal lattices with truncated Taylor series-based interatomic potentials. Comput. Mater. Sci. 2016, 120, 127–134. [Google Scholar] [CrossRef]
- Hafskjold, B.; Travis, K.P.; Hass, A.B.; Hammer, M.; Aasen, A.; Wilhelmsen, Ø. Thermodynamic properties of the 3D Lennard-Jones/spline model. Mol. Phys. 2019, 117, 3754–3769. [Google Scholar] [CrossRef]
- Magomedov, M.N. The Correlation of the Parameters of Interatomic Interaction in Crystals with the Position of Atom in the Periodic Table. High Temp. 2008, 46, 484–494. [Google Scholar] [CrossRef]
- Blazhynska, M.M.; Kyrychenko, A.; Kalugin, O.N. Molecular dynamics simulation of the size-dependent morphological stability of cubic shape silver nanoparticles. Mol. Simul. 2018, 44, 981–991. [Google Scholar] [CrossRef]
- Ramezani-Dakhel, H.; Ruan, L.; Huang, Y.; Heinz, H. Molecular Mechanism of Specific Recognition of Cubic Pt Nanocrystals by Peptides and of the Concentration-Dependent Formation from Seed Crystals. Adv. Funct. Mater. 2015, 25, 1374–1384. [Google Scholar] [CrossRef]
- Lutsko, J.F.; Schoonen, C. Classical density-functional theory applied to the solid state. Phys. Rev. E 2020, 102, 062136. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, J.; Smith, J.R.; Rose, J.H. Diatomic Molecules and Metallic Adhesion, Cohesion, and Chemisorption: A Single Binding-Energy Relation. Phys. Rev. Lett. 1986, 50, 1385. [Google Scholar] [CrossRef]
- Stacey, W.M. Nuclear Reactor Physics, 2nd ed.; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2007. [Google Scholar]
- Chai, Y.; Gao, X.; Liang, Y.; Wang, J.; Hu, W.; Wang, Y. Improvement and prediction on high temperature melting characteristics of coal ash. High Temp. Mater. Process. 2023, 42, 20220039. [Google Scholar] [CrossRef]
- Trachenko, K.; Monserrat, B.; Pickard, C.J.; Brazhkin, V.V. Speed of sound from fundamental physical constants. Sci. Adv. 2020, 6, eabc8662. [Google Scholar] [CrossRef]
- Erokhin, K.M.; Kalashnikov, N.P. Relationships of Macroscopic Characteristics of a Solid with the Binding Energy of an Ion in a Metal Lattice. Phys. Solid State 2021, 63, 973–977. [Google Scholar] [CrossRef]
- Vos, F.L.J.; Aalberts, D.P.; van Saarloos, W. Simple method for calculating the speed of sound in tight-binding models: Application to the Su-Schrieffer-Heeger model. Phys. Rev. B 1996, 53, R5986. [Google Scholar] [CrossRef]
- Khrapak, S.A. Sound Velocities of Lennard-Jones Systems Near the Liquid-Solid Phase Transition. Molecules 2020, 25, 3498. [Google Scholar] [CrossRef] [PubMed]
- Kinsler, L.E.; Frey, A.R.; Coppens, A.B.; Sanders, J.V. Fundamentals of Acoustics, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 1999. [Google Scholar]
- Lung, C.W.; March, N.H. Mechanical Properties of Metals: Atomistic and Fractal Continuum Approaches; World Scientific Pub. Co., Inc.: Singapore, 1999. [Google Scholar]
- Roylance, D. Mechanics of Materials; John Wiley & Sons: Hoboken, NJ, USA, 1996. [Google Scholar]
- Kittel, C. Introduction to Solid State Physics; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Stone, A. The Theory of Intermolecular Forces; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Magomedov, M.N. The Calculation of the Parameters of the Mie-Lennard-Jones Potential. High Temp. 2006, 44, 513–529. [Google Scholar] [CrossRef]
- Horton, G.K. Ideal Rare-Gas Crystals. Am. J. Phys. 1968, 36, 93–119. [Google Scholar] [CrossRef]
- Fürth, R. On the equation of state for solids. Proc. R. Soc. A 1994, 183, 87–110. [Google Scholar]
- Heinz, H.; Vaia, R.A.; Farmer, B.L.; Naik, R.R. Accurate Simulation of Surfaces and Interfaces of Face-Centered Cubic Metals Using 12-6 and 9-6 Lennard-Jones Potentials. J. Phys. Chem. C 2008, 112, 17281–17290. [Google Scholar] [CrossRef]
- Aleksandrov, I.V.; Goncharov, A.F.; Zisman, A.N.; Stishov, S.M. Diamond at high pressures: Raman scattering of light, equation of state, and high-pressure scale. Sov. Phys. JETP 1987, 66, 384–390. [Google Scholar]
- Niebel, K.F.; Venables, J.A. An explanation of the crystal structure of the rare gas solids. Proc. R. Soc. Lond. A 1974, 336, 365–377. [Google Scholar]
- Hakim, T.M.; Glyde, H.R. Dynamics of solid neon monolayers. Phys. Rev. B 1988, 37, 984. [Google Scholar] [CrossRef]
- Lawrence, D.J.; Neale, F.E. Longitudinal wave velocity in solid argon. Proc. Phys. Soc. 1965, 85, 1261. [Google Scholar] [CrossRef]
- Peter, H.; Korpiun, P.; Lüscher, E. Measurement of the longitudinal sound velocity in solid krypton at 4.2 °K, 77 °K and 90 °K. Phys. Lett. A 1968, 26, 207. [Google Scholar] [CrossRef]
- Weigel, P.L.R.; Hansen, E.V.; Dolinski, M.J. Development and Characterization of Solid Noble Bolometers. In Proceedings of the 2019 Meeting of the Division of Particles and Fields of the American Physical Society, DPF 2019, Boston, MA, USA, 29 July–2 August 2019. [Google Scholar]
- Lide, D.R. (Ed.) CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Samsonov, G.V. (Ed.) Handbook of the Physicochemical Properties of the Elements; IFI/Plenum: New York, NY, USA; Washington, DC, USA, 1968. [Google Scholar]
- Kanhaiya, K.; Kim, S.; Im, W.; Heinz, H. Accurate simulation of surfaces and interfaces of ten FCC metals and steel using Lennard-Jones potentials. NPJ Comput. Mater. 2021, 7, 17. [Google Scholar] [CrossRef]
- Jacobson, D.W.; Thompson, G.B. Revisting Lennard Jones, Morse, and N-M potentials for metals. Comput. Mater. Sci. 2022, 205, 111206. [Google Scholar] [CrossRef]
- Kube, C.M. Elastic anisotropy of crystals. AIP Adv. 2016, 6, 095209. [Google Scholar] [CrossRef]
- Landau, L.D.; Lifshitz, E.M. Theory of Elasticity, 2nd ed.; Pergamon Press: Oxford, UK, 1970. [Google Scholar]
- Ziegenhain, G.; Urbassek, H.M.; Hartmaier, A. Influence of crystal anisotropy on elastic deformation and onset of plasticity in nanoindentation: A simulational study. J. Appl. Phys. 2010, 107, 061807. [Google Scholar] [CrossRef]
Figure 1.
The Mie-Lennard-Jones potential.
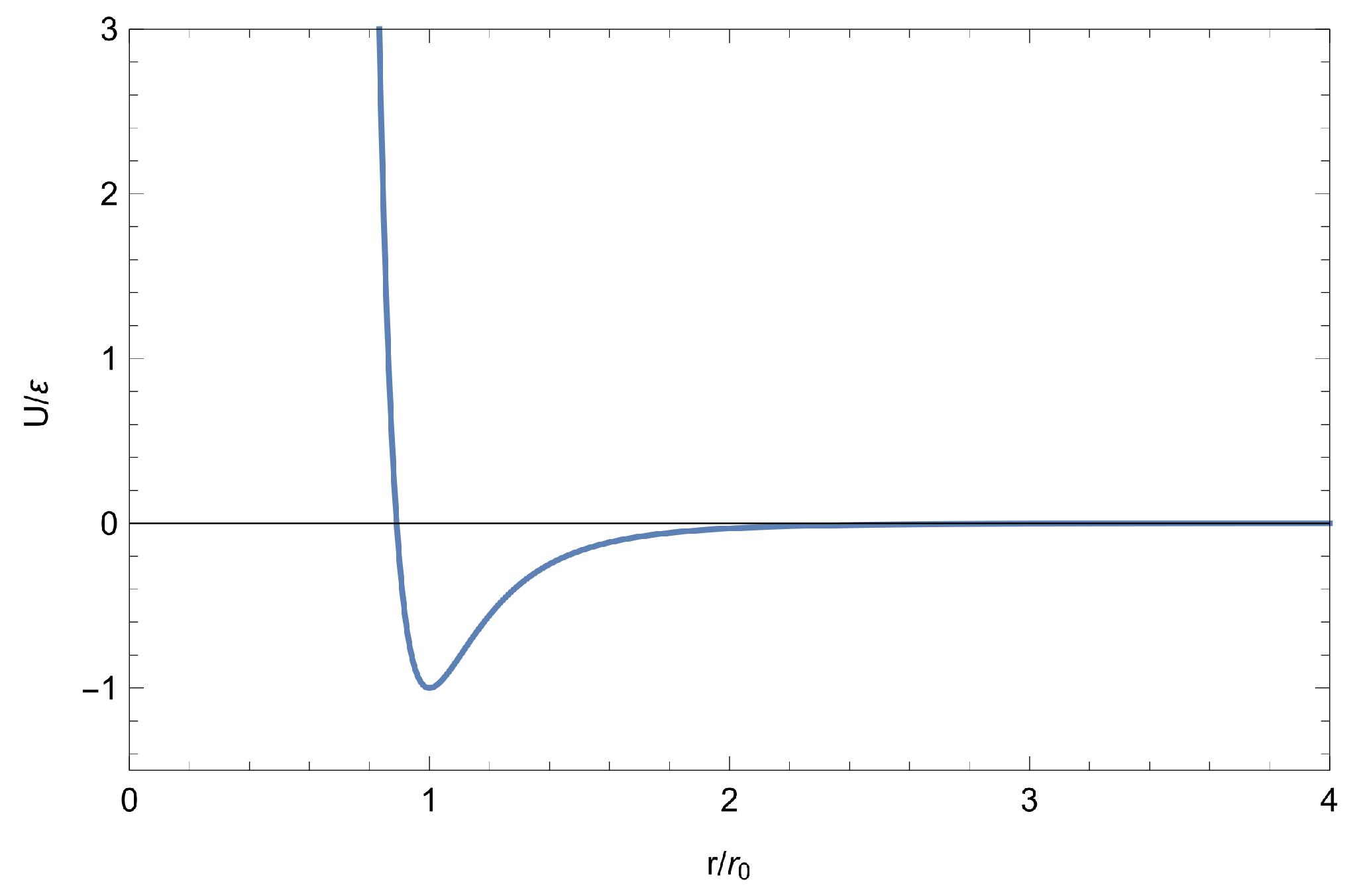
Figure 2.
Cubic lattices: simple, body centered and face centered.
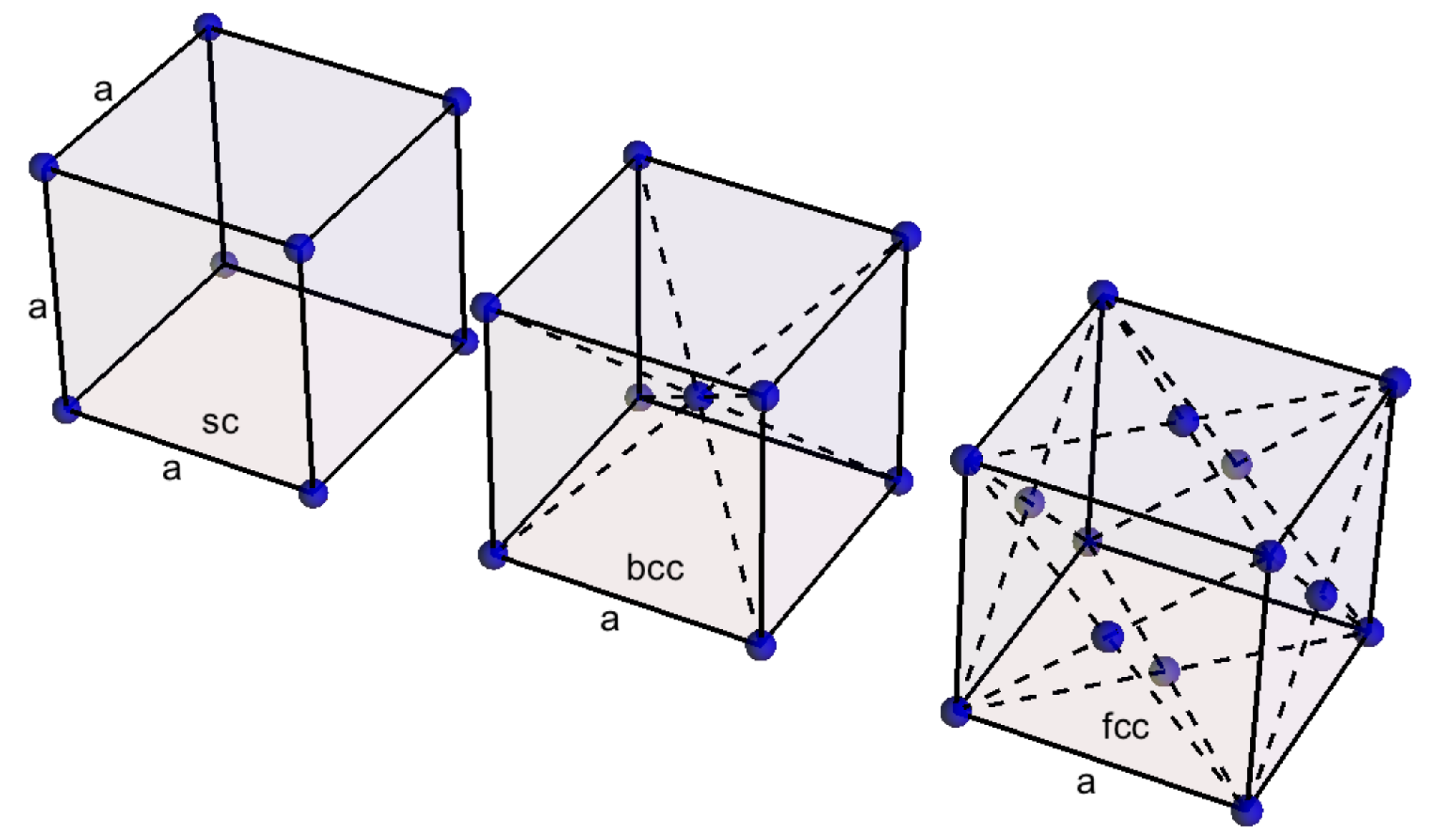
Table 1.
Data used in the evaluation of the binding energy for the elements studied.
| Atomic Weight | Crystal Structure | Speed of Sound (m/s) | Reference | |
|---|---|---|---|---|
| Ne | 20.149 | fcc | 1290 | [26] |
| Ar | 39.948 | fcc | 1630 | [27] |
| Kr | 83.798 | fcc | 1335 | [28] |
| Xe | 131.29 | fcc | 1150 | [29] |
| A1 | 26.981 | fcc | 6420 | [30] |
| Au | 196.97 | fcc | 3240 | [30] |
| Pb | 207.20 | fcc | 2160 | [30] |
| Ni | 58.693 | fcc | 6040 | [30] |
| Pt | 195.08 | fcc | 3260 | [30] |
| Ag | 107.87 | fcc | 3650 | [30] |
| Cu | 63.546 | fcc | 4760 | [30] |
| Fe | 55.845 | bcc | 5950 | [30] |
| Mo | 95.940 | bcc | 6250 | [30] |
| W | 183.84 | bcc | 5220 | [30] |
| Ce | 140.12 | fcc | 2100 | [31] |
| Yb | 173.04 | fcc | 1590 | [31] |
| Th | 232.04 | fcc | 2490 | [31] |
Table 2.
Binding energy (eV) for rare gases using the 12-6 Lennard-Jones potential and comparison with Horton’s results.
Table 2.
Binding energy (eV) for rare gases using the 12-6 Lennard-Jones potential and comparison with Horton’s results.
| This Work | Horton [21] | |
|---|---|---|
| Ne | 0.00483 | 0.00450 |
| Ar | 0.01528 | 0.01473 |
| Kr | 0.02150 | 0.02028 |
| Xe | 0.02500 | 0.02859 |
Table 3.
Binding energy (eV) for 10 metals using the 12-6 Lennard-Jones potential and comparison with Heinz’s, Kanhaiya’s (*) and Jacobson and Thompson’s results.
Table 3.
Binding energy (eV) for 10 metals using the 12-6 Lennard-Jones potential and comparison with Heinz’s, Kanhaiya’s (*) and Jacobson and Thompson’s results.
| This Work | Heinz et al. [23] | Jacobson and Thompson [33] | |
|---|---|---|---|
| Al | 0.1601 | 0.1743 | 0.1983 |
| Au | 0.2977 | 0.2294 | 0.2231 |
| Pb | 0.1392 | 0.1270 | 0.1188 |
| Ni | 0.3083 | 0.2450 | 0.2608 |
| Pt | 0.2985 | 0.3382 | 0.3427 |
| Ag | 0.2069 | 0.1977 | 0.1722 |
| Cu | 0.2073 | 0.2046 | 0.2050 |
| Fe | 0.2846 | 0.2601 * | 0.2625 |
| Mo | 0.5395 | - | - |
| W | 0.7211 | - | 0.5377 |
Table 4.
Binding energy (eV) for 3 fcc rare earth metals using the 12-6 Lennard-Jones potential and comparison with Kanhaiya’s results.
Table 4.
Binding energy (eV) for 3 fcc rare earth metals using the 12-6 Lennard-Jones potential and comparison with Kanhaiya’s results.
| This Work | Kanhaiya et al. [32] | |
|---|---|---|
| Ce | 0.0890 | 0.2766 |
| Yb | 0.0630 | 0.1175 |
| Th | 0.2071 | 0.3672 |
Table 5.
Binding energy (eV) for rare gases using the Mie potential and comparison with Magomedov’s results.
Table 5.
Binding energy (eV) for rare gases using the Mie potential and comparison with Magomedov’s results.
| This Work | Magomedov [20] | n | m | |
|---|---|---|---|---|
| Ne | 0.0028 | 0.0045 | 5.83 | 21.39 |
| Ar | 0.0100 | 0.0150 | 6.62 | 16.69 |
| Kr | 0.0148 | 0.0205 | 6.56 | 15.92 |
| Xe | 0.0173 | 0.0285 | 6.73 | 15.42 |
Table 6.
Binding energy (eV) for 10 metals using the Mie potential and comparison with Magomedov’s results.
Table 6.
Binding energy (eV) for 10 metals using the Mie potential and comparison with Magomedov’s results.
| This Work | Magomedov [20] | n | m | |
|---|---|---|---|---|
| Al | 0.4239 | 0.5714 | 2.49 | 10.92 |
| Au | 0.7027 | 0.6387 | 1.96 | 15.56 |
| Pb | 0.3100 | 0.3399 | 2.27 | 14.24 |
| Ni | 0.8149 | 0.7506 | 3.56 | 7.65 |
| Pt | 0.6372 | 0.9795 | 2.53 | 13.33 |
| Ag | 0.4673 | 0.4944 | 3.08 | 10.35 |
| Cu | 0.5884 | 0.5895 | 3.03 | 8.37 |
| Fe | 0.8975 | 1.0838 | 3.54 | 6.45 |
| Mo | 2.3635 | 1.7042 | 2.14 | 7.68 |
| W | 1.7695 | 2.2068 | 3.42 | 8.58 |
Table 7.
Binding energy (eV) for 3 fcc rare earth metals using the Mie potential and comparison with Magomedov’s results.
Table 7.
Binding energy (eV) for 3 fcc rare earth metals using the Mie potential and comparison with Magomedov’s results.
| This Work | Magomedov [20] | n | m | |
|---|---|---|---|---|
| Ce | 0.5390 | 0.7334 | 2.45 | 4.85 |
| Yb | 0.2374 | 0.2682 | 3.61 | 5.29 |
| Th | 0.8161 | 0.9970 | 3.02 | 6.05 |
Table 8.
Binding energy (eV) for 10 metals using the Mie potential and comparison with Fürth’s results.
Table 8.
Binding energy (eV) for 10 metals using the Mie potential and comparison with Fürth’s results.
| This Work | Fürth [22] | n | m | |
|---|---|---|---|---|
| Al | 0.4117 | 0.4886 | 4 | 7 |
| Au | 0.4871 | 0.6556 | 5.5 | 8 |
| Pb | 0.2783 | 0.3397 | 3 | 12 |
| Ni | 0.7926 | 0.7083 | 4 | 7 |
| Pt | 0.4884 | 0.9013 | 5.5 | 8 |
| Ag | 0.4729 | 0.5016 | 4.5 | 7 |
| Cu | 0.5330 | 0.5905 | 4 | 7 |
| Fe | 0.7319 | 1.0462 | 4 | 7 |
| Mo | 1.1099 | 1.6913 | 5 | 7 |
| W | 1.4835 | 2.2107 | 5 | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dall’Osso, A. On the Binding Energy of Atoms in Crystals of Noble Gases and Metals and the Speed of Sound. Crystals 2024, 14, 852. https://doi.org/10.3390/cryst14100852
AMA Style
Dall’Osso A. On the Binding Energy of Atoms in Crystals of Noble Gases and Metals and the Speed of Sound. Crystals. 2024; 14(10):852. https://doi.org/10.3390/cryst14100852
Chicago/Turabian StyleDall’Osso, Aldo. 2024. "On the Binding Energy of Atoms in Crystals of Noble Gases and Metals and the Speed of Sound" Crystals 14, no. 10: 852. https://doi.org/10.3390/cryst14100852
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.