
Association Mapping for Common Bunt Resistance in Wheat Landraces and Cultivars

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Phenotyping Reaction to Common Bunt
2.3. Statistical Analysis
2.4. Genotyping and Genome-Wide Association Analysis
3. Results
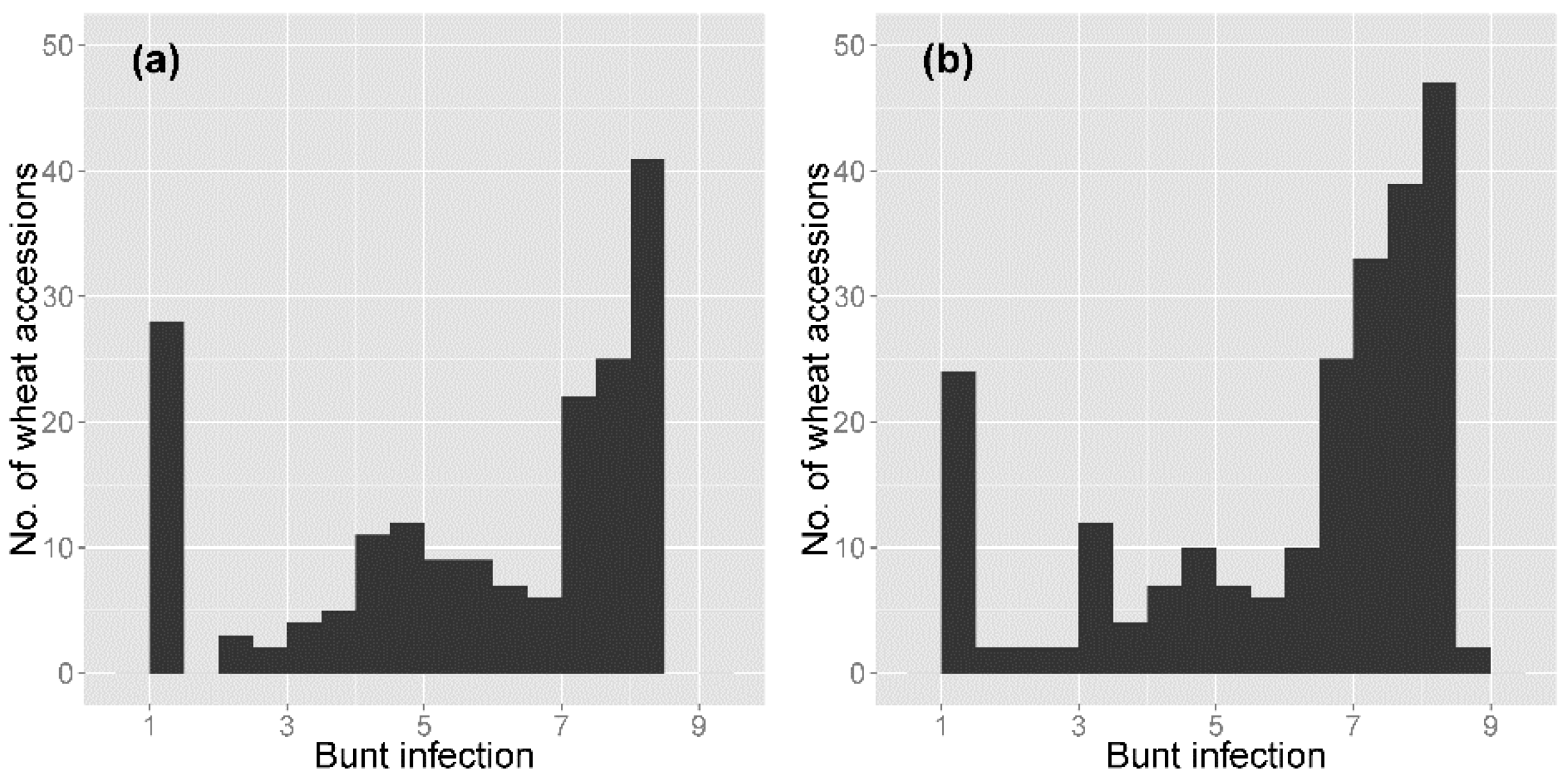
3.1. Common Bunt Infection Scoring
3.2. Genome-Wide Analysis of Common Bunt Resistance
4. Discussion
4.1. Reaction to Common Bunt in the Field
4.2. Distribution of Polymorphic Markers
4.3. Genome Wide Association Analysis
4.4. QTLs Identified for Common Bunt Resistance
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Blazkova, V.; Bartos, P. Virulence pattern of European bunt samples (Tilletia tritici and T-laevis) and sources of resistance. Cereal Res. Commun. 2002, 30, 335–342. [Google Scholar] [CrossRef]
- Martynov, S.P.; Dobrotvorskaya, T.V.; Sorokin, O.D. Comparative genealogical analysis of the resistance of winter wheat to common bunt. Russ. J. Genet. 2004, 4, 410–424. [Google Scholar] [CrossRef]
- Váñová, M.; Matusinsky, P.; Benada, J. Survey of incidence of bunts (Tilletia caries and Tilletia controversa) in the Czech Republic and susceptibility of winter wheat cultivars. Plant Prot. Sci. 2006, 42, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Waldow, F.; Jahn, M. Investigations in the regulation of common bunt (Tilletia tritici) of winter wheat with regard to threshold values, cultivar susceptibility and non-chemical protection measures. J. Plant Dis. Prot. 2007, 114, 269–275. [Google Scholar] [CrossRef]
- Ciuca, M.; Saulescu, N.N. Screening Romanian Winter Wheat Germplasm for Presence of Bt10 Bunt Resistance Gene, Using Molecular Markers. Rom. Agric. Res. 2008, 25, 1–5. [Google Scholar]
- Slinkard, A.E.; Elliott, F.C. The Effect of Bunt Incidence on the Yield of Wheat in Eastern Washington. Agron. J. 1954, 46, 439–441. [Google Scholar] [CrossRef]
- Swinburne, T.R. Infection of wheat by Tilletia caries (DC.) TUL.; the causal organism of bunt. Trans. Br. Mycol. Soc. 1963, 46, 145-IN12. [Google Scholar] [CrossRef]
- Fernandez, J.A.; Duran, R.; Schafer, J.F. Histological Aspects of Dwarf Bunt Resistance in Wheat. Phytopathology 1978, 68, 1417–1421. [Google Scholar] [CrossRef]
- Padel, S.; Orsini, S.; Solfanelli, F.; Zanoli, R. Can the Market Deliver 100% Organic Seed and Varieties in Europe? Sustainability 2021, 13, 10305. [Google Scholar] [CrossRef]
- Skovmand, B.; Fox, P.N.; Villareal, R.L. Triticale in Commercial Agriculture: Progress and Promise. In Advances in Agronomy; Brady, N.C., Ed.; Academic Press: Cambridge, MA, USA, 1984; Volume 37, pp. 1–45. [Google Scholar] [CrossRef]
- Dumalasova, V.; Bartos, P. Reaction of wheat, alternative wheat and triticale cultivars to common bunt. Czech J. Genet. Plant Breed. 2010, 46, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Goates, B.J. Identification of New Pathogenic Races of Common Bunt and Dwarf Bunt Fungi, and Evaluation of Known Races Using an Expanded Set of Differential Wheat Lines. Plant Dis. 2012, 96, 361–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muellner, A.E.; Buerstmayr, M.; Eshonkulov, B.; Hole, D.; Michel, S.; Hagenguth, J.F.; Pachler, B.; Pernold, R.; Buerstmayr, H. Comparative mapping and validation of multiple disease resistance QTL for simultaneously controlling common and dwarf bunt in bread wheat. Theor. Appl. Genet. 2021, 134, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Fofana, B.; Humphreys, D.G.; Cloutier, S.; McCartney, C.A.; Somers, D.J. Mapping quantitative trait loci controlling common bunt resistance in a doubled haploid population derived from the spring wheat cross RL4452 x AC Domain. Mol. Breed. 2008, 21, 317–325. [Google Scholar] [CrossRef]
- Wang, S.; Knox, R.E.; DePauw, R.M.; Clarke, F.R.; Clarke, J.M.; Thomas, J.B. Markers to a common bunt resistance gene derived from ‘Blizzard’ wheat (Triticum aestivum L.) and mapped to chromosome arm 1BS. Theor. Appl. Genet. 2009, 119, 541–553. [Google Scholar] [CrossRef]
- Singh, A.; Knox, R.E.; DePauw, R.M.; Singh, A.K.; Cuthbert, R.D.; Kumar, S.; Campbell, H.L. Genetic mapping of common bunt resistance and plant height QTL in wheat. Theor. Appl. Genet. 2016, 129, 243–256. [Google Scholar] [CrossRef]
- Dumalasova, V.; Simmonds, J.; Bartos, P.; Snape, J. Location of genes for common bunt resistance in the European winter wheat cv. Trintella. Euphytica 2012, 186, 257–264. [Google Scholar] [CrossRef]
- Zou, J.; Semagn, K.; Chen, H.; Iqbal, M.; Asif, M.; N’Diaye, A.; Navabi, A.; Perez-Lara, E.; Pozniak, C.; Yang, R.C.; et al. Mapping of QTLs associated with resistance to common bunt, tan spot, leaf rust, and stripe rust in a spring wheat population. Mol. Breed. 2017, 37, 144. [Google Scholar] [CrossRef]
- Bokore, F.E.; Cuthbert, R.D.; Knox, R.E.; Singh, A.; Campbell, H.L.; Pozniak, C.; N’Diaye, A.; Sharpe, A.G.; Ruan, Y. Mapping quantitative trait loci associated with common bunt resistance in a spring wheat (Triticum aestivum L.) variety Lillian. Theor. Appl. Genet. 2019, 132, 3023–3033. [Google Scholar] [CrossRef] [Green Version]
- Knox, R.E.; Campbell, H.L.; Depauw, R.M.; Gaudet, D.; Puchalski, B.; Clarke, F.C. DNA markers for resistance to common bunt in ‘McKenzie’ wheat. Can. J. Plant Pathol. 2013, 35, 328–337. [Google Scholar] [CrossRef]
- Muellner, A.E.; Eshonkulov, B.; Hagenguth, J.; Pachler, B.; Michel, S.; Buerstmayr, M.; Hole, D.; Buerstmayr, H. Genetic mapping of the common and dwarf bunt resistance gene Bt12 descending from the wheat landrace PI119333. Euphytica 2020, 216, 83. [Google Scholar] [CrossRef]
- Goates, B.J. Bunt and Smut Disease of Wheat. In Bunt and Smut Disease of Wheat: Concepts and Methods of Disease Management; Wilcoxson, R.D., Saari, E.E., Eds.; CIMMYT: El Batán, Mexico, 1996; pp. 12–25. [Google Scholar]
- Orabi, J.; Jahoor, A.; Backes, G. Changes in allelic frequency over time in European bread wheat (Triticum aestivum L.) varieties revealed using DArT and SSR markers. Euphytica 2014, 197, 447–462. [Google Scholar] [CrossRef]
- Akbari, M.; Wenzl, P.; Caig, V.; Carling, J.; Xia, L.; Yang, S.Y.; Uszynski, G.; Mohler, V.; Lehmensiek, A.; Kuchel, H.; et al. Diversity arrays technology (DArT) for high-throughput profiling of the hexaploid wheat genome. Theor. Appl. Genet. 2006, 113, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. 2013. Available online: http://www.R-project.org/ (accessed on 16 December 2016).
- Schwender, H.; Fritsch, A. scrime: Analysis of High-Dimensional Categorical Data such as SNP Data. 2013. Available online: http://CRAN.R-project.org/package=scrime (accessed on 16 December 2016).
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Lipka, A.E.; Tian, F.; Wang, Q.S.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z.W. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.M.; Zaitlen, N.A.; Wade, C.M.; Kirby, A.; Heckerman, D.; Daly, M.J.; Eskin, E. Efficient control of population structure in model organism association mapping. Genetics 2008, 178, 1709–1723. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.W.; Ersoz, E.; Lai, C.Q.; Todhunter, R.J.; Tiwari, H.K.; Gore, M.A.; Bradbury, P.J.; Yu, J.M.; Arnett, D.K.; Ordovas, J.M.; et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 2010, 42, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Churchill, G.A.; Doerge, R.W. Empirical Threshold Values for Quantitative Trait Mapping. Genetics 1994, 138, 963–971. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. Roy. Stat. Soc. B Met. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [Green Version]
- Warnes, G.; Gorjanc, G.; Leisch, F.; Man, M. Genetics: Population Genetics. 2013. Available online: http://CRAN.R-project.org/package=genetics (accessed on 16 December 2016).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Breseghello, F.; Sorrells, M.E. Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 2006, 172, 1165–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danmarks Meteorologisk Institut. 2014. Available online: http://www.dmi.dk/vejr/arkiver/vejrarkiv/ (accessed on 4 April 2014).
- McIntosh, R.; Hart, G.E.; Devos, K.M.; Gale, M.D.; Rogers, W.J. Catalogue of Gene Symbols for Wheat. 1998. Available online: http://wheat.pw.usda.gov/ggpages/wgc/98/ (accessed on 29 June 2014).
- Semagn, K.; Bjornstad, A.; Skinnes, H.; Maroy, A.G.; Tarkegne, Y.; William, M. Distribution of DArT, AFLP, and SSR markers in a genetic linkage map of a doubled-haploid hexaploid wheat population. Genome 2006, 49, 545–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marone, D.; Panio, G.; Ficco, D.B.M.; Russo, M.A.; De Vita, P.; Papa, R.; Rubiales, D.; Cattivelli, L.; Mastrangelo, A.M. Characterization of wheat DArT markers: Genetic and functional features. Mol. Genet. Genom. 2012, 287, 741–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, A.L.; Zaitlen, N.A.; Reich, D.; Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 2010, 11, 459–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, K.Y.; Aranzana, M.J.; Kim, S.; Lister, C.; Shindo, C.; Tang, C.L.; Toomajian, C.; Zheng, H.G.; Dean, C.; Marjoram, P.; et al. An Arabidopsis example of association mapping in structured samples. PLoS Genet. 2007, 3, e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.M.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Muller, B.U.; Stich, B.; Piepho, H.P. A general method for controlling the genome-wide type I error rate in linkage and association mapping experiments in plants. Heredity 2011, 106, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Steffan, P.M.; Torp, A.M.; Borgen, A.; Backes, G.; Rasmussen, S.K. Mapping of common bunt resistance gene Bt9 in wheat. Theor. Appl. Genet. 2017, 130, 1031–1040. [Google Scholar] [CrossRef] [Green Version]
- Menzies, J.G.; Knox, R.E.; Popovic, Z.; Procunier, J.D. Common bunt resistance gene Bt10 located on wheat chromosome 6D. Can. J. Plant Sci. 2006, 86, 1409–1412. [Google Scholar] [CrossRef]
- Christensen, D.; Borgen, A. Is Bt8 located at chromosome 6D and closely linked to Bt10? Virtual Bunt and Smut Workshop 5–6 May 2021. p. 14. Available online: https://bunt.boku.ac.at (accessed on 11 January 2022).
- Pont, C.; Murat, F.; Guizard, S.; Flores, R.; Foucrier, S.; Bidet, Y.; Quraishi, U.M.; Alaux, M.; Dolezel, J.; Fahima, T.; et al. Wheat syntenome unveils new evidences of contrasted evolutionary plasticity between paleo- and neoduplicated subgenomes. Plant J. 2013, 76, 1030–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Guttieri, M.J.; Zhang, J.; Hole, D.; Souza, E.; Goates, B. A novel QTL associated with dwarf bunt resistance in Idaho 444 winter wheat. Theor. Appl. Genet. 2016, 129, 2313–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, E.R.; Schaller, C.W.; Briggs, F.N. Identification of the chromosome carrying the martin gene for resistance of wheat to bunT. Can. J. Genet. Cytol. 1960, 2, 262–267. [Google Scholar] [CrossRef]
- Bhatta, M.; Morgounov, A.; Belamkar, V.; Yorgancılar, A.; Baenziger, P.S. Genome-wide association study reveals favorable alleles associated with common bunt resistance in synthetic hexaploid wheat. Euphytica 2018, 214, 200. [Google Scholar] [CrossRef]
- Mourad, A.M.I.; Sallam, A.; Belamkar, V.; Mahdy, E.; Bakheit, B.; El-Wafaa, A.A.; Baenziger, P.S. Genetic architecture of common bunt resistance in winter wheat using genome-wide association study. BMC Plant Biol. 2018, 18, 280. [Google Scholar] [CrossRef]
- Gordon, T.; Wang, R.; Hole, D.; Bockelman, H.; Bonman, J.M.; Chen, J. Genetic characterization and genome-wide association mapping for dwarf bunt resistance in bread wheat accessions from the USDA National Small Grains Collection. Theor. Appl. Genet. 2020, 133, 1069–1080. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Infection | Type | 2011 (%) | 2012 (%) | Average (%) |
|---|---|---|---|---|
| Minimum | spring | 12 | 4 | 19 |
| winter | 0 | 0 | 0 | |
| Maximum | spring | 92 | 54 | 60 |
| winter | 100 | 73 | 82 | |
| Mean | spring | 49 | 25 | 36 |
| winter | 43 | 27 | 35 | |
| all | 44 | 27 | 35 |
| Chromo-Some | n | Average Distance (cM) | Maximum Distance (cM) |
|---|---|---|---|
| 1A | 130 | 1.2 | 19.6 |
| 2A | 63 | 1.9 | 15.1 |
| 3A | 61 | 3.5 | 29.5 |
| 4A | 60 | 1.8 | 54.5 |
| 5A | 25 | 5.0 | 60.9 |
| 6A | 125 | 0.9 | 34.5 |
| 7A | 85 | 2.1 | 28.9 |
| 1B | 93 | 1.2 | 15.1 |
| 2B | 124 | 1.1 | 9.2 |
| 3B | 132 | 1.0 | 12.9 |
| 4B | 33 | 3.5 | 27.1 |
| 5B | 76 | 2.1 | 20.7 |
| 6B | 104 | 1.3 | 20.0 |
| 7B | 66 | 3.5 | 69.6 |
| 1D | 36 | 2.6 | 14.6 |
| 2D | 41 | 2.8 | 27.8 |
| 3D | 46 | 3.5 | 95.4 |
| 4D | 3 | 24.3 | 0 |
| 5D | 2 | 80.9 | 76.4 |
| 6D | 20 | 6.7 | 94.1 |
| 7D | 127 | 1.4 | 56.3 |
| A genome | 549 | 2.3 | 60.9 |
| B genome | 628 | 2.0 | 69.6 |
| D genome | 275 | 17.5 | 95.4 |
| QTL | Marker | Chr. | Pos. cM | Pos. Mbp | MAF | Year | Scale | p | q | P perm. | r2 | Effect |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QCbt.cph-2B | wPt.744022 | 2B | 10.88 | 1591 | 0.41 | 2011 | Log1–9 | 0.00001 | 0.016 | 0.00011 | 0.07 | 0.76 |
| QCbt.cph-2B | wPt.0100 | 2B | 4.21 | 655 | 0.49 | 2011 | Log1–9 | 0.00006 | 0.042 | 0.00011 | 0.06 | 0.68 |
| QCbt.cph-7A | tPt.6221 | 7A | 55.13 | 4369 | 0.05 | Mean | Log1–2 | 0.000004 | 0.005 | 0.00003 | 0.08 | 0.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steffan, P.M.; Borgen, A.; Torp, A.M.; Backes, G.; Rasmussen, S.K. Association Mapping for Common Bunt Resistance in Wheat Landraces and Cultivars. Agronomy 2022, 12, 642. https://doi.org/10.3390/agronomy12030642
Steffan PM, Borgen A, Torp AM, Backes G, Rasmussen SK. Association Mapping for Common Bunt Resistance in Wheat Landraces and Cultivars. Agronomy. 2022; 12(3):642. https://doi.org/10.3390/agronomy12030642
Chicago/Turabian StyleSteffan, Philipp Matthias, Anders Borgen, Anna Maria Torp, Gunter Backes, and Søren K. Rasmussen. 2022. "Association Mapping for Common Bunt Resistance in Wheat Landraces and Cultivars" Agronomy 12, no. 3: 642. https://doi.org/10.3390/agronomy12030642