
The Genetic Diversity Assessment of Broomcorn Millet (Panicum miliaceum) and the Construction of a Mini-Core Collection
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction, PCR Amplification, and Electrophoresis
2.3. Molecular Data Analysis
2.4. Construction of the Mini-Core Collection from the Core Collection
3. Results
3.1. SSR Characterization of Broomcorn Millet Core Collection
3.2. Genetic Diversity of the Broomcorn Millet Core Collection
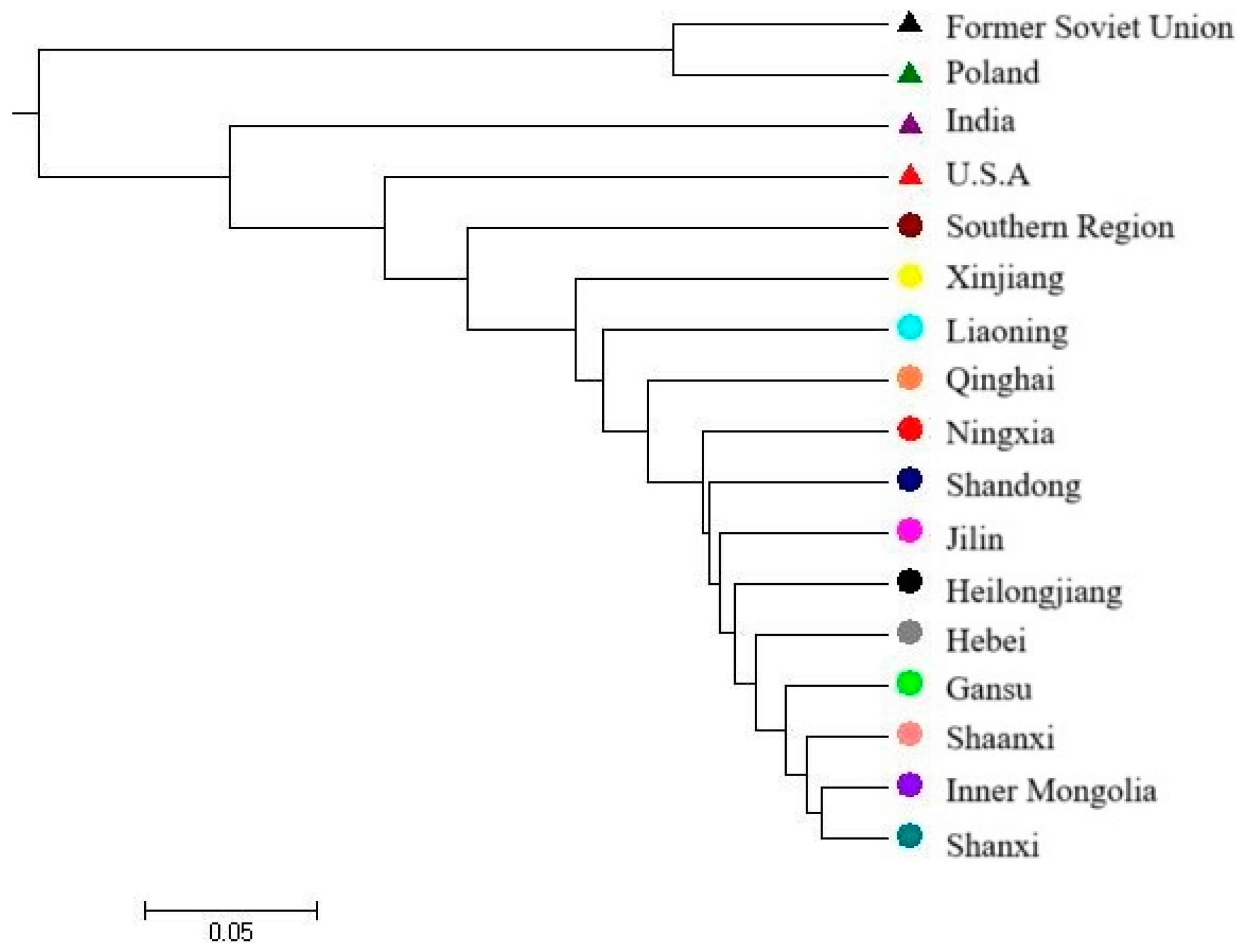
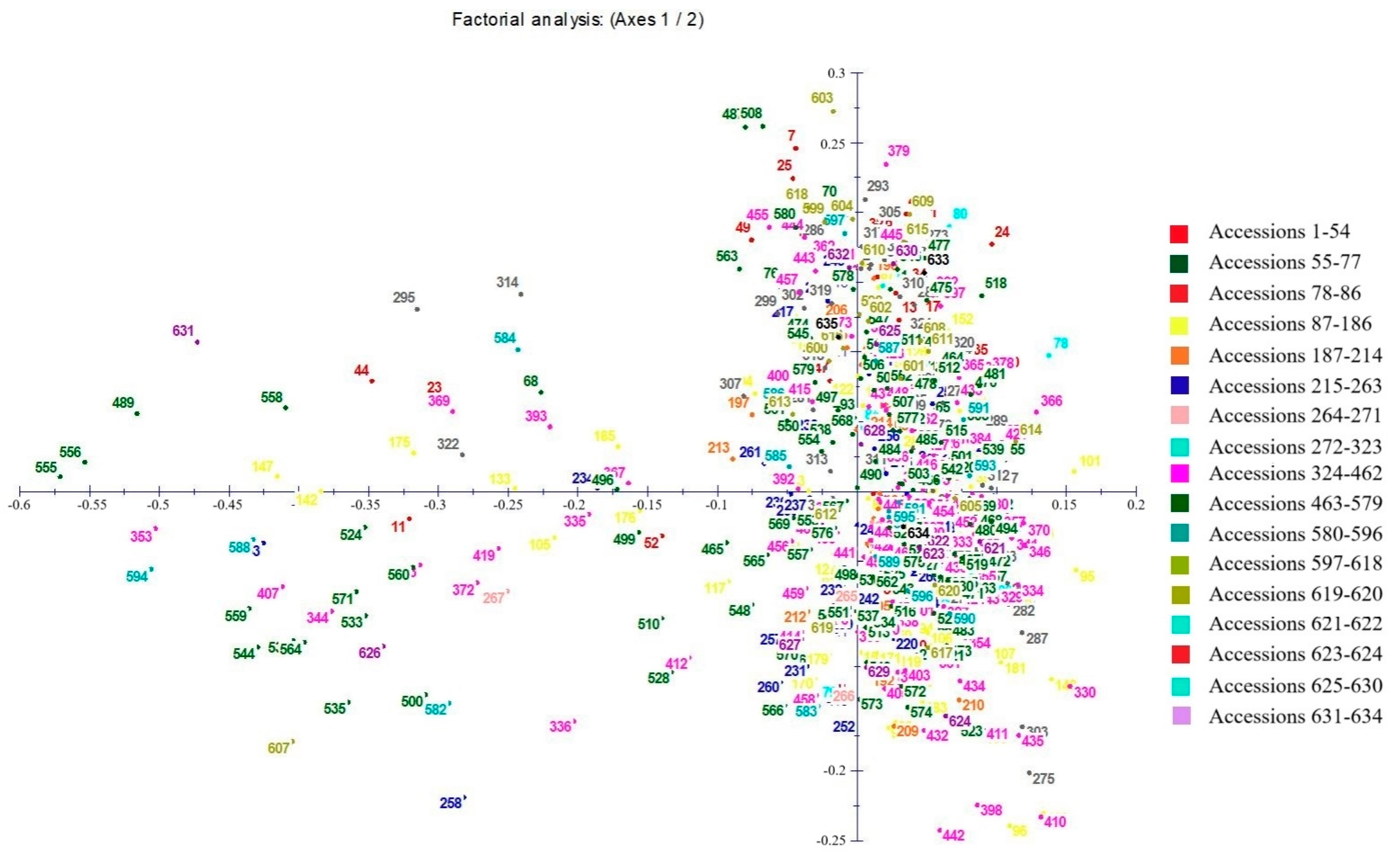
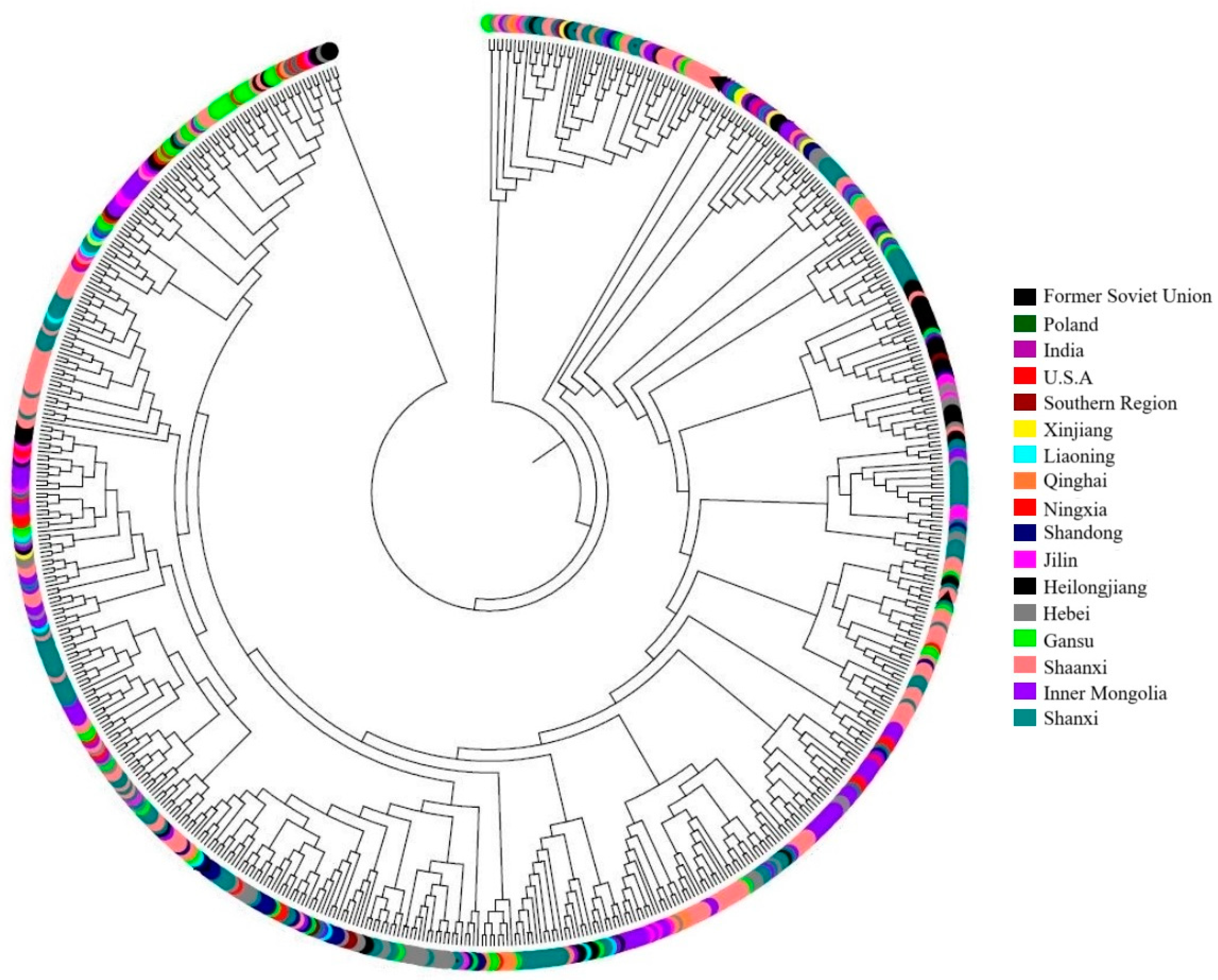
3.3. Genetic Similarity and Cluster Analysis of the Broomcorn Millet Core Collection
3.4. Mini-Core Collection Construction and Comparison with Core Collection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Crawford, G.W. Agricultural origins in North China pushed back to the Pleistocene–Holocene boundary. Proc. Natl. Acad. Sci. USA 2009, 106, 7271–7272. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Liu, S. Study on protein content and amino acid composition of Chinese foxtail millet. Acta Agric. Boreali-Sin. 1989, 4, 8–15. (In Chinese) [Google Scholar]
- Lágler, R.; Gyulai, G.; Humphreys, M.; Szabó, Z.; Horváth, L.; Bittsánszky, A.; Kiss, J.; Holly, L.; Heszky, L. Morphological and molecular analysis of common millet (P. miliaceum) cultivars compared to a DNA sample from the 15th century (Hungary). Euphytica 2005, 146, 77–85. [Google Scholar] [CrossRef]
- Liu, M.X.; Zhang, Z.W.; Wu, B.; Lu, P. Evaluation of Mixed Salt-Tolerance at Germination Stage and Seedling Stage and the Related Physiological Characteristics of Panicum miliaceum L. Sci. Agric. Sin. 2012, 18, 3733–3743. [Google Scholar]
- Liu, M.X.; Qiao, Z.J.; Zhang, S.; Wang, Y.Y.; Lu, P. Response of broomcorn millet (Panicum miliaceum L.) genotypes from semiarid regions of China to salt stress. Crop J. 2015, 3, 57–66. [Google Scholar] [CrossRef]
- Diao, X.M. Production and genetic improvement of minor cereals in China. Crop J. 2017, 5, 103–114. [Google Scholar] [CrossRef]
- Hunt, H.V.; Badakshi, F.; Romanova, O.; Howe, C.J.; Jones, M.K.; Heslop-Harrison, J.S.P. Reticulate evolution in Panicum (Poaceae): The origin of tetraploid broomcorn millet, P. miliaceum. J. Exp. Bot. 2014, 65, 3165–3175. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.Y.; Wen, Q.F. Research and utilization of proso millet germplasm resource in China. J. Plant Genet. Resour. 2005, 6, 471–474. [Google Scholar]
- Xiang, J.; Zhang, H.; Liu, H.; Suo, L.; Ja, S.; Zhang, Y.; Shi, J.; Hu, L.; Cal, Y. Comparison of phenotypic traits of foxtail millet germplasm resources in different ecological regions. J. Agric. Sci. Technol. 2020, 22, 31–41. (In Chinese) [Google Scholar]
- Dong, Y.C.; Zheng, D.S. Crops and Their Wild Relatives in China; China Agriculture Press: Beijing, China, 2006; pp. 331–359. [Google Scholar]
- Holden, J.H.W. The second ten years. In Crop Genetic Resources Conservation and Evaluation; Allen & Unwin: London, UK, 1984; pp. 277–285. [Google Scholar]
- Frankel, O.H. Genetic Manipulation: Impact on Man and Society; Cambridge University Press: Cambridge, UK, 1984; pp. 161–170. [Google Scholar]
- Zhang, H.L.; Zhang, D.L.; Wang, M.X.; Sun, J.L.; Qi, Y.W.; Li, J.J.; Wei, X.H.; Han, L.Z.; Qiu, Z.E.; Tang, S.X.; et al. A core collection and mini core collection of Oryza sativa L. in China. Theor. Appl. Genet. 2011, 122, 49–61. [Google Scholar] [CrossRef]
- Hintum, T.J.L. Comparison of marker systems and construction of a core collection in a pedigree of European spring barley. Theor. Appl. Genet. 1995, 89, 991–997. [Google Scholar] [CrossRef]
- Upadhyaya, H.D.; Ortiz, R.; Bramel, P.J.; Singh, S. Development of a groundnut core collection using taxonomical, geographical and morphological descriptors. Genet. Resour. Crop Evol. 2003, 50, 139–148. [Google Scholar] [CrossRef]
- Wang, L.X.; Guan, Y.; Guan, R.X.; Li, Y.H.; Ma, Y.S.; Dong, Z.M.; Liu, X.; Zhang, H.Y.; Zhang, Y.Q.; Liu, Z.X.; et al. Establishment of Chinese soybean (Glycine max) core collections with agronomic traits and SSR markers. Euphytica 2006, 151, 215–223. [Google Scholar] [CrossRef]
- Jiang, J.Y.; Yang, T.; Wang, F.; Fang, L.; Zhong, W.W.; Guan, J.P.; Zong, X.X. Genetic Diversity Analysis of Germplasm Resources and Construction of Mini-core Collections for Vicia faba L. at Home and Abroad. Acta Agron. Sin. 2014, 40, 1311–1319. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Zhang, X.R.; Che, Z.; Wang, L.H.; Wei, W.L.; Li, D.H. Genetic diversity assessment of sesame core collection in China by phenotype and molecular markers and extraction of a mini-core collection. BMC Genet. 2012, 13, 102–115. [Google Scholar] [CrossRef]
- Upadhyaya, H.D.; Ortiz, R. A mini core subset for capturing diversity and promoting utilization of chickpea genetic resources in crop improvement. Theor. Appl. Genet. 2001, 102, 1292–1298. [Google Scholar] [CrossRef]
- Upadhyaya, H.D.; Reddy, L.J.; Gowda, C.L.L.; Reddy, K.N.; Singh, S. Development of a mini core subset for enhanced and diversified utilization of pigeon pea germplasm resources. Crop Sci. 2006, 46, 2127–2132. [Google Scholar] [CrossRef]
- Upadhyaya, H.D.; Pundir, R.P.S.; Dwivedi, S.L.; Gowda, C.L.L.; Reddy, V.G.; Singh, S. Developing a minicore collection of sorghum for diversified utilization of germplasm. Crop Sci. 2009, 49, 1769–1780. [Google Scholar] [CrossRef]
- Agrama, H.A.; Yan, W.G.; Lee, F.; Fjellstrom, R.; Chen, M.H.; Jia, M.; McClung, A. Genetic assessment of a mini–core subset developed from the USDA rice Genebank. Crop Sci. 2009, 49, 1336–1346. [Google Scholar] [CrossRef]
- Hu, X.Y.; Wang, L.; Zhang, Z.W.; Lu, P.; Zhang, H.S. Establishment of Broomcorn Millet Core Collection in China. Sci. Agric. Sin. 2008, 41, 3489–3502. [Google Scholar]
- Hu, X.Y.; Wang, J.F.; Lu PZhang, H.S. Assessment of genetic diversity in broomcorn millet (Panicum miliaceum L.) using SSR markers. J. Genet. Genom. 2009, 36, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.X.; He, D.H.; Zhang, X.L.; Nie, Y.C.; Guo XPFeng, C.D.; Stewart, J.M. Linkage map construction and mapping QTL for cotton fiber quality using SRAP, SSR and RAPD. Plant Breed. 2005, 124, 180–187. [Google Scholar] [CrossRef]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.C.; Yang, R.C.; Boyle, T. POPGENE Software Package Version 1.31 for Population Genetic Analysis; University of Alberta: Edmonton, AB, Canada, 1999. [Google Scholar]
- Kumar, S.; Tamura, K.; Nei, M. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 2004, 5, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Belkhir, K.; Borsa, P.; Chikhi, L.; Raufaste, N.; Bonhomme, F. Genetix Version 4.05.2. 2001. Available online: http://www.univ-montp2.fr/*genetix/genetix/genetix.htm (accessed on 5 May 2004).
- Kim, K.W.; Chung, H.K.; Cho, G.T.; Ma, K.H.; Chandrabalan, D.; Gwag, J.G.; Kim, T.S.; Cho, E.G.; Park, Y.J. PowerCore: A program applying the advanced M strategy with a heuristic search for establishing core sets. Bioinformatics 2007, 23, 2155–2162. [Google Scholar] [CrossRef]
- Zhao, W.G.; Chung, J.W.; Ma, K.H.; Kim, T.S.; Kim, S.M.; Shin, D.I.; Kim, C.H.; Koo, H.M.; Park, Y.J. Analysis of genetic diversity and population structure of rice cultivars from Korea, China and Japan using SSR markers. Genes Genom. 2009, 31, 283–292. [Google Scholar] [CrossRef]
- Spagnoletti, Z.P.L.; Qualset, C.Q. Geographical diversity for quantitative spike characters in a worked collection of durum wheat. Crop Sci. 1987, 27, 235–241. [Google Scholar] [CrossRef]
- Li, Y.C.; Korol, A.B.; Fathima, T.; Belles, A.; Nevo, E. Microsatellites: Genomic distribution, putative functions and mutational mechanisms: A review. Mol. Ecol. 2002, 11, 2453–2465. [Google Scholar] [CrossRef]
- Ellis, P.R.; Pink, D.A.C.; Phelps, K.; Jukes, P.L.; Breeds, S.E.; Pinnegar, A.E. Evaluation of a core collection of Brassica oleracea accessions for resistance to Brevicoryne brassicae, the cabbage aphid. Euphytica 1998, 103, 149–160. [Google Scholar] [CrossRef]
- Miklas, P.N.; Delorme, R.; Hannan, R.; Dickson, M.H. Using a sub sample of the core collection to identify new sources of resistance to white mold in common bean. Crop Sci. 1999, 39, 569–573. [Google Scholar] [CrossRef]
- Santos, M.R.; Dias, J.S. Evaluation of a core collection of Brassica oleracea accessions for resistance to white rust of crucifers (Albugo candida) at the cotyledon stage. Genet. Resour. Crop Evol. 2004, 51, 713–722. [Google Scholar] [CrossRef]
- Qiu, L.J.; Li, Y.H.; Guan, R.X.; Liu, Z.X.; Wang, L.X.; Chang, R.Z. Establishment, representative testing and research progress of soybean core collection and mini core collection. Acta Agron. Sin. 2009, 35, 571–579. [Google Scholar] [CrossRef]
- Mi, S.J.; Qiu, L.J.; Chang, R.Z.; Hao, Z.B.; Guan, R.X. Genetic diversity analysis of varieties of Glycine max (L.) merr resistant to Soybean mosaic virus by SSR fingerprints. Acta Phytopathol. Sin. 2004, 34, 244–253. [Google Scholar] [CrossRef]
- Jiang, H.F.; Ren, X.P.; Liao, B.S.; Huang, J.Q.; Lei, Y.; Chen, B.Y.; Guo, B.Z.; Holbrook, C.C.; Upadhyaya, H.D. Peanut core collection established in China and compared with ICRISAT mini core collection. Acta Agron. Sin. 2008, 34, 25–30. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Acc. No. | Origin | Ecotype |
|---|---|---|---|
| 1 | 54 | Heilongjiang | Northeast |
| 2 | 23 | Jilin | Northeast |
| 3 | 9 | Liaoning | Northeast |
| 4 | 100 | Inner Monogolia | Mongolian Plateau |
| 5 | 28 | Ningxia | Northwest |
| 6 | 48 | Gansu | Northwest |
| 7 | 8 | Xinjiang | Northwest |
| 8 | 52 | Hebei, Beijing | North China Plain |
| 9 | 139 | Shanxi | Loess Plateau |
| 10 | 118 | Shannxi | Loess Plateau |
| 11 | 16 | QingHai, Xizang | Qinghai–Tibetan Plateau |
| 12 | 23 | Shandong | North China Plain |
| 13 | 2 | The Soviet Union | Foreign |
| 14 | 2 | Poland | Foreign |
| 15 | 2 | India | Foreign |
| 16 | 6 | USA | Foreign |
| 17 | 4 | Anhui, Jiangsu, Hubei | South region |
| Number of Alleles | Number of SSR Loci | Polymorphic Loci (%) |
|---|---|---|
| 2 | 12 | 35.3 |
| 3 | 13 | 38.2 |
| 4 | 7 | 20.6 |
| 5 | 2 | 5.9 |
| SSR Primer Pair | Na a | Ne b | I c | Ho d | He e | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| CC | MC | CC | MC | CC | MC | CC | MC | CC | MC | |
| lmx334 | 2.000 | 2.000 | 1.746 | 1.735 | 0.618 | 0.615 | 0.064 | 0.425 | 0.428 | 0.424 |
| lmx503 | 3.000 | 3.000 | 2.823 | 2.875 | 1.069 | 1.078 | 0.126 | 0.654 | 0.647 | 0.652 |
| lmx510 | 3.000 | 3.000 | 2.394 | 2.388 | 0.981 | 0.978 | 0.276 | 0.583 | 0.583 | 0.581 |
| lmx515 | 4.000 | 3.000 | 1.666 | 1.730 | 0.722 | 0.746 | 0.078 | 0.423 | 0.400 | 0.422 |
| lmx619 | 3.000 | 3.000 | 1.218 | 1.282 | 0.365 | 0.443 | 0.053 | 0.221 | 0.179 | 0.220 |
| lmx621 | 3.000 | 3.000 | 1.899 | 2.002 | 0.826 | 0.866 | 0.367 | 0.502 | 0.474 | 0.501 |
| lmx630 | 3.000 | 3.000 | 2.181 | 2.226 | 0.899 | 0.917 | 0.631 | 0.552 | 0.542 | 0.551 |
| lmx632 | 3.000 | 3.000 | 1.399 | 1.502 | 0.554 | 0.629 | 0.136 | 0.335 | 0.286 | 0.334 |
| lmx746 | 3.000 | 3.000 | 1.757 | 1.971 | 0.771 | 0.857 | 0.172 | 0.494 | 0.431 | 0.493 |
| lmx780 | 2.000 | 2.000 | 1.976 | 1.988 | 0.687 | 0.690 | 0.253 | 0.498 | 0.494 | 0.497 |
| lmx836 | 3.000 | 3.000 | 2.070 | 2.089 | 0.831 | 0.857 | 0.037 | 0.522 | 0.517 | 0.521 |
| lmx845 | 3.000 | 3.000 | 2.204 | 2.258 | 0.875 | 0.902 | 0.045 | 0.558 | 0.547 | 0.557 |
| lmx1065 | 3.000 | 3.000 | 2.206 | 2.295 | 0.909 | 0.930 | 0.257 | 0.566 | 0.547 | 0.564 |
| lmx1072 | 5.000 | 5.000 | 4.141 | 4.132 | 1.487 | 1.482 | 0.992 | 0.760 | 0.759 | 0.758 |
| lmx1080 | 4.000 | 4.000 | 2.040 | 2.221 | 0.892 | 0.960 | 0.046 | 0.551 | 0.510 | 0.550 |
| lmx1380 | 2.000 | 2.000 | 1.859 | 1.900 | 0.655 | 0.667 | 0.014 | 0.475 | 0.463 | 0.474 |
| lmx1429 | 2.000 | 2.000 | 2.000 | 1.997 | 0.693 | 0.692 | 0.000 | 0.500 | 0.500 | 0.499 |
| lmx1553 | 4.000 | 4.000 | 3.593 | 3.633 | 1.327 | 1.335 | 0.883 | 0.726 | 0.722 | 0.725 |
| lmx1610 | 2.000 | 2.000 | 1.989 | 1.999 | 0.690 | 0.693 | 0.206 | 0.501 | 0.498 | 0.500 |
| lmx1625 | 2.000 | 2.000 | 1.981 | 1.964 | 0.688 | 0.684 | 0.397 | 0.492 | 0.496 | 0.491 |
| lmx1672 | 4.000 | 4.000 | 3.051 | 3.094 | 1.224 | 1.235 | 0.882 | 0.678 | 0.673 | 0.677 |
| lmx1703 | 4.000 | 4.000 | 3.383 | 3.425 | 1.295 | 1.304 | 0.108 | 0.710 | 0.705 | 0.708 |
| lmx1761 | 4.000 | 4.000 | 2.705 | 2.683 | 1.123 | 1.115 | 0.994 | 0.629 | 0.631 | 0.627 |
| lmx1940 | 2.000 | 2.000 | 1.194 | 1.228 | 0.301 | 0.333 | 0.000 | 0.186 | 0.163 | 0.186 |
| lmx1959 | 3.000 | 3.000 | 2.277 | 2.314 | 0.895 | 0.911 | 0.165 | 0.569 | 0.561 | 0.568 |
| lmx2068 | 4.000 | 4.000 | 2.896 | 2.864 | 1.177 | 1.170 | 0.953 | 0.653 | 0.655 | 0.651 |
| lmx2074 | 2.000 | 2.000 | 1.600 | 1.523 | 0.562 | 0.527 | 0.041 | 0.344 | 0.375 | 0.343 |
| lmx2239 | 2.000 | 2.000 | 1.760 | 1.793 | 0.623 | 0.634 | 0.243 | 0.443 | 0.432 | 0.442 |
| lmx2281 | 2.000 | 2.000 | 1.652 | 1.733 | 0.584 | 0.614 | 0.004 | 0.424 | 0.395 | 0.423 |
| lmx2288 | 3.000 | 3.000 | 2.866 | 2.881 | 1.076 | 1.079 | 0.979 | 0.654 | 0.652 | 0.653 |
| lmx2551 | 2.000 | 2.000 | 1.240 | 1.293 | 0.344 | 0.387 | 0.117 | 0.227 | 0.194 | 0.227 |
| lmx2734 | 5.000 | 5.000 | 3.243 | 3.351 | 1.336 | 1.357 | 0.874 | 0.703 | 0.692 | 0.702 |
| lmx2782 | 2.000 | 2.000 | 2.000 | 2.000 | 0.693 | 0.693 | 0.848 | 0.501 | 0.500 | 0.500 |
| lmx2979 | 3.000 | 3.000 | 2.105 | 2.148 | 0.893 | 0.903 | 0.119 | 0.536 | 0.525 | 0.535 |
| Total | 101.000 | 100.000 | 75.114 | 76.515 | 28.665 | 29.281 | 11.360 | 17.593 | 17.176 | 17.553 |
| Mean | 2.971 | 2.941 | 2.209 | 2.250 | 0.843 | 0.861 | 0.334 | 0.517 | 0.505 | 0.516 |
| St. | 0.904 | 0.886 | 0.698 | 0.691 | 0.294 | 0.286 | 0.358 | 0.142 | 0.150 | 0.141 |
| Population | Na * | Ne * | I * | Ho | He | Nei ** | PIC | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CC | MC | CC | MC | CC | MC | CC | MC | CC | MC | CC | MC | CC | MC | |
| Heilongjiang | 2.794 | 2.677 | 2.209 | 2.165 | 0.799 | 0.785 | 0.327 | 0.343 | 0.484 | 0.490 | 0.479 | 0.475 | 0.542 | 0.568 |
| Jilin | 2.824 | 2.765 | 2.225 | 2.249 | 0.821 | 0.829 | 0.343 | 0.355 | 0.511 | 0.533 | 0.500 | 0.506 | 0.549 | 0.565 |
| Liaoning | 2.618 | 2.559 | 1.998 | 2.006 | 0.715 | 0.711 | 0.296 | 0.315 | 0.456 | 0.466 | 0.430 | 0.431 | 0.526 | 0.522 |
| Inner Mongolia | 2.941 | 2.912 | 2.181 | 2.145 | 0.834 | 0.827 | 0.332 | 0.350 | 0.509 | 0.510 | 0.507 | 0.501 | 0.473 | 0.506 |
| Ningxia | 2.706 | 2.677 | 2.004 | 2.037 | 0.716 | 0.737 | 0.313 | 0.317 | 0.439 | 0.461 | 0.431 | 0.445 | 0.132 | 0.132 |
| Gansu | 2.941 | 2.853 | 2.065 | 2.114 | 0.775 | 0.795 | 0.357 | 0.361 | 0.470 | 0.491 | 0.464 | 0.480 | 0.088 | 0.088 |
| Xinjiang | 2.618 | 2.588 | 1.979 | 1.987 | 0.729 | 0.723 | 0.392 | 0.376 | 0.483 | 0.483 | 0.445 | 0.442 | 0.199 | 0.199 |
| Heibei | 2.882 | 2.735 | 2.137 | 2.216 | 0.799 | 0.825 | 0.312 | 0.311 | 0.488 | 0.525 | 0.483 | 0.509 | 0.416 | 0.315 |
| Shanxi | 2.912 | 2.824 | 2.141 | 2.200 | 0.807 | 0.824 | 0.346 | 0.358 | 0.486 | 0.503 | 0.484 | 0.497 | 0.250 | 0.200 |
| Shannxi | 2.912 | 2.912 | 2.041 | 2.059 | 0.772 | 0.783 | 0.337 | 0.334 | 0.464 | 0.476 | 0.461 | 0.468 | 0.521 | 0.525 |
| Qingzang | 2.471 | 2.441 | 1.951 | 1.957 | 0.679 | 0.677 | 0.332 | 0.347 | 0.438 | 0.443 | 0.420 | 0.419 | 0.422 | 0.422 |
| Shandong | 2.735 | 2.500 | 2.112 | 2.125 | 0.786 | 0.753 | 0.293 | 0.302 | 0.490 | 0.500 | 0.477 | 0.472 | 0.548 | 0.566 |
| The Soviet Union | 1.546 | 1.546 | 1.449 | 1.449 | 0.346 | 0.346 | 0.364 | 0.364 | 0.333 | 0.333 | 0.242 | 0.242 | 0.441 | 0.439 |
| Poland | 1.594 | 1.594 | 1.544 | 1.544 | 0.374 | 0.374 | 0.406 | 0.406 | 0.354 | 0.354 | 0.258 | 0.258 | 0.506 | 0.550 |
| India | 2.000 | 2.000 | 1.875 | 1.875 | 0.560 | 0.560 | 0.455 | 0.455 | 0.480 | 0.480 | 0.360 | 0.360 | 0.480 | 0.469 |
| USA | 2.063 | 1.724 | 1.804 | 1.612 | 0.570 | 0.433 | 0.328 | 0.310 | 0.411 | 0.409 | 0.364 | 0.290 | 0.506 | 0.543 |
| South region | 1.882 | 1.647 | 1.619 | 1.518 | 0.466 | 0.374 | 0.228 | 0.201 | 0.345 | 0.301 | 0.298 | 0.249 | 0.556 | 0.554 |
| Mean | 2.496 | 2.409 | 1.961 | 1.956 | 0.679 | 0.668 | 0.339 | 0.341 | 0.449 | 0.456 | 0.418 | 0.414 | 0.421 | 0.421 |
| Pop ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.9592 | 0.9004 | 0.9326 | 0.8892 | 0.9258 | 0.915 | 0.927 | 0.939 | 0.9107 | 0.92 | 0.9143 | 0.59 | 0.508 | 0.75 | 0.857 | 0.8928 | |
| 2 | 0.0417 | 0.9272 | 0.9733 | 0.9375 | 0.9518 | 0.932 | 0.946 | 0.955 | 0.939 | 0.939 | 0.937 | 0.621 | 0.561 | 0.797 | 0.8492 | 0.8645 | |
| 3 | 0.1049 | 0.0755 | 0.939 | 0.9371 | 0.9295 | 0.892 | 0.945 | 0.961 | 0.955 | 0.92 | 0.9404 | 0.596 | 0.568 | 0.809 | 0.7786 | 0.8173 | |
| 4 | 0.0698 | 0.0271 | 0.063 | 0.965 | 0.9698 | 0.941 | 0.954 | 0.962 | 0.964 | 0.945 | 0.9377 | 0.631 | 0.591 | 0.82 | 0.8471 | 0.8434 | |
| 5 | 0.1174 | 0.0646 | 0.065 | 0.0356 | 0.9749 | 0.878 | 0.914 | 0.929 | 0.9701 | 0.932 | 0.9367 | 0.547 | 0.51 | 0.805 | 0.8073 | 0.8382 | |
| 6 | 0.0771 | 0.0494 | 0.0731 | 0.0306 | 0.0254 | 0.9 | 0.921 | 0.938 | 0.9727 | 0.95 | 0.9348 | 0.609 | 0.559 | 0.816 | 0.8457 | 0.86 | |
| 7 | 0.0893 | 0.0708 | 0.1143 | 0.061 | 0.1302 | 0.1051 | 0.918 | 0.935 | 0.9152 | 0.87 | 0.8729 | 0.668 | 0.619 | 0.759 | 0.8458 | 0.7801 | |
| 8 | 0.0761 | 0.0555 | 0.0564 | 0.0472 | 0.0905 | 0.0825 | 0.086 | 0.952 | 0.9373 | 0.946 | 0.9452 | 0.611 | 0.576 | 0.821 | 0.8369 | 0.8717 | |
| 9 | 0.0629 | 0.0461 | 0.0402 | 0.0391 | 0.0737 | 0.0636 | 0.067 | 0.049 | 0.9582 | 0.918 | 0.9396 | 0.637 | 0.575 | 0.773 | 0.8458 | 0.8479 | |
| 10 | 0.0935 | 0.0629 | 0.046 | 0.0366 | 0.0303 | 0.0277 | 0.089 | 0.065 | 0.043 | 0.924 | 0.9415 | 0.614 | 0.584 | 0.795 | 0.8336 | 0.8506 | |
| 11 | 0.0832 | 0.0635 | 0.0833 | 0.0568 | 0.0708 | 0.0517 | 0.14 | 0.056 | 0.085 | 0.079 | 0.9357 | 0.598 | 0.531 | 0.822 | 0.8331 | 0.8418 | |
| 12 | 0.0896 | 0.0651 | 0.0614 | 0.0644 | 0.0654 | 0.0674 | 0.136 | 0.056 | 0.062 | 0.0603 | 0.066 | 0.633 | 0.595 | 0.821 | 0.8253 | 0.8824 | |
| 13 | 0.5279 | 0.4763 | 0.5171 | 0.4599 | 0.6029 | 0.4959 | 0.404 | 0.492 | 0.451 | 0.4871 | 0.514 | 0.4578 | 0.947 | 0.625 | 0.529 | 0.4389 | |
| 14 | 0.6778 | 0.5785 | 0.5657 | 0.5253 | 0.6743 | 0.5815 | 0.48 | 0.551 | 0.553 | 0.5385 | 0.633 | 0.5196 | 0.055 | 0.628 | 0.5126 | 0.3791 | |
| 15 | 0.2873 | 0.2268 | 0.2121 | 0.1989 | 0.2166 | 0.2029 | 0.276 | 0.197 | 0.257 | 0.2289 | 0.196 | 0.1977 | 0.469 | 0.465 | 0.6776 | 0.6937 | |
| 16 | 0.1543 | 0.1635 | 0.2502 | 0.166 | 0.214 | 0.1676 | 0.168 | 0.178 | 0.168 | 0.182 | 0.183 | 0.192 | 0.637 | 0.668 | 0.389 | 0.8436 | |
| 17 | 0.1134 | 0.1456 | 0.2018 | 0.1704 | 0.1765 | 0.1508 | 0.248 | 0.137 | 0.165 | 0.1618 | 0.172 | 0.1251 | 0.823 | 0.97 | 0.366 | 0.1701 |
| Group | Core Collection | Mini-Core Collection | |||
|---|---|---|---|---|---|
| Number | Percentage in CC (%) | Number | Percentage in MC (%) | Percentage in Population of CC (%) | |
| Heilongjiang | 54 | 8.49 | 18 | 8.8 | 37.0 |
| Jilin | 23 | 3.62 | 11 | 3.9 | 39.1 |
| Liaoning | 9 | 1.42 | 7 | 1.8 | 44.4 |
| Inner Mongolia | 100 | 15.72 | 35 | 14.9 | 34.0 |
| Ningxia | 28 | 4.40 | 16 | 4.8 | 39.3 |
| Gansu | 48 | 7.55 | 24 | 8.3 | 39.6 |
| Xinjiang | 8 | 1.26 | 7 | 2.6 | 75.0 |
| Hebei | 52 | 8.18 | 21 | 8.3 | 36.5 |
| Shanxi | 139 | 21.86 | 48 | 19.3 | 31.7 |
| Shannxi | 118 | 18.55 | 37 | 16.2 | 31.4 |
| Qingzang | 16 | 2.52 | 12 | 3.5 | 50.0 |
| Shandong | 23 | 3.62 | 9 | 3.9 | 39.1 |
| The Soviet Union | 2 | 0.31 | 2 | 0.4 | 50.0 |
| Poland | 2 | 0.31 | 2 | 0.4 | 50.0 |
| India | 2 | 0.31 | 2 | 0.4 | 50.0 |
| USA | 6 | 0.94 | 3 | 0.9 | 33.3 |
| South region | 4 | 0.63 | 2 | 1.3 | 75.0 |
| Total | 634 | 100% | 256 | 100% | |
| Genetic Parameter | t | df | p-Value | Mean Difference | Se Value | 95% Confidence Interval | ||
|---|---|---|---|---|---|---|---|---|
| Lower Limit | Limit | |||||||
| SSR | Na | 0.136 | 66.0 | 0.893 | 0.029 | 0.217 | −0.404 | 0.463 |
| Ne | −0.244 | 66.0 | 0.808 | −0.041 | 0.169 | −0.378 | 0.295 | |
| I | −0.258 | 66.0 | 0.797 | −0.018 | 0.070 | −0.158 | 0.122 | |
| Ho | −2.775 | 66.0 | 0.007 | −0.183 | 0.066 | −0.315 | −0.051 | |
| He | −0.314 | 66.0 | 0.754 | −0.011 | 0.035 | −0.082 | 0.059 | |
| Nei | 2.695 | 66.0 | 0.009 | 0.095 | 0.035 | 0.025 | 0.165 | |
| Population | Na | 0.520 | 32.0 | 0.607 | 0.087 | 0.168 | −0.255 | 0.430 |
| Ne | 0.051 | 32.0 | 0.959 | 0.004 | 0.085 | −0.170 | 0.178 | |
| I | 0.195 | 32.0 | 0.846 | 0.011 | 0.058 | −0.106 | 0.129 | |
| Ho | −0.149 | 32.0 | 0.882 | −0.003 | 0.018 | −0.038 | 0.033 | |
| He | −0.318 | 32.0 | 0.753 | −0.007 | 0.021 | −0.050 | 0.037 | |
| Nei | 0.110 | 32.0 | 0.913 | 0.003 | 0.031 | −0.060 | 0.066 | |
| PIC | 0.110 | 32.0 | 0.913 | 0.003 | 0.031 | −0.060 | 0.066 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, J.; Yu, X.; Wang, X.; Wang, Y.; Xin, X.; Wang, R.; Zhang, Y.; Liu, M.; Xiang, J. The Genetic Diversity Assessment of Broomcorn Millet (Panicum miliaceum) and the Construction of a Mini-Core Collection. Agronomy 2024, 14, 2226. https://doi.org/10.3390/agronomy14102226
Ren J, Yu X, Wang X, Wang Y, Xin X, Wang R, Zhang Y, Liu M, Xiang J. The Genetic Diversity Assessment of Broomcorn Millet (Panicum miliaceum) and the Construction of a Mini-Core Collection. Agronomy. 2024; 14(10):2226. https://doi.org/10.3390/agronomy14102226
Chicago/Turabian StyleRen, Jiandong, Xiaohan Yu, Xiaoxing Wang, Yue Wang, Xuxia Xin, Ruonan Wang, Yingxing Zhang, Minxuan Liu, and Jishan Xiang. 2024. "The Genetic Diversity Assessment of Broomcorn Millet (Panicum miliaceum) and the Construction of a Mini-Core Collection" Agronomy 14, no. 10: 2226. https://doi.org/10.3390/agronomy14102226