

Colloidal Characteristics of Poly(L-Lactic Acid)-b-Poly (ε-Caprolactone) Block Copolymer-Based Nanoparticles Obtained by an Emulsification/Evaporation Method

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Block Copolymers
2.3. Preparation of NPs by an Emulsification/Evaporation Method
2.4. Characterization Techniques
3. Results
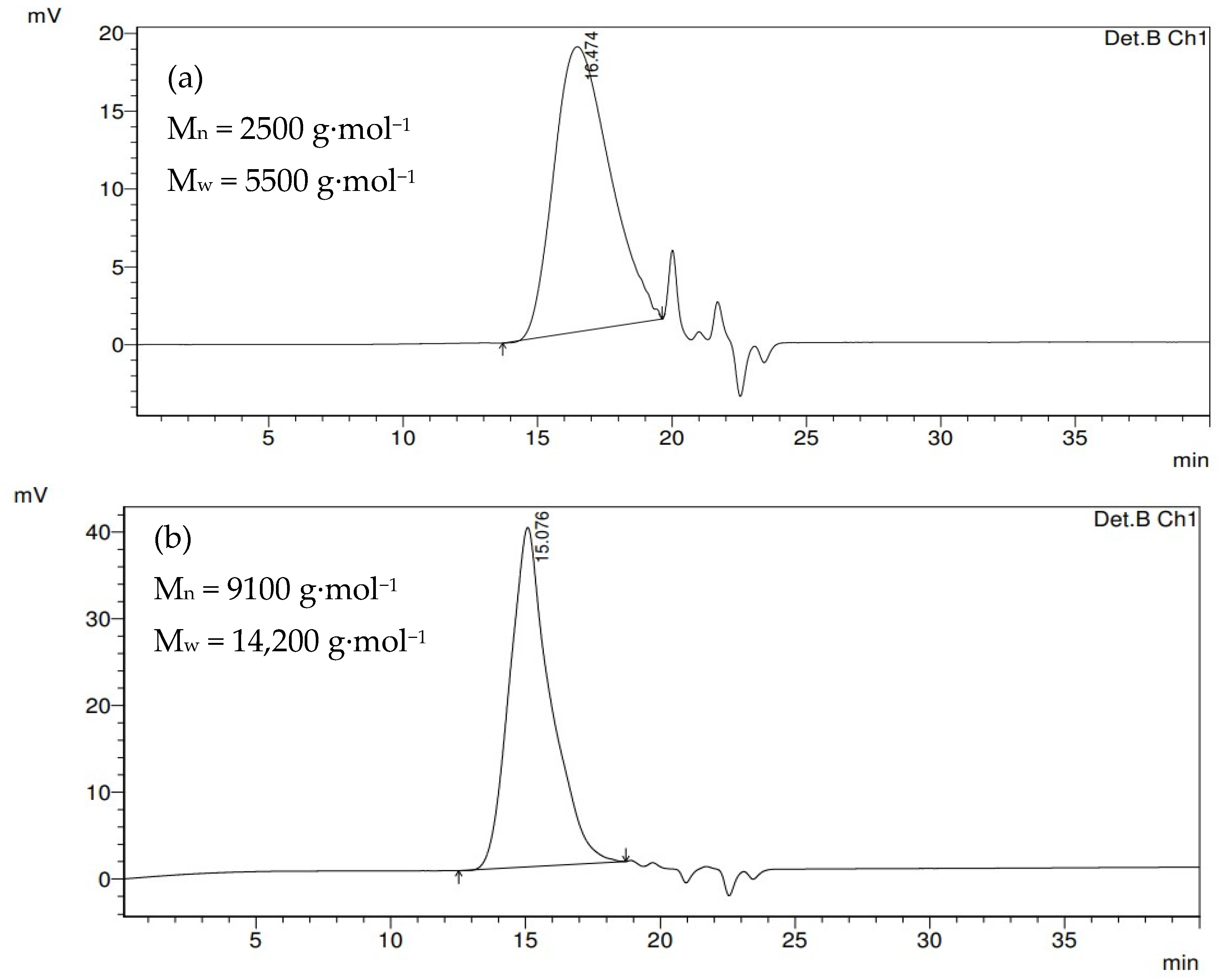
3.1. Diblock Copolymer Synthesis
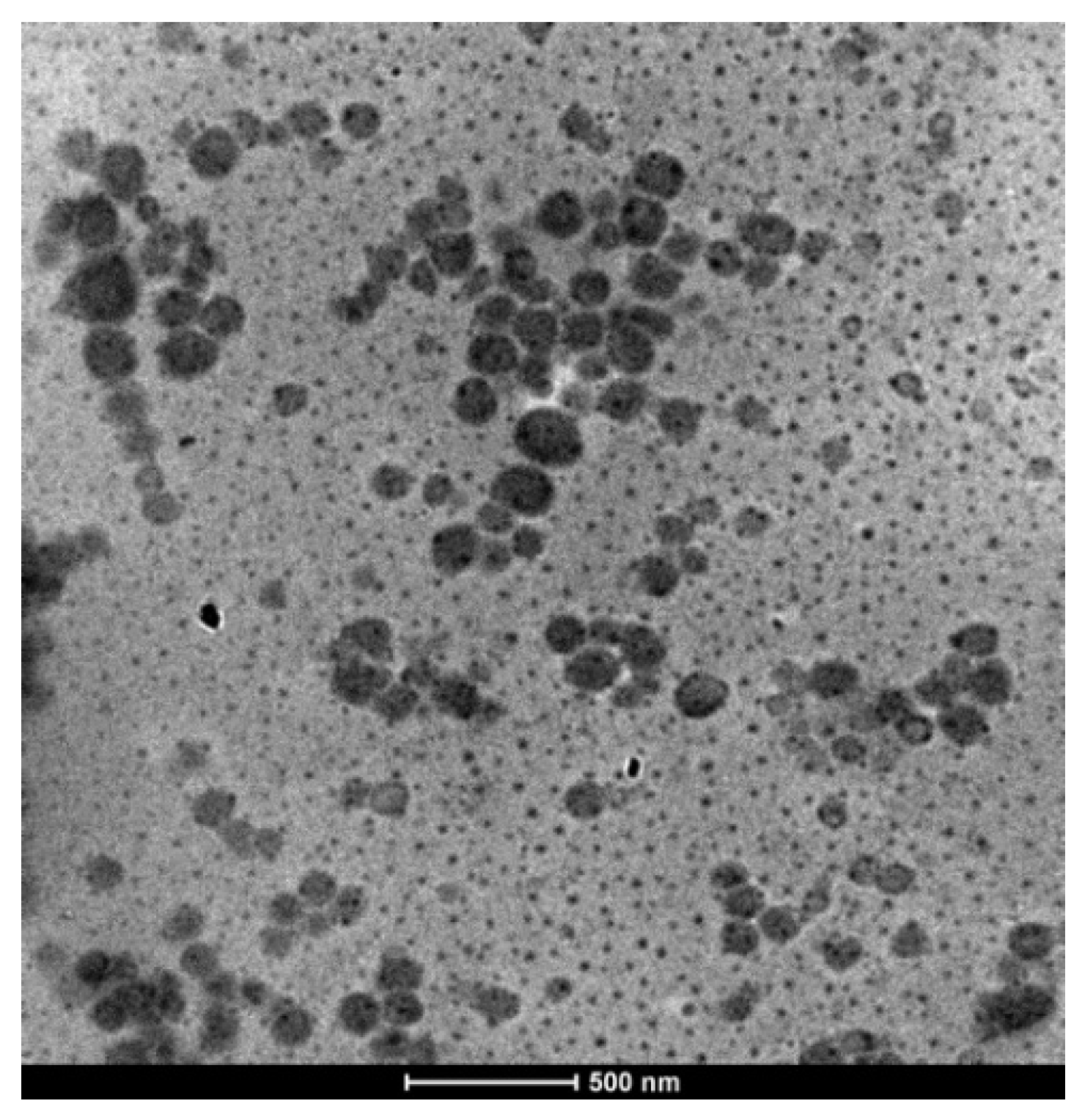
3.2. From Controlled Block Copolymers to Nanoparticles
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Curcio, M.; Diaz-Gomez, L.; Cirillo, G.; Nicoletta, F.P.; Leggio, A.; Iemma, F. Dual-Targeted Hyaluronic Acid/Albumin Micelle-Like Nanoparticles for the Vectorization of Doxorubicin. Pharmaceutics 2021, 13, 304. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Santos, B.; Silva, P.B.; Balansin Rigon, R.; Rillo Sato, M.; Chorilli, M. Formulating SLN and NLC as Innovative Drug Delivery Systems for Non-Invasive Routes of Drug Administration. Curr. Med. Chem. 2020, 27, 3623–3656. [Google Scholar] [CrossRef] [PubMed]
- Toro-Córdova, A.; Sanz, B.; Goya, G.F. A Concise Review of Nanomaterials for Drug Delivery and Release. Curr. Nanosci. 2020, 16, 399–412. [Google Scholar] [CrossRef]
- Ladurantie, C.; Coustets, M.; Czaplicki, G.; Demange, P.; Mazères, S.; Dauvillier, S.; Teissié, J.; Rols, M.P.; Milon, A.; Ecochard, V.; et al. A protein nanocontainer targeting epithelial cancers: Rational engineering, biochemical characterization, drug loading and cell delivery. Nanoscale 2019, 11, 3248–3260. [Google Scholar] [CrossRef]
- Washington, K.E.; Kularatne, R.N.; Karmegam, V.; Biewer, M.C.; Stefan, M.C. Recent advances in aliphatic polyesters for drug delivery applications. Nanomed. Nanobiotechnol. 2017, 9, 1446. [Google Scholar] [CrossRef]
- Mohamed, F.; van der Walle, C.F. Engineering biodegradable polyester particles with specific drug targeting and drug release properties. J. Pharm. Sci. 2008, 97, 71–87. [Google Scholar] [CrossRef]
- Shive, M.S.; Anderson, J.M. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [CrossRef]
- Tokiwa, Y.; Calabia, B.P. Biodegradability and biodegradation of poly(lactide). Appl. Microbiol. Biotechnol. 2006, 72, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Moreira, T.D.; Martins, V.B.; da Silva Junior, A.H.; Sayer, C.; de Araujo, P.H.H.; Immich, A.P.S. New insights into biomaterials for wound dressings and care: Challenges and trends. Prog. Org. Coat. 2024, 187, 108118. [Google Scholar] [CrossRef]
- Engelberg, I.; Kohn, J. Physico-mechanical properties of degradable polymers used in medical applications: A comparative study. Biomaterials 1991, 12, 292–304. [Google Scholar] [CrossRef]
- Kubisa, P.; Penczek, S. Cationic activated monomer polymerization of heterocyclic monomers. Prog. Polym. Sci. 1999, 24, 1409–1437. [Google Scholar] [CrossRef]
- Schwach, G.; Coudane, J.; Engel, R.; Vert, M. More about the polymerization of lactides in the presence of stannous octoate. J. Polym. Sci. A Polym. Chem. 1997, 35, 3431–3440. [Google Scholar] [CrossRef]
- Florczak, M.; Mosnacek, J.; Duda, A. L,L-Lactide and ε-Caprolactone Block Copolymers by a ‘Poly(L,L-lactide) Block First’ Route. Macromol. Rapid Commun. 2007, 28, 1385–1391. [Google Scholar] [CrossRef]
- Peponi, L.; Navarro-Baena, I.; Báez, J.E.; Kenny, J.M.; Marcos-Fernández, A. Effect of the molecular weight on the crystallinity of PCL-b-PLLA di-block copolymers. Polymer 2012, 53, 4561–4568. [Google Scholar] [CrossRef]
- Simões, C.L.; Viana, J.C.; Cunha, A.M. Mechanical properties of poly(ε-caprolactone) and poly(lactic acid) blends. J. Appl. Polym. Sci. 2009, 112, 345–352. [Google Scholar] [CrossRef]
- Feng, X.D.; Song, C.X.; Chen, W.Y. Synthesis and evaluation of biodegradable block copolymers of epsilon -caprolactone and D L-lactide. J. Polym. Sci. Polym. Lett. 1983, 21, 593–600. [Google Scholar] [CrossRef]
- Ye, W.P.; Du, F.S.; Jin, W.H.; Yang, J.Y.; Xu, Y. In vitro degradation of poly(caprolactone), poly(lactide) and their block copolymers: Influence of composition, temperature and morphology. React. Funct. Polym. 1997, 32, 161–168. [Google Scholar] [CrossRef]
- Ye, W.P.; Chien, Y.W. Dual-controlled drug delivery across biodegradable copolymer. II. Delivery kinetics of levonorgestrel and estradiol from (matrix/matrix) laminate drug delivery system. J. Control Release 1996, 41, 259–269. [Google Scholar] [CrossRef]
- Atanase, L.I.; Salhi, S.; Cucoveica, O.; Ponjavic, M.; Nikodinovic-Runic, J.; Delaite, C. Biodegradability Assessment of Polyester Copolymers Based on Poly(ethylene adipate) and Poly(ε-caprolactone). Polymers 2022, 14, 3736. [Google Scholar] [CrossRef]
- Kuperkar, K.; Atanase, L.I.; Bahadur, A.; Crivei, I.C.; Bahadur, P. Degradable Polymeric Bio(nano)materials and Their Biomedical Applications: A Comprehensive Overview and Recent Updates. Polymers 2024, 16, 206. [Google Scholar] [CrossRef]
- Tehrani, S.F.; Bharadwaj, P.; Leblond, C.J.; Roullin, V.G. Purification processes of polymeric nanoparticles: How to improve their clinical translation? J. Control Release 2023, 360, 591–612. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Bhattacharyya, C.; Sahu, R.; Dua, T.K.; Kandimalla, R.; Dewanjee, S. Polymeric nanotherapeutics: An emerging therapeutic approach for the management of neurodegenerative disorders. J. Drug Deliver. Sci. Technol. 2024, 91, 105267. [Google Scholar] [CrossRef]
- Pulingam, T.; Foroozandeh, P.; Chuah, J.-A.; Sudesh, K. Exploring Various Techniques for the Chemical and Biological Synthesis of Polymeric Nanoparticles. Nanomaterials 2022, 12, 576. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, H.; Jangde, R.K. Current updated review on preparation of polymeric nanoparticles for drug delivery and biomedical applications. Next Nanotechnol. 2023, 2, 100013. [Google Scholar] [CrossRef]
- Hernandez-Giottonini, K.Y.; Rodriguez-Cordova, R.J.; Gutierez-Valenzuela, C.A.; Penunuri-Miranda, O.; Zavala-Rivera, P.; Guerrero-German, P.; Lucero-Acuna, A. PLGA nanoparticles preparations by emulsification and nanoprecipitation techniques: Effects of formulation parameters. RSC Adv. 2020, 10, 4218–4231. [Google Scholar] [CrossRef] [PubMed]
- Crescenzi, V.; Manzini, G.; Calzolari, G.; Borri, C. Thermodynamics of fusion of poly-beta-propiolactone and poly-ε-caprolactone. comparative analysis of the melting of aliphatic polylactone and polyester chains. Eur. Polym. J. 1972, 8, 449–463. [Google Scholar] [CrossRef]
- Kowalski, A.; Duda, A.; Penczek, S. Kinetics and mechanism of cyclic esters polymerizationinitiated with tin(II) octoate. 3. Polymerization of l,l-dilactide. Macromolecules 2000, 33, 7359–7370. [Google Scholar] [CrossRef]
- Zhang, R.; Du, F.; Jariyavidyanont, K.; Zhuravlev, E.; Schick, C.; Androsch, R. Glass transition temperature of poly(d,l-lactic acid) of different molar mass. Thermochim. Acta 2022, 718, 179387. [Google Scholar] [CrossRef]
- McKeen, L. The effect of heat aging on the properties of sustainable polymers. In Plastics Design Library, The Effect of Long Term Thermal Exposure on Plastics and Elastomers, 2nd ed.; McKeen, L., Ed.; William Andrew Publishing: Norwich, NY, USA, 2021; Chapter 11; pp. 313–332. [Google Scholar] [CrossRef]
- Fernández-Tena, A.; Pérez-Camargo, R.; Coulembier, O.; Sangroniz, L.; Aranburu, N.; Guerrica-Echevarria, G.; Liu, G.; Wang, D.; Cavallo, D.; Müller, A. Effect of Molecular Weight on the Crystallization and Melt Memory of Poly(ε-caprolactone) (PCL). Macromolecules 2023, 56, 4602–4620. [Google Scholar] [CrossRef]
- Witt, S.; Scheper, T.; Walter, J.G. Production of polycaprolactone nanoparticles with hydrodynamic diameters below 100 nm. Eng. Life Sci. 2019, 19, 658–665. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
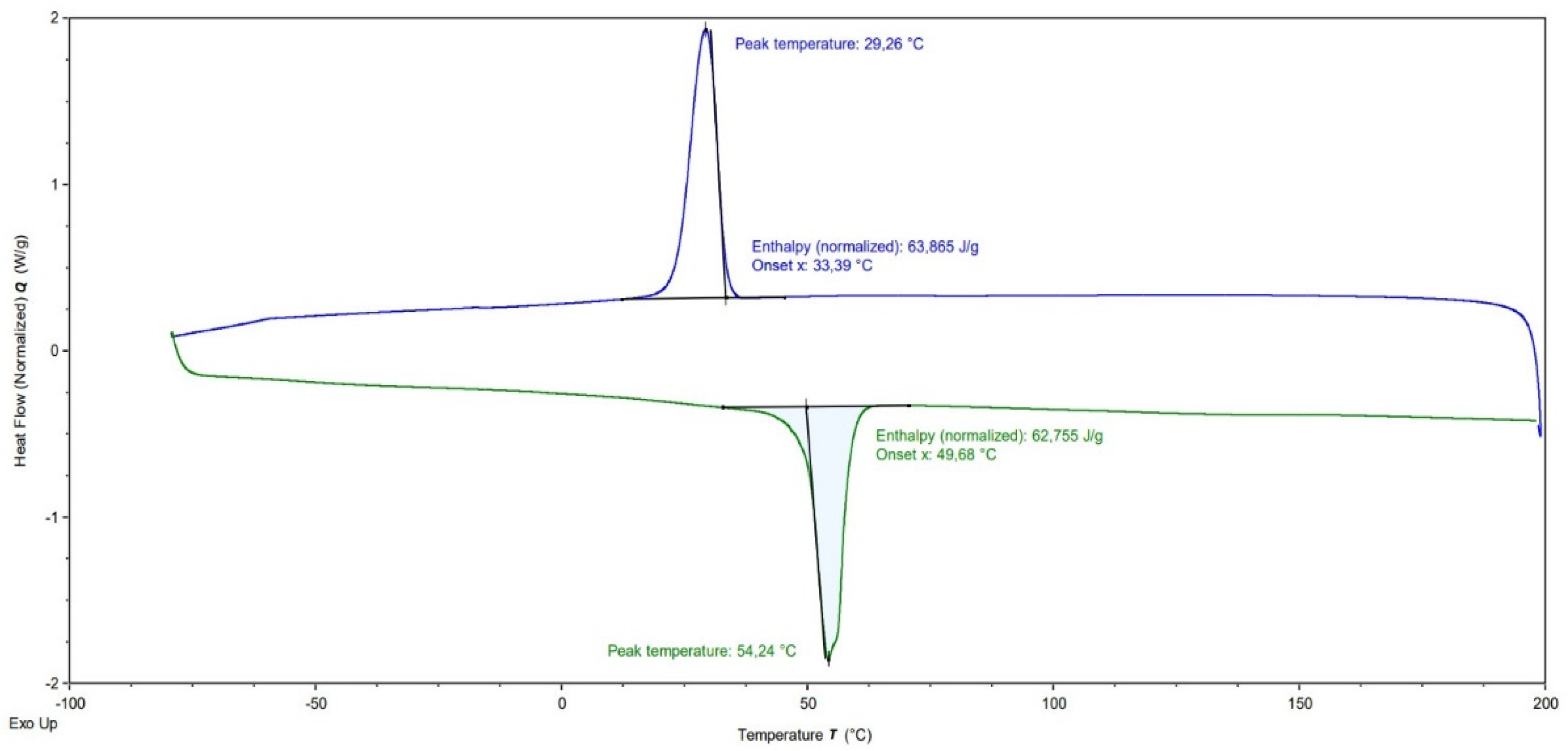
| Sample | Mn PLLA b (g·mol−1) | Theor. DPn PCL | Exp. DPn PCLa | Mn PCL a (g·mol−1) | Mn Cop. a (g·mol−1) | Mn Cop. b (g·mol−1) | Ð b | Tm (°C) ΔHm (J.g−1) c | TC c (°C) | χc c (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| DB1 | 2500 | 18 | 16 | 1800 | 4300 | 5700 | 1.45 | 48.2 17.7 | 16.0 | 30.8 |
| DB2 | 53 | 77 | 8800 | 11,300 | 9100 | 1.56 | 52.8 41.8 | 30.3 | 38.5 | |
| DB3 | 88 | 85 | 9800 | 12,300 | 10,200 | 1.46 | 52.0 57.2 | 28.0 | 51.5 | |
| DB4 | 123 | 143 | 16,300 | 18,800 | 12,700 | 1.41 | 53.4 63.5 | 30.3 | 52.5 | |
| DB5 | 175 | 162 | 18,500 | 21,000 | 13,200 | 1.41 | 49.7 62.8 | 29.3 | 51.0 |
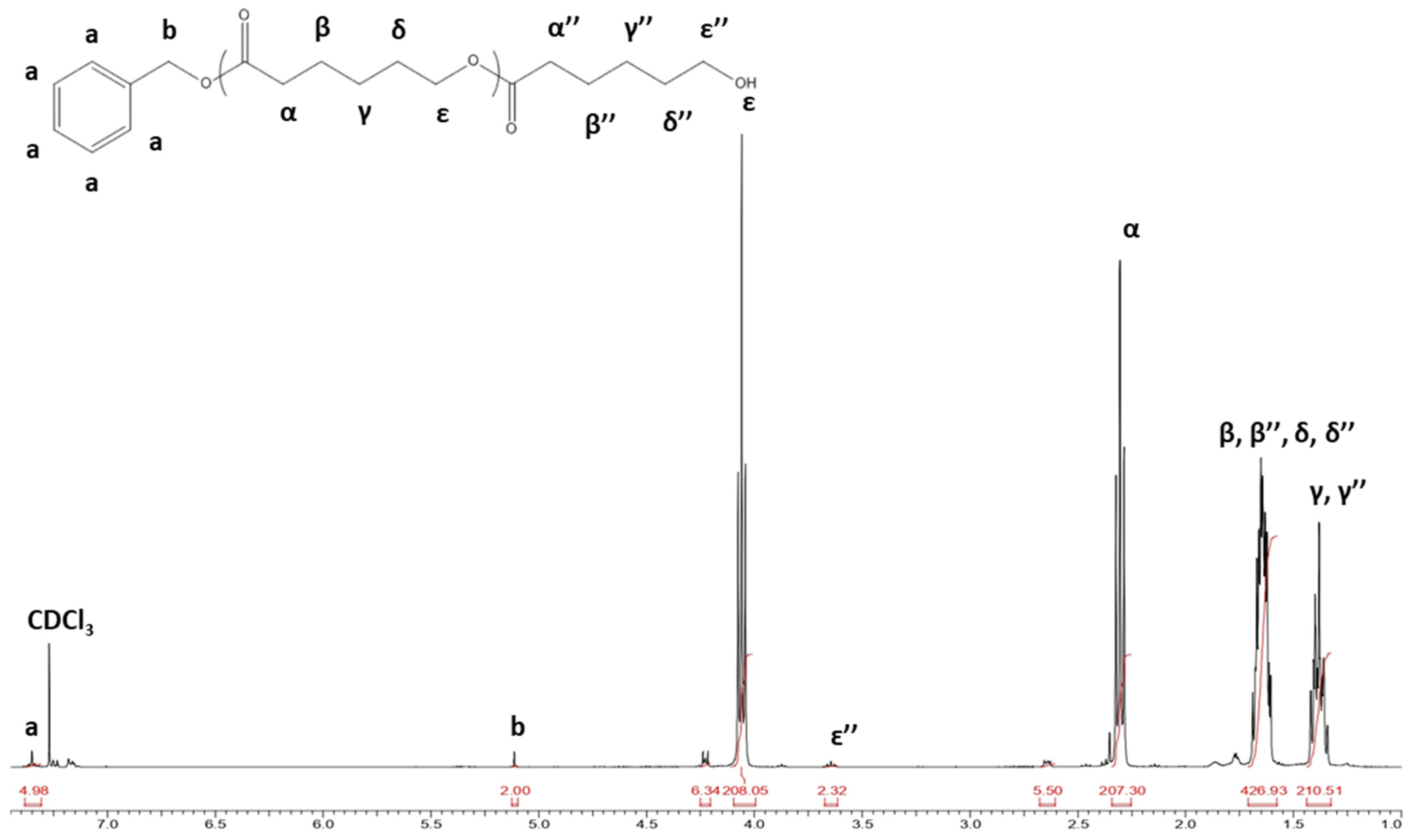
| Signal Groups | Type of Proton | Assigned Letter | δ (ppm) |
|---|---|---|---|
| Initiator’s groups | Bn-CH2-O- O-CH2-Bn-CH2-O- | a | 7.35 |
| Bn-CH2-O- O-CH2-Bn-CH2-O- | b | 5.10 | |
| Repetitive units | -(CO-CH(CH3)-O)n- | c | 5.15 |
| -(CO-CH(CH3)-O)n- | d | 1.50 | |
| Intermediary groups | -(CO-CH(CH3)-O)- | c′ | 5.15 |
| -(CO-CH(CH3)-O)- | d′ | 1.50 | |
| -(CO-CH2-CH2-CH2-CH2-CH2-O)- | α′ | 2.40 | |
| -(CO-CH2-CH2-CH2-CH2-CH2-O)- | β′, γ′, δ′ | 1.42/1.65 | |
| -(CO-CH2-CH2-CH2-CH2-CH2-O)- | ε′ | 4.15 | |
| Repetitive units | -(CO-CH2-CH2-CH2-CH2-CH2-O)n- | α | 2.30 |
| -(CO-CH2-CH2-CH2-CH2-CH2-O)n | β, γ, δ | 1.42/1.65 | |
| -(CO-CH2-CH2-CH2-CH2-CH2-O)n- | ε | 4.05 | |
| Terminal groups | -CO-CH2-CH2-CH2-CH2-CH2-OH | α″ | N/A |
| -CO-CH2-CH2-CH2-CH2-CH2-OH | β″, γ″, δ″ | N/A | |
| -CO-CH2-CH2-CH2-CH2-CH2-OH | ε″ | 3.65 | |
| Residual groups | α-CL | N/A | 2.63 |
| ε-CL | N/A | 4.23 | |
| CH-LA | N/A | 5.03 |
| Temperature (°C) | Parameters | After Solvent Evaporation | After Addition of Trehalose | After Freeze-Drying in the Absence of Trehalose | After Freeze-Drying in the Presence of Trehalose |
|---|---|---|---|---|---|
| 25 | Z-average (nm) | 128.5 ± 0.1 | 139.2 ± 0.3 | 173.3 ± 0.8 | 151.4 ± 0.4 |
| PDI | 0.066 ± 0.01 | 0.071 ± 0.3 | 0.134 ± 0.02 | 0.069 ± 0.01 | |
| 37 | Z-average (nm) | 127.8 ± 0.2 | 138.9 ± 0.2 | 172.4 ± 0.4 | 150.3 ± 0.2 |
| PDI | 0.064 ± 0.01 | 0.078 ± 0.2 | 0.127 ± 0.01 | 0.099 ± 0.01 |
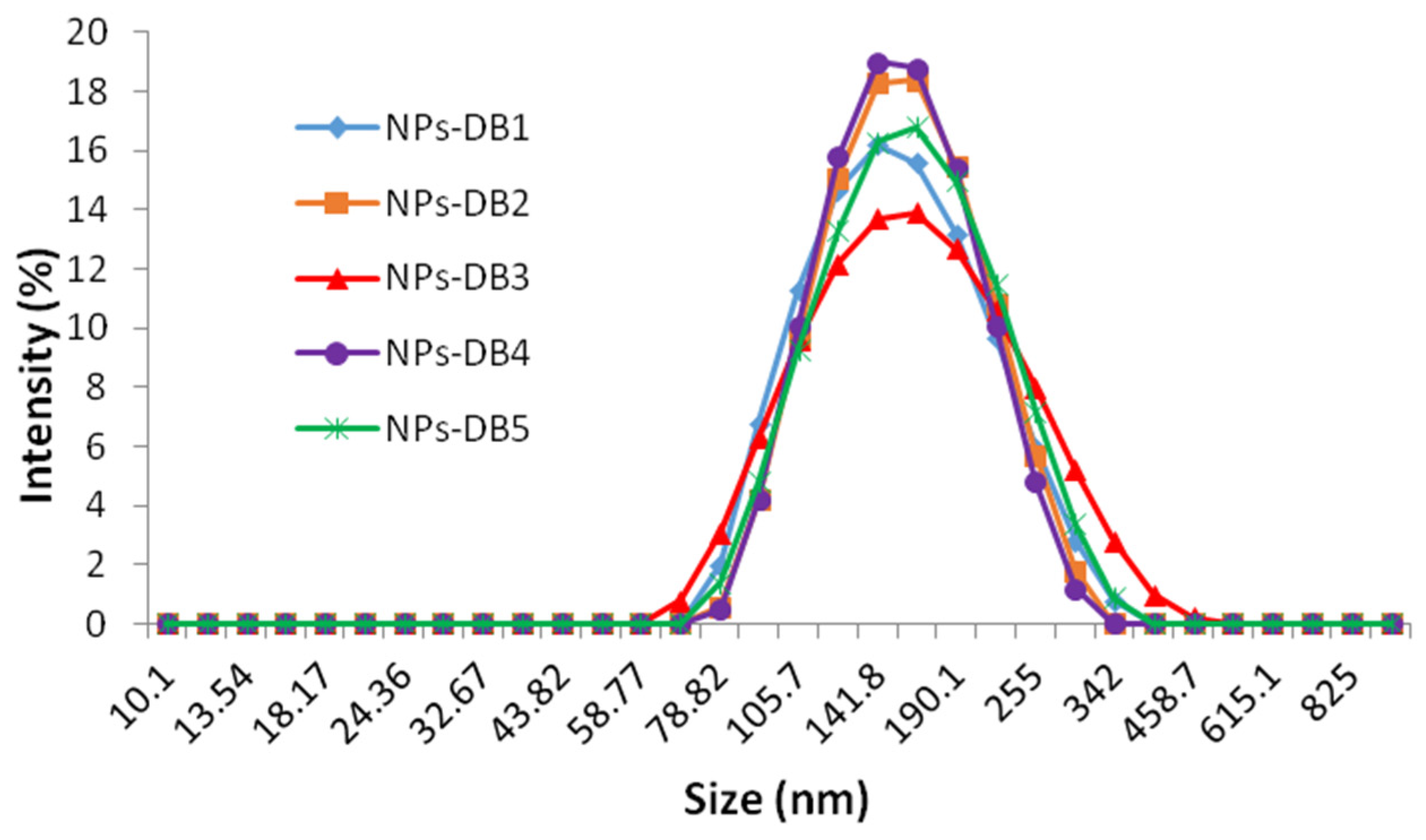
| T (°C) | Parameters | NPs-DB1 | NPs-DB2 | NPs-DB3 | NPs-DB4 | NPs-DB5 |
|---|---|---|---|---|---|---|
| 25 | Z-average (nm) | 144.3 ± 0.2 | 148.9 ± 0.3 | 151.4 ± 0.4 | 151.7 ± 0.4 | 153.1 ± 0.3 |
| Dv (nm) | 143.8 ± 0.3 | 148.2 ± 0.2 | 150.9 ± 0.3 | 151.2 ± 0.4 | 153.0 ± 0.2 | |
| PDI | 0.114 ± 0.02 | 0.078 ± 0.01 | 0.069 ± 0.01 | 0.078 ± 0.01 | 0.088 ± 0.01 | |
| 37 | Z-average (nm) | 143.5 ± 0.4 | 148.0 ± 0.1 | 150.3 ± 0.2 | 150.9 ± 0.5 | 152.8 ± 0.4 |
| Dv (nm) | 143.2 ± 0.3 | 147.5 ± 0.2 | 149.5 ± 0.3 | 150.2 ± 0.3 | 152.5 ± 0.3 | |
| PDI | 0.097 ± 0.01 | 0.072 ± 0.01 | 0.099 ± 0.01 | 0.070 ± 0.01 | 0.072 ± 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cucoveica, O.; Stadoleanu, C.; Bertsch, C.; Triaud, R.; Condriuc, I.P.; Atanase, L.I.; Delaite, C. Colloidal Characteristics of Poly(L-Lactic Acid)-b-Poly (ε-Caprolactone) Block Copolymer-Based Nanoparticles Obtained by an Emulsification/Evaporation Method. Polymers 2024, 16, 2748. https://doi.org/10.3390/polym16192748
Cucoveica O, Stadoleanu C, Bertsch C, Triaud R, Condriuc IP, Atanase LI, Delaite C. Colloidal Characteristics of Poly(L-Lactic Acid)-b-Poly (ε-Caprolactone) Block Copolymer-Based Nanoparticles Obtained by an Emulsification/Evaporation Method. Polymers. 2024; 16(19):2748. https://doi.org/10.3390/polym16192748
Chicago/Turabian StyleCucoveica, Oana, Carmen Stadoleanu, Christelle Bertsch, Romain Triaud, Iustina Petra Condriuc, Leonard Ionut Atanase, and Christelle Delaite. 2024. "Colloidal Characteristics of Poly(L-Lactic Acid)-b-Poly (ε-Caprolactone) Block Copolymer-Based Nanoparticles Obtained by an Emulsification/Evaporation Method" Polymers 16, no. 19: 2748. https://doi.org/10.3390/polym16192748