
HSP70s in Breast Cancer: Promoters of Tumorigenesis and Potential Targets/Tools for Therapy


Abstract
:1. Introduction
2. Members of HSP70 Subfamily: General Characteristics, Localization, Functioning
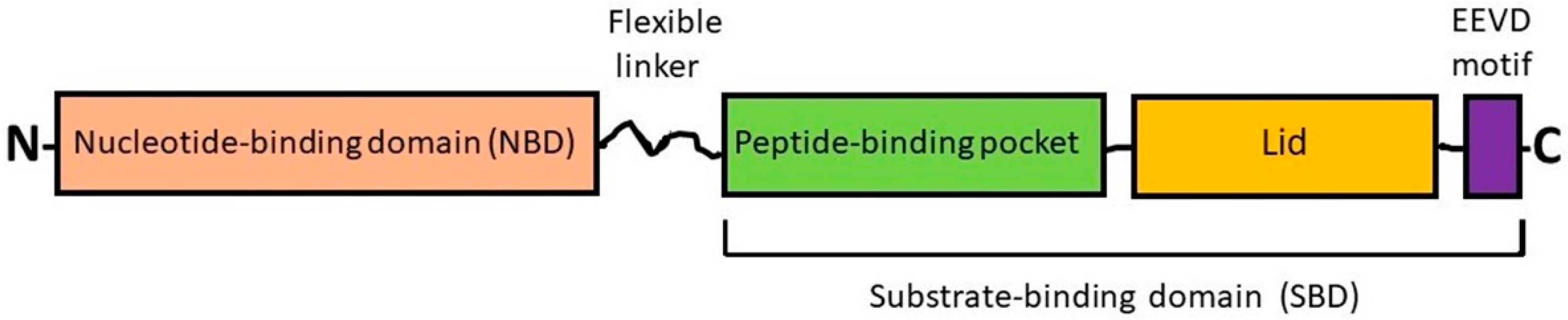
2.1. Heat Shock Cognate Protein 70 (HSC70) and Inducible HSP70
2.2. Glucose-Regulated Protein 78 (GRP78)
2.3. Glucose-Regulated Protein 75 (GRP75 or Mortalin)
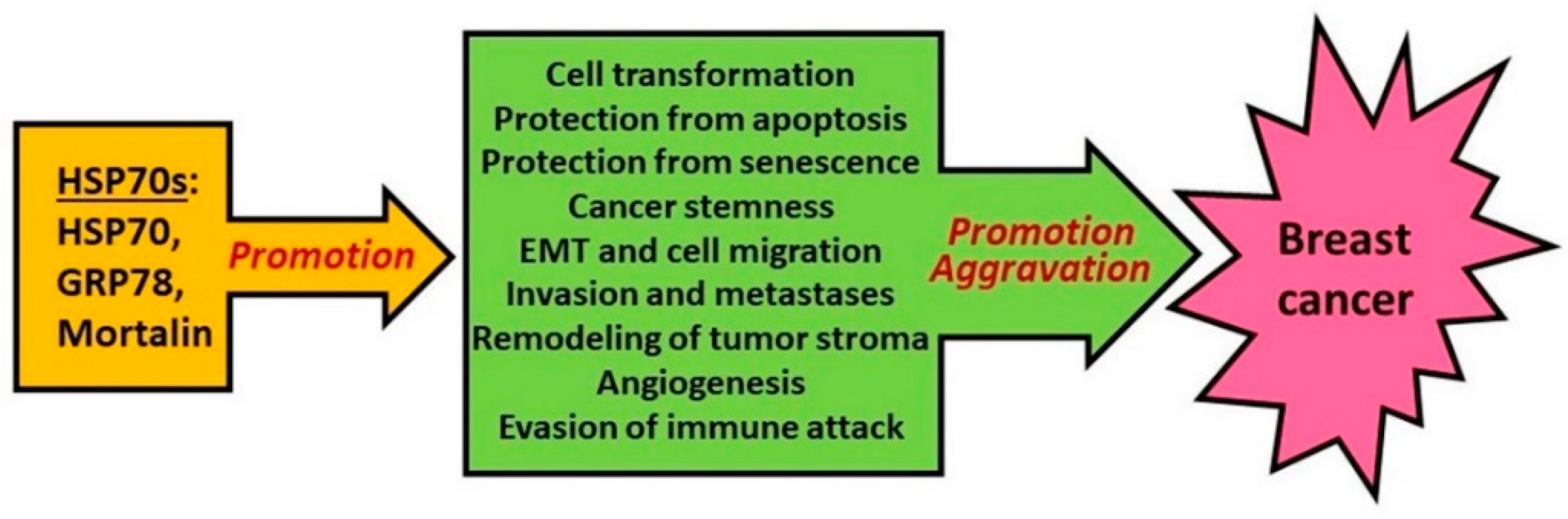
3. Role of HSP70 in Oncogenesis and Tumor Progression
3.1. HSP70 and Cell Transformation
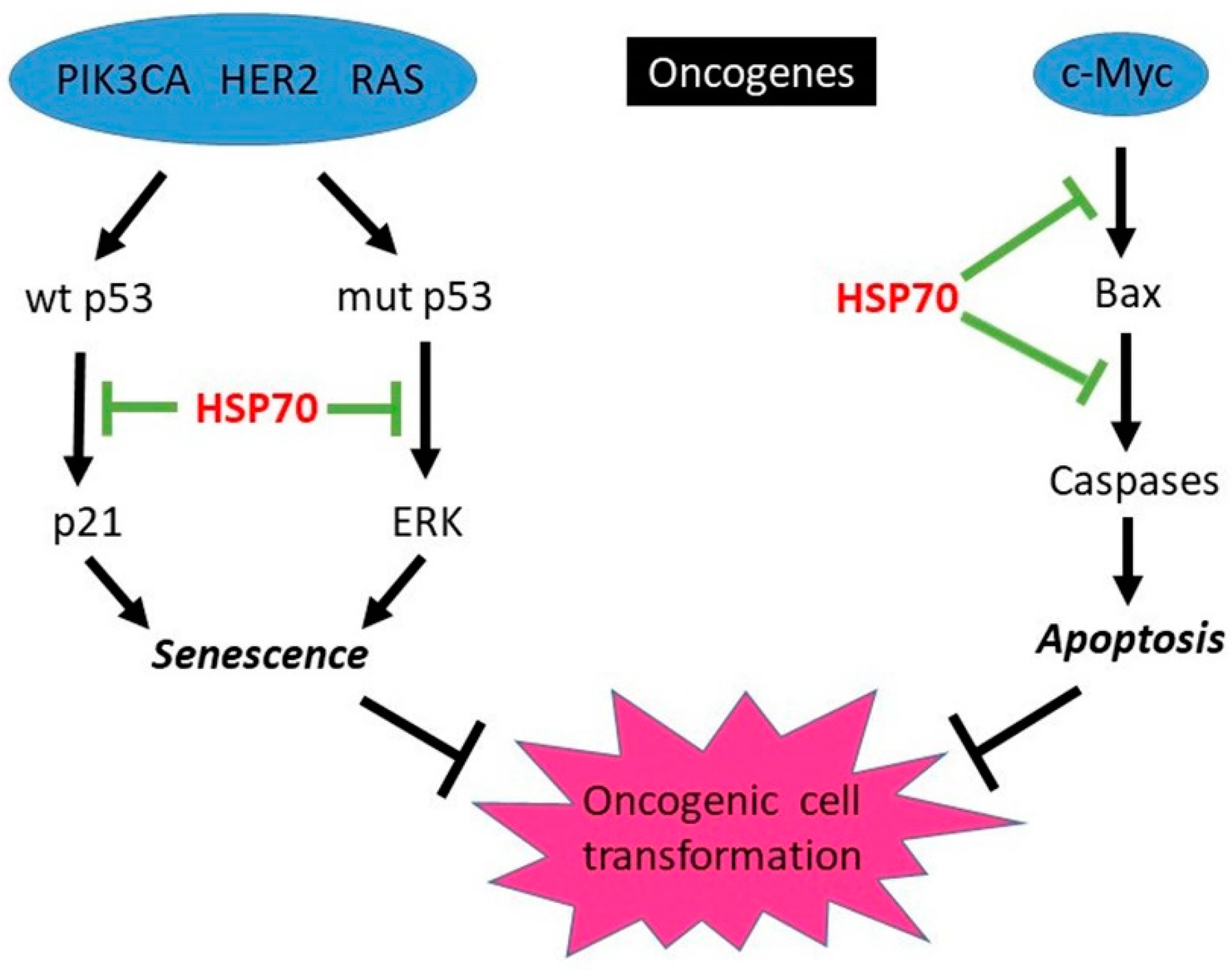
3.1.1. HSP70-Mediated Protection from Oncogene-Induced Apoptosis
3.1.2. HSP70-Mediated Protection from Oncogene-Induced Senescence
3.1.3. HSP70 and Tumor Evasion of Immune System
3.2. HSP70 and Tumor Progression
3.2.1. HSP70-Mediated Promotion of Angiogenesis
3.2.2. HSP70-Mediated Promotion of Epithelial-to-Mesenchymal Transition (EMT), Migration, and Invasion
3.2.3. HSP70-Mediated Promotion of Metastases
3.2.4. HSP70 in Tumor Stroma
4. Role of other HSP70 Subfamily Members in Tumorigenesis
4.1. HSPA2 (HSP70-2)
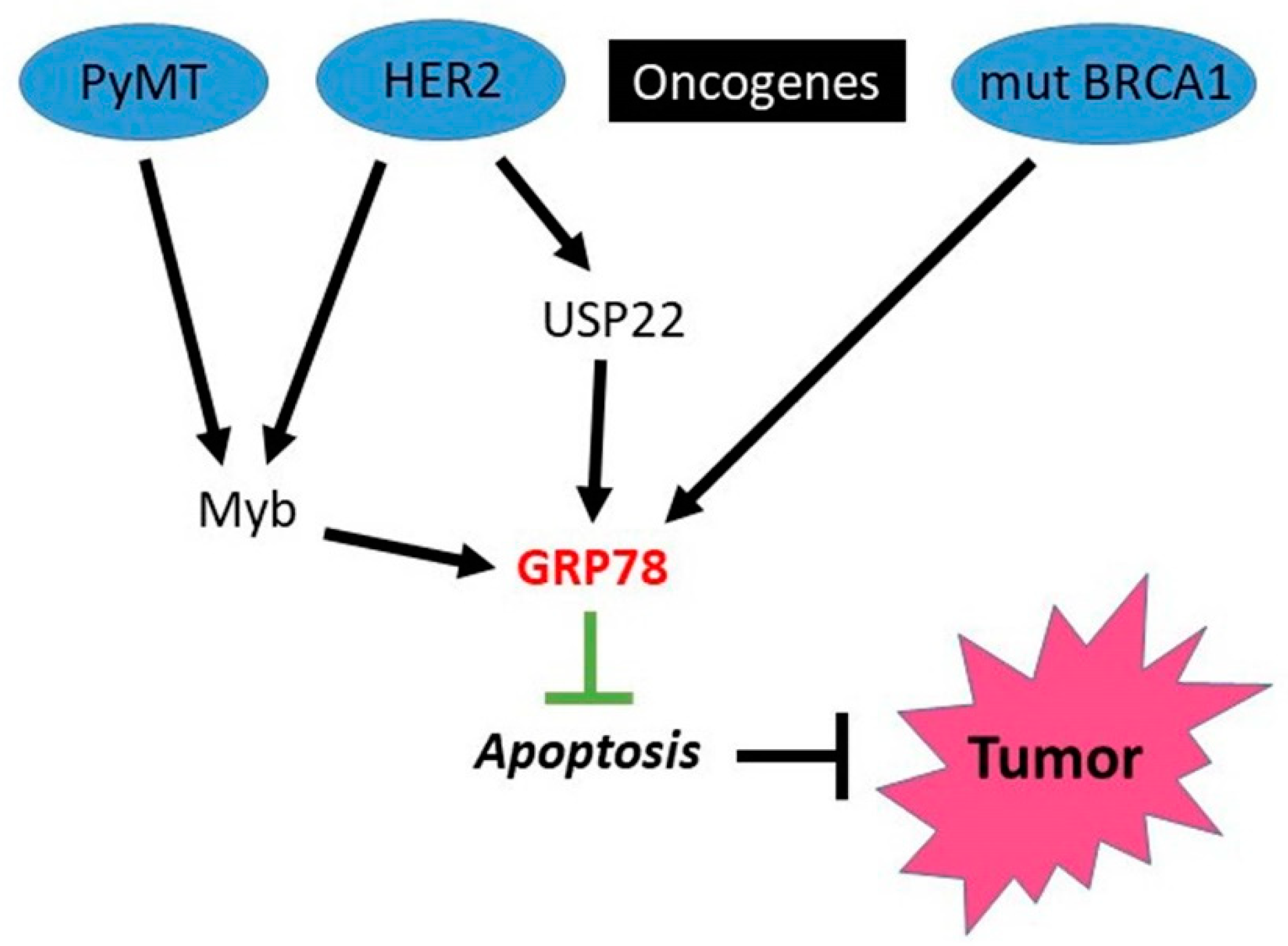
4.2. HSPA5 (GRP78)
4.2.1. Intracellular HSPA5
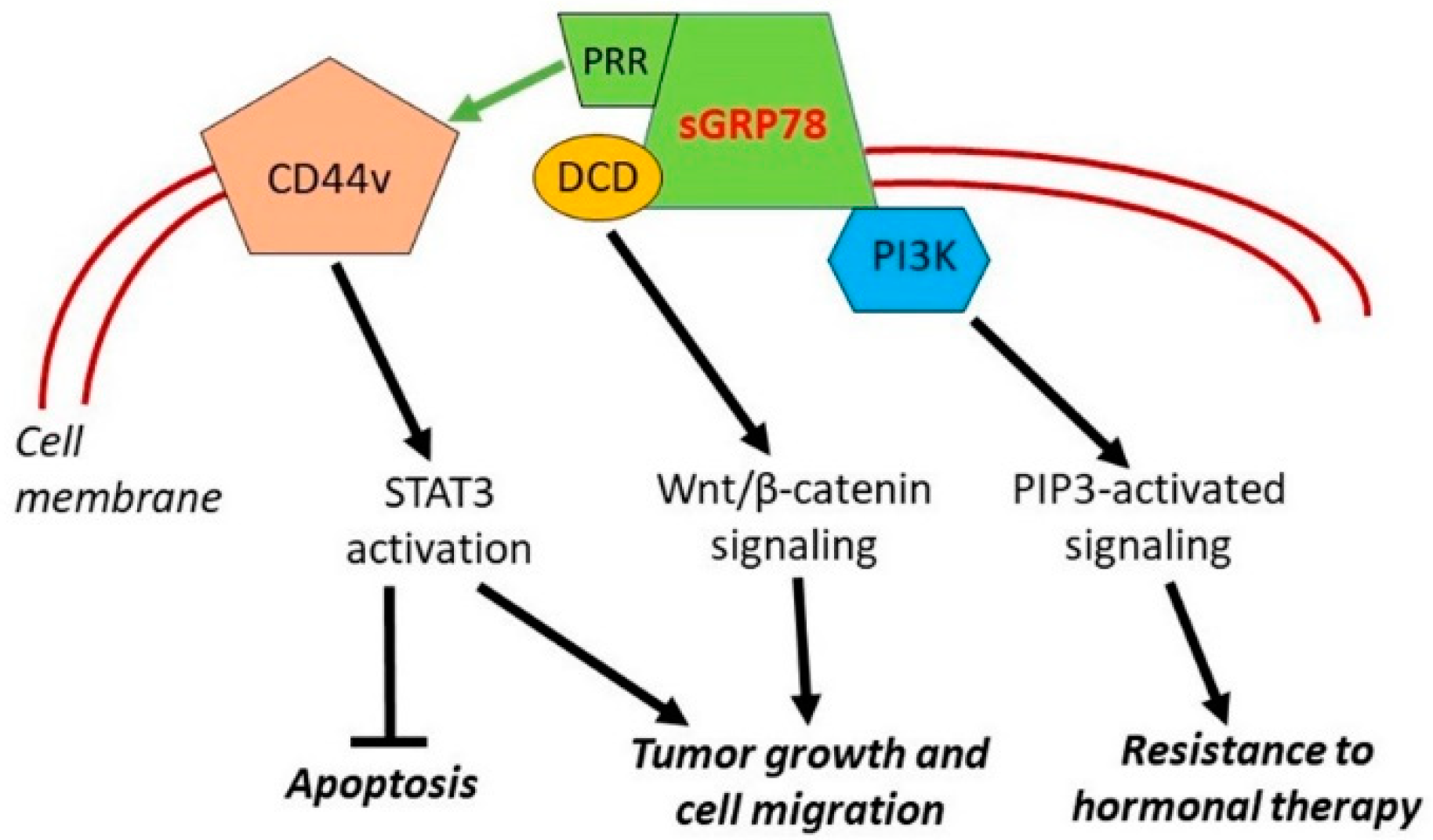
4.2.2. Cell Surface HSPA5
4.2.3. GRP78 and Breast Tumor Evasion of Host Immunity
4.2.4. Clinical Data
4.3. HSPA9 (GRP75 or Mortalin)
4.4. Conclusions on above Two Sections
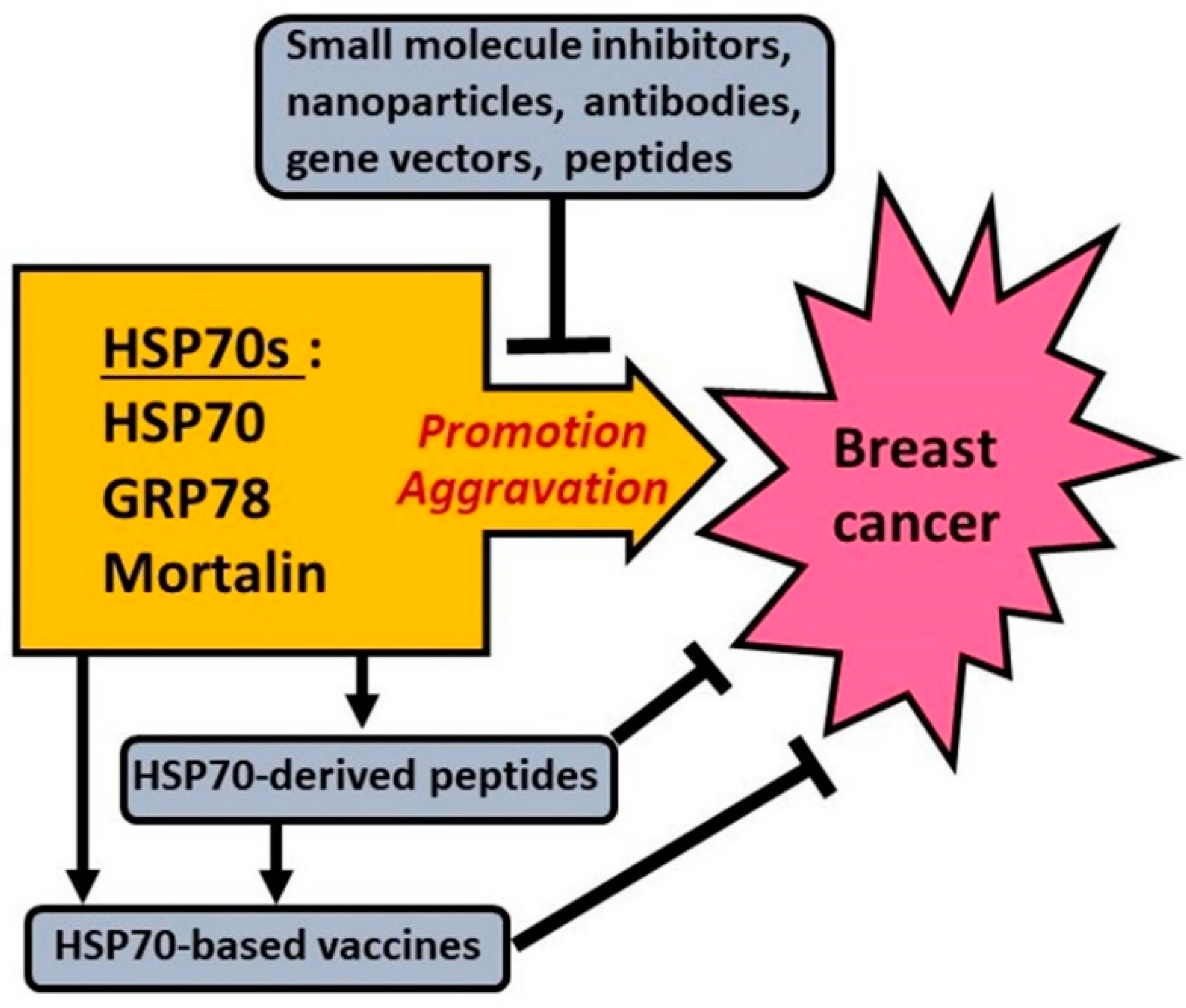
5. Approaches to Targeting HSP70s to Fight Breast Cancer
5.1. Targeting HSP70 in/on Breast Cancer Cells
5.2. Targeting GRP78 (HSPA5) in Breast Cancer Cells
5.2.1. Targeting GRP78 Inside Breast Cancer Cells
5.2.2. Targeting Breast Cancer Cell Surface-Exposed GRP78
5.3. Targeting GRP75 (HSPA9 or Mortalin) in Breast Cancer Cells
6. HSP70s as Potential Targets or Tools for Immunotherapy of Breast Cancer
6.1. Inducible HSP70 as a Potential Target or Tool for Immunotherapy of Breast Cancer
6.1.1. Released and Cancer Cell Surface-Exposed HSP70
6.1.2. HSP70-Peptide Complexes and HSP70-Derived Peptides as Adjuvants or Enhancers of Immune Response against Breast Cancer
6.2. GRP78 (HSPA5) as a Potential Target for Immunotherapy of Breast Cancer
6.2.1. Targeting GRP78 Inside Breast Cancer Cells to Enhance the Antitumor Immune Response
6.2.2. Targeting Breast Cancer Cell Surface-Exposed GRP78 with Anti-GRP78 Antibodies
6.3. GRP75 (Mortalin) as a Potential Target for Immunotherapy of Breast Cancer Cells
7. General Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Coughlin, S.S. Epidemiology of Breast Cancer in Women. Adv. Exp. Med. Biol. 2019, 1152, 9–29. [Google Scholar] [PubMed]
- Lei, S.; Zheng, R.; Zhang, S.; Wang, S.; Chen, R.; Sun, K.; Zeng, H.; Zhou, J.; Wei, W. Global patterns of breast cancer incidence and mortality: A population-based cancer registry data analysis from 2000 to 2020. Cancer Commun. 2021, 41, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.S.; Tse, G.M. Molecular Classification of Breast Cancer. Adv. Anat. Pathol. 2020, 27, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Barzaman, K.; Karami, J.; Zarei, Z.; Hosseinzadeh, A.; Kazemi, M.H.; Moradi-Kalbolandi, S.; Safari, E.; Farahmand, L. Breast cancer: Biology, biomarkers, and treatments. Int. Immunopharmacol. 2020, 84, 106535. [Google Scholar] [CrossRef] [PubMed]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.-P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmer, J. Breast cancer stem cells: Features, key drivers and treatment options. Semin. Cancer Biol. 2018, 53, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Butti, R.; Gunasekaran, V.P.; Kumar, T.V.S.; Banerjee, P.; Kundu, G.C. Breast cancer stem cells: Biology and therapeutic implications. Int. J. Biochem. Cell Biol. 2019, 107, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, H.; Ren, G. Epithelial-mesenchymal transition and drug resistance in breast cancer (Review). Int. J. Oncol. 2015, 47, 840–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gooding, A.J.; Schiemann, W.P. Epithelial-Mesenchymal Transition Programs and Cancer Stem Cell Phenotypes: Mediators of Breast Cancer Therapy Resistance. Mol. Cancer Res. 2020, 18, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Buttacavoli, M.; Di Cara, G.; D’Amico, C.; Geraci, F.; Pucci-Minafra, I.; Feo, S.; Cancemi, P. Prognostic and Functional Significant of Heat Shock Proteins (HSPs) in Breast Cancer Unveiled by Multi-Omics Approaches. Biology 2021, 10, 247. [Google Scholar] [CrossRef] [PubMed]
- Zoppino, F.C.M.; Guerrero-Gimenez, M.E.; Castro, G.N.; Ciocca, D.R. Comprehensive transcriptomic analysis of heat shock proteins in the molecular subtypes of human breast cancer. BMC Cancer 2018, 18, 700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vostakolaei, M.A.; Hatami-Baroogh, L.; Babaei, G.; Molavi, O.; Kordi, S.; Abdolalizadeh, J. Hsp70 in cancer: A double agent in the battle between survival and death. J. Cell. Physiol. 2021, 236, 3420–3444. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef] [PubMed]
- Nitika; Porter, C.M.; Truman, A.W.; Truttmann, M.C. Post-translational modifications of Hsp70 family proteins: Expanding the chaperone code. J. Biol. Chem. 2020, 295, 10689–10708. [Google Scholar] [CrossRef] [PubMed]
- Takakuwa, J.E.; Nitika; Knighton, L.E.; Truman, A.W. Oligomerization of Hsp70: Current Perspectives on Regulation and Function. Front. Mol. Biosci. 2019, 6, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, W.; Fan, M.; Huang, M.; Li, J.J.; Wang, Y. Targeted Profiling of Heat Shock Proteome in Radioresistant Breast Cancer Cells. Chem. Res. Toxicol. 2019, 32, 326–332. [Google Scholar] [CrossRef]
- Dayalan Naidu, S.; Dinkova-Kostova, A.T. Regulation of the mammalian heat shock factor 1. FEBS J. 2017, 284, 1606–1627. [Google Scholar] [CrossRef] [Green Version]
- Pincus, D. Regulation of Hsf1 and the Heat Shock Response. Adv. Exp. Med. Biol. 2020, 1243, 41–50. [Google Scholar] [PubMed]
- Hageman, J.; van Waarde, M.A.W.H.; Zylicz, A.; Walerych, D.; Kampinga, H.H. The diverse members of the mammalian HSP70 machine show distinct chaperone-like activities. Biochem. J. 2011, 435, 127–142. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef]
- Albakova, Z.; Armeev, G.A.; Kanevskiy, L.M.; Kovalenko, E.I.; Sapozhnikov, A.M. HSP70 Multi-Functionality in Cancer. Cells 2020, 9, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesa, L.C.; Shao, H.; Srinivasan, S.R.; Tse, E.; Jain, C.; Zuiderweg, E.R.P.; Southworth, D.R.; Mapp, A.K.; Gestwicki, J.E. X-linked inhibitor of apoptosis protein (XIAP) is a client of heat shock protein 70 (Hsp70) and a biomarker of its inhibition. J. Biol. Chem. 2018, 293, 2370–2380. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Fernández, M.R.; Gragera, M.; Ochoa-Ibarrola, L.; Quintana-Gallardo, L.; Valpuesta, J.M. Hsp70—A master regulator in protein degradation. FEBS Lett. 2017, 591, 2648–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabakov, A.E.; Yakimova, A.O. Hypoxia-Induced Cancer Cell Responses Driving Radioresistance of Hypoxic Tumors: Approaches to Targeting and Radiosensitizing. Cancers 2021, 13, 1102. [Google Scholar] [CrossRef] [PubMed]
- Elmallah, M.I.Y.; Cordonnier, M.; Vautrot, V.; Chanteloup, G.; Garrido, C.; Gobbo, J. Membrane-anchored heat-shock protein 70 (Hsp70) in cancer. Cancer Lett. 2020, 469, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lee, J.; Liem, D.; Ping, P. HSPA5 Gene encoding Hsp70 chaperone BiP in the endoplasmic reticulum. Gene 2017, 618, 14–23. [Google Scholar] [CrossRef]
- Chang, Y.-W.; Chen, H.-A.; Tseng, C.-F.; Hong, C.-C.; Ma, J.-T.; Hung, M.C.; Wu, C.-H.; Huang, M.-T.; Su, J.-L. De-acetylation and degradation of HSPA5 is critical for E1A metastasis suppression in breast cancer cells. Oncotarget 2014, 5, 10558–10570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conner, C.; Lager, T.W.; Guldner, I.H.; Wu, M.-Z.; Hishida, Y.; Hishida, T.; Ruiz, S.; Yamasaki, A.E.; Gilson, R.C.; Belmonte, J.C.I.; et al. Cell surface GRP78 promotes stemness in normal and neoplastic cells. Sci. Rep. 2020, 10, 3474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabakov, A.; Yakimova, A.; Matchuk, O. Molecular Chaperones in Cancer Stem Cells: Determinants of Stemness and Potential Targets for Antitumor Therapy. Cells 2020, 9, 892. [Google Scholar] [CrossRef] [Green Version]
- Grkovic, S.; O’Reilly, V.C.; Han, S.; Hong, M.; Baxter, R.C.; Firth, S.M. IGFBP-3 binds GRP78, stimulates autophagy and promotes the survival of breast cancer cells exposed to adverse microenvironments. Oncogene 2013, 32, 2412–2420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havalová, H.; Ondrovičová, G.; Keresztesová, B.; Bauer, J.A.; Pevala, V.; Kutejová, E.; Kunová, N. Mitochondrial HSP70 Chaperone System—The Influence of Post-Translational Modifications and Involvement in Human Diseases. Int. J. Mol. Sci. 2021, 22, 8077. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Vishwanathan, V.; Birje, A.; Sinha, D.; D’Silva, P. Evolving paradigms on the interplay of mitochondrial Hsp70 chaperone system in cell survival and senescence. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Meng, Z.; Wu, X.; Zhang, M.; Zhang, S.; Jin, T. Mortalin promotes breast cancer malignancy. Exp. Mol. Pathol. 2021, 118, 104593. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Cao, J.; Tian, J.-H.; Yu, C.-Y.; Huang, Q.; Yu, J.-J.; Ma, R.; Wang, J.; Xu, F.; Wang, L.-B. Mortalin maintains breast cancer stem cells stemness via activation of Wnt/GSK3β/β-catenin signaling pathway. Am. J. Cancer Res. 2021, 11, 2696–2716. [Google Scholar] [PubMed]
- Huang, M.-B.; Xia, M.; Gao, Z.; Zhou, H.; Liu, M.; Huang, S.; Zhen, R.; Wu, J.Y.; Roth, W.W.; Bond, V.C.; et al. Characterization of Exosomes in Plasma of Patients with Breast, Ovarian, Prostate, Hepatic, Gastric, Colon, and Pancreatic Cancers. J. Cancer Ther. 2019, 10, 382–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, M.Y.; Gabai, V.L. Hsp70 in cancer: Back to the future. Oncogene 2014, 34, 4153–4161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volloch, V.Z.; Sherman, M.Y. Oncogenic potential of hsp72. Oncogene 1999, 18, 3648–3651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabakov, A.E.; Molotkov, A.O.; Budagova, K.R.; Makarova, Y.M.; Mosin, A.F.; Gabai, V.L. Adaptation of Ehrlich ascites carcinoma cells to energy deprivation in vivo can be associated with heat shock protein accumulation. J. Cell. Physiol. 1995, 165, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gabai, V.L.; Mosina, V.A.; Budagova, K.R.; Kabakov, A.E. Spontaneous overexpression of heat-shock proteins in Ehrlich ascites carcinoma cells during in vivo growth. Biochem. Mol. Biol. Int. 1995, 35, 95–102. [Google Scholar] [PubMed]
- Pastorek, M.; Muller, P.; Coates, P.J.; Vojtesek, B. Intrinsic proteotoxic stress levels vary and act as a predictive marker for sensitivity of cancer cells to hsp90 inhibition. PLoS ONE 2018, 13, e0202758. [Google Scholar] [CrossRef] [Green Version]
- Colvin, T.A.; Gabai, V.L.; Sherman, M.Y. Proteotoxicity is not the reason for the dependence of cancer cells on the major chaperone hsp70. Cell Cycle 2014, 13, 2306–2310. [Google Scholar] [CrossRef] [Green Version]
- Lang, B.J.; Guerrero-Giménez, M.E.; Prince, T.L.; Ackerman, A.; Bonorino, C.; Calderwood, S.K. Heat shock proteins are essential components in transformation and tumor progression: Cancer cell intrinsic pathways and beyond. Int. J. Mol. Sci. 2019, 20, 4507. [Google Scholar] [CrossRef] [Green Version]
- Afanasyeva, E.A.; Komarova, E.Y.; Larsson, L.-G.; Bahram, F.; Margulis, B.A.; Guzhova, I.V. Drug-induced myc-mediated apoptosis of cancer cells is inhibited by stress protein hsp70. Int. J. Cancer 2007, 121, 2615–2621. [Google Scholar] [CrossRef] [PubMed]
- Gennaro, V.J.; Wedegaertner, H.; McMahon, S.B. Interaction between the BAG1S isoform and HSP70 mediates the stability of anti-apoptotic proteins and the survival of osteosarcoma cells expressing oncogenic MYC. BMC Cancer 2019, 19, 258. [Google Scholar] [CrossRef] [PubMed]
- Jamerson, M.H.; Johnson, M.D.; Korsmeyer, S.J.; Furth, P.A.; Dickson, R.B. Bax regulates c-Myc-induced mammary tumour apoptosis but not proliferation in MMTV-c-myc transgenic mice. Br. J. Cancer 2004, 91, 1372–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhooren, V.; Liu, X.-E.; Desmyter, L.; Fan, Y.-D.; Vanwalleghem, L.; Van Molle, W.; Dewaele, S.; Praet, M.; Contreras, R.; Libert, C.; et al. Over-expression of heat shock protein 70 in mice is associated with growth retardation, tumor formation, and early death. Rejuvenation Res. 2008, 11, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Braig, M.; Schmitt, C.A. Oncogene-induced senescence: Putting the brakes on tumor development. Cancer Res. 2006, 66, 2881–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, J.P.; Li, Y. Oncogene-induced senescence and its role in tumor suppression. J. Mammary Gland Biol. Neoplasia 2011, 16, 247–256. [Google Scholar] [CrossRef]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar] [PubMed]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A senescence program controlled by p53 and p16ink4a contributes to the outcome of cancer therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.D.; Broude, E.V.; Dokmanovic, M.; Zhu, H.; Ruth, A.; Xuan, Y.; Kandel, E.S.; Lausch, E.; Christov, K.; Roninson, I.B. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999, 59, 3761–3767. [Google Scholar] [PubMed]
- O’Callaghan-Sunol, C.; Gabai, V.L. Involvement of heat shock proteins in protection of tumor cells from genotoxic stresses. In Heat Shock Proteins in Cancer, 1st ed.; Calderwood, S., Sherman, M.Y., Ciocca, D.R., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 169–190. [Google Scholar]
- Gabai, V.L.; Sherman, M.Y.; Yaglom, J.A. Hsp72 depletion suppresses gamma h2ax activation by genotoxic stresses via p53/p21 signaling. Oncogene 2010, 29, 1952–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astle, M.V.; Hannan, K.M.; Ng, P.Y.; Lee, R.S.; George, A.J.; Hsu, A.K.; Haupt, Y.; Hannan, R.D.; Pearson, R.B. Akt induces senescence in human cells via mtorc1 and p53 in the absence of DNA damage: Implications for targeting mTOR during malignancy. Oncogene 2012, 31, 1949–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, P.D.; Fluck, M.F.Z.; Pedersen, K.; Parra-Palau, J.L.; Guiu, M.; Bernadó Morales, C.; Vicario, R.; Luque-García, A.; Navalpotro, N.P.; Giralt, J.; et al. Constitutive HER2 signaling promotes breast cancer metastasis through cellular senescence. Cancer Res. 2013, 73, 450–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacarias-Fluck, M.F.; Morancho, B.; Vicario, R.; Luque Garcia, A.; Escorihuela, M.; Villanueva, J.; Rubio, I.T.; Arribas, J. Effect of cellular senescence on the growth of HER2-positive breast cancers. J. Natl. Cancer Inst. 2015, 107, djv020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bihani, T.; Chicas, A.; Lo, C.P.-K.; Lin, A.W. Dissecting the senescence-like program in tumor cells activated by Ras signaling. J. Biol. Chem. 2007, 282, 2666–2675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabai, V.L.; Yaglom, J.A.; Waldman, T.; Sherman, M.Y. Heat shock protein hsp72 controls oncogene-induced senescence pathways in cancer cells. Mol. Cell. Biol. 2009, 29, 559–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, L.; Hunt, C.; Yaglom, J.A.; Gabai, V.L.; Sherman, M.Y. Heat shock protein HSP72 plays an essential role in HER2-induced mammary tumorigenesis. Oncogene 2011, 30, 2836–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, C.R.; Dix, D.J.; Sharma, G.G.; Pandita, R.K.; Gupta, A.; Funk, M.; Pandita, T.K. Genomic instability and enhanced radiosensitivity in hsp70.1- and hsp70.3-deficient mice. Mol. Cell. Biol. 2004, 24, 899–911. [Google Scholar] [CrossRef] [Green Version]
- Muller, W.J.; Arteaga, C.L.; Muthuswamy, S.K.; Siegel, P.M.; Webster, M.A.; Cardiff, R.D.; Meise, K.S.; Li, F.; Halter, S.A.; Coffey, R.J. Synergistic interaction of the neu proto-oncogene product and transforming growth factor alpha in the mammary epithelium of transgenic mice. Mol. Cell. Biol. 1996, 16, 5726–5736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Weng, D.; Eguchi, T.; Murshid, A.; Sherman, M.Y.; Song, B.; Calderwood, S.K. Targeting the hsp70 gene delays mammary tumor initiation and inhibits tumor cell metastasis. Oncogene 2015, 34, 5460–5471. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.D.; Rai, S.K.; Salvato, M.S.; Band, H.; Malkovsky, M. Hsp72-mediated augmentation of MHC class I surface expression and endogenous antigen presentation. Int. Immunol. 1998, 10, 609–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dressel, R.; Grzeszik, C.; Kreiss, M.; Lindemann, D.; Herrmann, T.; Walter, L.; Günther, E. Differential effect of acute and permanent heat shock protein 70 overexpression in tumor cells on lysability by cytotoxic T lymphocytes. Cancer Res. 2003, 63, 8212–8220. [Google Scholar] [PubMed]
- Dressel, R.; Lübbers, M.; Walter, L.; Herr, W.; Günther, E. Enhanced susceptibility to cytotoxic T lymphocytes without increase of MHC class I antigen expression after conditional overexpression of heat shock protein 70 in target cells. Eur. J. Immunol. 1999, 29, 3925–3935. [Google Scholar] [CrossRef]
- Multhoff, G.; Pfister, K.; Botzler, C.; Jordan, A.; Scholz, R.; Schmetzer, H.; Burgstahler, R.; Hiddemann, W. Adoptive transfer of human natural killer cells in mice with severe combined immunodeficiency inhibits growth of hsp70-expressing tumors. Int. J. Cancer 2000, 88, 791–797. [Google Scholar] [CrossRef]
- Gastpar, R.; Gehrmann, M.; Bausero, M.A.; Asea, A.; Gross, C.; Schroeder, J.A.; Multhoff, G. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005, 65, 5238–5247. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Deepak, P.; Acharya, A. Hsp70 induces Th1 polarization through tumor-associated macrophages in a T-cell lymphoma. Neoplasma 2007, 54, 113–122. [Google Scholar]
- Gautam, P.K.; Kumar, S.; Deepak, P.; Acharya, A. Morphological effects of autologous hsp70 on peritoneal macrophages in a murine T cell lymphoma. Tumor Biol. 2013, 34, 3407–3415. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.-P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated HSP72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Gobbo, J.; Marcion, G.; Cordonnier, M.; Dias, A.M.M.; Pernet, N.; Hammann, A.; Richaud, S.; Mjahed, H.; Isambert, N.; Clausse, V.; et al. Restoring anticancer immune response by targeting tumor-derived exosomes with a hsp70 peptide aptamer. J. Natl. Cancer Inst. 2015, 108, djv330. [Google Scholar] [CrossRef]
- Jäättelä, M. Over-expression of hsp70 confers tumorigenicity to mouse fibrosarcoma cells. Int. J. Cancer 1995, 60, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Jäättelä, M.; Wissing, D. Heat-shock proteins protect cells from monocyte cytotoxicity: Possible mechanism of self-protection. J. Exp. Med. 1993, 177, 231–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemons, N.J.; Buzzard, K.; Steel, R.; Anderson, R.L. Hsp72 inhibits Fas-mediated apoptosis upstream of the mitochondria in type II cells. J. Biol. Chem. 2005, 280, 9005–9012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nylandsted, J.; Gyrd-Hansen, M.; Danielewicz, A.; Fehrenbacher, N.; Lademann, U.; Hoyer-Hansen, M.; Weber, E.; Multhoff, G.; Rohde, M.; Jaattela, M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J. Exp. Med. 2004, 200, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, H.; Jiang, W.; Zhang, X.; Qiu, F.; Gan, Z.; Cheng, W.; Zhang, J.; Guan, S.; Tang, B.; Huang, Q.; et al. Suppression of HSP70 expression sensitizes NSCLC cell lines to TRAIL-induced apoptosis by upregulating DR4 and DR5 and downregulating c-FLIP-L expressions. J. Mol. Med. 2013, 91, 219–235. [Google Scholar] [CrossRef]
- Monma, H.; Harashima, N.; Inao, T.; Okano, S.; Tajima, Y.; Harada, M. The hsp70 and autophagy inhibitor pifithrin-μ enhances the antitumor effects of TRAIL on human pancreatic cancer. Mol. Cancer Ther. 2013, 12, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Bellmann, K.; Jaattela, M.; Wissing, D.; Burkart, V.; Kolb, H. Heat shock protein hsp70 overexpression confers resistance against nitric oxide. FEBS Lett. 1996, 391, 185–188. [Google Scholar] [CrossRef] [Green Version]
- Burkart, V.; Liu, H.; Bellmann, K.; Wissing, D.; Jaattela, M.; Cavallo, M.G.; Pozzilli, P.; Briviba, K.; Kolb, H. Natural resistance of human beta cells toward nitric oxide is mediated by heat shock protein 70. J. Biol. Chem. 2000, 275, 19521–19528. [Google Scholar] [CrossRef] [Green Version]
- Gurbuxani, S.; Bruey, J.M.; Fromentin, A.; Larmonier, N.; Parcellier, A.; Jaattela, M.; Martin, F.; Solary, E.; Garrido, C. Selective depletion of inducible hsp70 enhances immunogenicity of rat colon cancer cells. Oncogene 2001, 20, 7478–7485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nylandsted, J.; Wick, W.; Hirt, U.A.; Brand, K.; Rohde, M.; Leist, M.; Weller, M.; Jaattela, M. Eradication of glioblastoma, and breast and colon carcinoma xenografts by hsp70 depletion. Cancer Res. 2002, 62, 7139–7142. [Google Scholar]
- Barnoud, T.; Leung, J.C.; Leu, J.I.-J.; Basu, S.; Poli, A.N.R.; Parris, J.L.D.; Indeglia, A.; Martynyuk, T.; Good, M.; Gnanapradeepan, K.; et al. A novel inhibitor of hsp70 induces mitochondrial toxicity and immune cell recruitment in tumors. Cancer Res. 2020, 80, 5270–5281. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.; Maingret, L.; Puig, P.-E.; Rerole, A.-L.; Ghiringhelli, F.; Hammann, A.; Solary, E.; Kroemer, G.; Garrido, C. Heat shock protein 70 neutralization exerts potent antitumor effects in animal models of colon cancer and melanoma. Cancer Res. 2006, 66, 4191–4197. [Google Scholar] [CrossRef] [Green Version]
- Rérole, A.-L.; Gobbo, J.; De Thonel, A.; Schmitt, E.; Pais de Barros, J.P.; Hammann, A.; Lanneau, D.; Fourmaux, E.; Deminov, O.; Micheau, O.; et al. Peptides and aptamers targeting hsp70: A novel approach for anticancer chemotherapy. Cancer Res. 2011, 71, 484–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colvin, T.A.; Gabai, V.L.; Gong, J.; Calderwood, S.K.; Li, H.; Gummuluru, S.; Matchuk, O.N.; Smirnova, S.G.; Orlova, N.V.; Zamulaeva, E.A.; et al. HSP70-BAG3 module regulates cancer-related signaling networks. Cancer Res. 2014, 74, 4731–4740. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Schmid, T.; Frank, R.; Brüne, B. PI3K/Akt is required for heat shock proteins to protect hypoxia-inducible factor 1alpha from pVHL-independent degradation. J. Biol. Chem. 2004, 279, 13506–13513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Li, J.; Costa, M.; Gao, J.; Huang, C. JNK1 mediates degradation HIF-1α by a VHL-independent mechanism that involves the chaperones Hsp90/Hsp70. Cancer Res. 2010, 70, 813–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.L.; Chung, T.-W.; Kim, S.; Hwang, B.; Kim, J.M.; Lee, H.M.; Cha, H.-J.; Seo, Y.; Choe, S.Y.; Ha, K.-T.; et al. HSP70-1 is required for interleukin-5-induced angiogenic responses through eNOS pathway. Sci. Rep. 2017, 7, 44687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.-K.; Na, H.J.; Lee, W.R.; Jeoung, M.H.; Lee, S. Heat shock protein 70-1a is a novel angiogenic regulator. Biochem. Biophys. Res. Commun. 2016, 469, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Kim, M.R.; Kim, T.-K.; Lee, W.R.; Kim, J.H.; Heo, K.; Lee, S. CLEC14a-HSP70-1A interaction regulates HSP70-1A-induced angiogenesis. Sci. Rep. 2017, 7, 10666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasioumi, P.; Vrazeli, P.; Vezyraki, P.; Zerikiotis, S.; Katsouras, C.; Damalas, A.; Angelidis, C. Hsp70 (hsp70a1a) downregulation enhances the metastatic ability of cancer cells. Int. J. Oncol. 2019, 54, 821–832. [Google Scholar] [CrossRef]
- Li, H.; Li, Y.; Liu, D.; Sun, H.; Su, D.; Yang, F.; Liu, J. Extracellular HSP70/HSP70-PCs promote epithelial-mesenchymal transition of hepatocarcinoma cells. PLoS ONE 2013, 8, e84759. [Google Scholar] [CrossRef] [Green Version]
- Boroughs, L.K.; Antonyak, M.A.; Johnson, J.L.; Cerione, R.A. A unique role for heat shock protein 70 and its binding partner tissue transglutaminase in cancer cell migration. J. Biol. Chem. 2011, 286, 37094–37107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, J.D.; McCready, J.; Jay, D.G. Extracellular heat shock protein (hsp)70 and hsp90α assist in matrix metalloproteinase-2 activation and breast cancer cell migration and invasion. PLoS ONE 2011, 6, e18848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perconti, G.; Maranto, C.; Romancino, D.P.; Rubino, P.; Feo, S.; Bongiovanni, A.; Giallongo, A. Pro-invasive stimuli and the interacting protein hsp70 favour the route of alpha-enolase to the cell surface. Sci. Rep. 2017, 7, 3841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, A.; Mauro, L.; Giordano, F.; Panza, S.; Iannacone, R.; Liuzzi, G.M.; Aquila, S.; De Amicis, F.; Cellini, F.; Indiveri, C.; et al. Recombinant Arabidopsis Hsp70 sustains cell survival and metastatic potential of breast cancer cells. Mol. Cancer Ther. 2016, 15, 1063–1073. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Ngoka, L.; Mei, Y.; Lesoon, L.; Cowell, J.K. HSP90 and HSP70 proteins are essential for stabilization and activation of WASF3 metastasis-promoting protein. J. Biol. Chem. 2012, 287, 10051–10059. [Google Scholar] [CrossRef] [Green Version]
- Budina-Kolomets, A.; Webster, M.R.; Leu, J.I.-J.; Jennis, M.; Krepler, C.; Guerrini, A.; Kossenkov, A.V.; Xu, W.; Karakousis, G.; Schuchter, L.; et al. HSP70 inhibition limits FAK-dependent invasion and enhances the response to melanoma treatment with BRAF inhibitors. Cancer Res. 2016, 76, 2720–2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluger, H.M.; Chelouche Lev, D.; Kluger, Y.; McCarthy, M.M.; Kiriakova, G.; Camp, R.L.; Rimm, D.L.; Price, J.E. Using a xenograft model of human breast cancer metastasis to find genes associated with clinically aggressive disease. Cancer Res. 2005, 65, 5578–5587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahim, H.H.; Mohamed, G.; Safwat, G.; Abo-Bakr, A.; Ibraheem, M.H.; Al-Mofty, S.; Kamel, M.M.; Abdel-Moneim, A.S.; Gameel, A.M. Hsp70 as a diagnostic and prognostic marker in Egyptian women with breast cancer. Clin. Breast Cancer 2021, 21, e177–e188. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhang, S.; Zhang, D.; Li, Y.; Zhao, X.; Luo, Y.; Guo, Y. Identification of metastasis-related proteins and their clinical relevance to triple-negative human breast cancer. Clin. Cancer Res. 2008, 14, 7050–7059. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Ouzounova, M.; Piranlioglu, R.; Ma, M.T.; Guzel, M.; Marasco, D.; Chadli, A.; Gestwicki, J.E.; Cowell, J.K.; Wicha, M.S.; et al. The pleiotropic effects of TNFα in breast cancer subtypes is regulated by TNFAIP3/A20. Oncogene 2019, 38, 469–482. [Google Scholar] [CrossRef]
- Gabai, V.L.; Yaglom, J.A.; Wang, Y.; Meng, L.; Shao, H.; Kim, G.; Colvin, T.; Gestwicki, J.; Sherman, M.Y. Anticancer effects of targeting hsp70 in tumor stromal cells. Cancer Res. 2016, 76, 5926–5932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scieglinska, D.; Krawczyk, Z. Expression, function, and regulation of the testis-enriched heat shock HSPA2 gene in rodents and humans. Cell Stress Chaperones 2015, 20, 221–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, B.; Bromfield, E.G.; Cui, J.; De Iuliis, G.N. Heat shock protein A2 (HSPA2): Regulatory roles in germ cell development and sperm function. In The Role of Heat Shock Proteins in Reproductive System Development and Function; MacPhee, D.J., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 67–93. [Google Scholar]
- Scieglinska, D.; Gogler-Piglowska, A.; Butkiewicz, D.; Chekan, M.; Malusecka, E.; Harasim, J.; Habryka, A.; Krawczyk, Z. HSPA2 is expressed in human tumors and correlates with clinical features in non-small cell lung carcinoma patients. Anticancer Res. 2014, 34, 2833–2840. [Google Scholar] [PubMed]
- Rohde, M.; Daugaard, M.; Jensen, M.H.; Helin, K.; Nylandsted, J.; Jäättelä, M. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 2005, 19, 570–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugaard, M.; Kirkegaard-Sørensen, T.; Ostenfeld, M.S.; Aaboe, M.; Høyer-Hansen, M.; Ørntoft, T.F.; Rohde, M.; Jäättelä, M. Lens epithelium-derived growth factor is an hsp70-2 regulated guardian of lysosomal stability in human cancer. Cancer Res. 2007, 67, 2559–2567. [Google Scholar] [CrossRef] [Green Version]
- Jagadish, N.; Agarwal, S.; Gupta, N.; Fatima, R.; Devi, S.; Kumar, V.; Suri, V.; Kumar, R.; Suri, V.; Sadasukhi, T.C.; et al. Heat shock protein 70-2 (hsp70-2) overexpression in breast cancer. J. Exp. Clin. Cancer Res. 2016, 35, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sojka, D.R.; Gogler-Pigłowska, A.; Klarzyńska, K.; Klimczak, M.; Zylicz, A.; Głowala-Kosińska, M.; Krawczyk, Z.; Scieglinska, D. HSPA2 chaperone contributes to the maintenance of epithelial phenotype of human bronchial epithelial cells but has non-essential role in supporting malignant features of non-small cell lung carcinoma, MCF7, and HeLa cancer cells. Cancers 2020, 12, 2749. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-L.; Zhang, Y.; Li, D.-D.; Zhang, F.-L.; Liu, H.-Y.; Liao, X.-H.; Xie, H.-Y.; Lu, Q.; Zhang, L.; Hong, Q.; et al. RNF144A functions as a tumor suppressor in breast cancer through ubiquitin ligase activity-dependent regulation of stability and oncogenic functions of HSPA2. Cell Death Differ. 2020, 27, 1105–1118. [Google Scholar] [CrossRef]
- Fernandez, P.M.; Tabbara, S.O.; Jacobs, L.K.; Manning, F.C.; Tsangaris, T.N.; Schwartz, A.M.; Kennedy, K.A.; Patierno, S.R. Overexpression of the glucose-regulated stress gene grp78 in malignant but not benign human breast lesions. Breast Cancer Res. Treat. 2000, 59, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Bini, L.; Magi, B.; Marzocchi, B.; Arcuri, F.; Tripodi, S.; Cintorino, M.; Sanchez, J.C.; Frutiger, S.; Hughes, G.; Pallini, V.; et al. Protein expression profiles in human breast ductal carcinoma and histologically normal tissue. Electrophoresis 1997, 18, 2832–2841. [Google Scholar] [CrossRef] [PubMed]
- Scriven, P.; Coulson, S.; Haines, R.; Balasubramanian, S.; Cross, S.; Wyld, L. Activation and clinical significance of the unfolded protein response in breast cancer. Br. J. Cancer 2009, 101, 1692–1698. [Google Scholar] [CrossRef] [Green Version]
- Gazit, G.; Lu, J.; Lee, A.S. De-regulation of grp stress protein expression in human breast cancer cell lines. Breast Cancer Res. Treat 1999, 54, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ni, M.; Li, J.; Xiong, S.; Ye, W.; Virrey, J.J.; Mao, C.; Ye, R.; Wang, M.; Pen, L.; et al. Critical role of the stress chaperone Grp78/Bip in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008, 68, 498–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. Mol. Cell. Biol. 1992, 12, 954–961. [Google Scholar] [PubMed] [Green Version]
- Miao, R.Y.; Drabsch, Y.; Cross, R.S.; Cheasley, D.; Carpinteri, S.; Pereira, L.; Malaterre, J.; Gonda, T.J.; Anderson, R.L.; Ramsay, R.G. MYB is essential for mammary tumorigenesis. Cancer Res. 2011, 71, 7029–7037. [Google Scholar] [CrossRef] [Green Version]
- Croci, S.; Recktenwald, C.V.; Lichtenfels, R.; Nicoletti, G.; Dressler, S.P.; De Giovanni, C.; Astolfi, A.; Palladini, A.; Shin-ya, K.; Landuzzi, L.; et al. Proteomic and proteomex profiling of mammary cancer progression in a HER-2/neu oncogene-driven animal model system. Proteomics 2010, 10, 3835–3853. [Google Scholar] [CrossRef] [PubMed]
- Prokakis, E.; Dyas, A.; Grün, R.; Fritzsche, S.; Bedi, U.; Kazerouni, Z.B.; Kosinsky, R.L.; Johnsen, S.A.; Wegwitz, F. USP22 promotes HER2-driven mammary carcinoma aggressiveness by suppressing the unfolded protein response. Oncogene 2021, 40, 4004–4018. [Google Scholar] [CrossRef] [PubMed]
- Yeung, B.H.Y.; Kwan, B.W.Y.; He, Q.Y.; Lee, A.S.; Liu, J.; Wong, A.S.T. Glucose-regulated protein 78 as a novel effector of BRCA1 for inhibiting stress-induced apoptosis. Oncogene 2008, 27, 6782–6789. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.A.; Chang, Y.W.; Tseng, C.F.; Chiu, C.F.; Hong, C.C.; Wang, W.; Wang, M.Y.; Hsiao, M.; Ma, J.T.; Chen, C.H.; et al. E1A-mediated inhibition of HSPA5 suppresses cell migration and invasion in triple-negative breast cancer. Ann. Surg. Oncol. 2015, 22, 889–898. [Google Scholar] [CrossRef]
- Chang, Y.W.; Tseng, C.F.; Wang, M.Y.; Chang, W.C.; Lee, C.C.; Chen, L.T.; Hung, M.C.; Su, J.L. Deacetylation of HSPA5 by HDAC6 leads to GP78-mediated HSPA5 ubiquitination at K447 and suppresses metastasis of breast cancer. Oncogene 2016, 35, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Kuang, X.-Y.; Jiang, H.-S.; Li, K.; Zheng, Y.-Z.; Liu, Y.-R.; Qiao, F.; Li, S.; Hu, X.; Shao, Z.-M. The phosphorylation-specific association of STMN1 with GRP78 promotes breast cancer metastasis. Cancer Lett. 2016, 377, 87–96. [Google Scholar] [CrossRef]
- Sun, L.L.; Chen, C.M.; Zhang, J.; Wang, J.; Yang, C.Z.; Lin, L.Z. Glucose-regulated protein 78 signaling regulates hypoxia-induced epithelial-mesenchymal transition in A549 cells. Front. Oncol. 2019, 9, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, D.; Katoch, A.; Sharma, D.; Faheem, M.M.; Chakraborty, S.; Sahu, P.K.; Chikan, N.A.; Amin, H.; Gupta, A.P.; Gandhi, S.G.; et al. Indolylkojyl methane analogue IKM5 potentially inhibits invasion of breast cancer cells via attenuation of GRP78. Breast Cancer Res. Treat. 2019, 177, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.R.; Eckhardt, B.L.; Cao, Y.; Pasqualini, R.; Argani, P.; Arap, W.; Ramsay, R.G.; Anderson, R.L. Inhibition of established micrometastases by targeted drug delivery via cell surface–associated grp78. Clin. Cancer Res. 2013, 19, 2107–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, D.; Stapleton, C.; Luo, B.; Xiong, S.; Ye, W.; Zhang, Y.; Jhaveri, N.; Zhu, G.; Ye, R.; Liu, Z.; et al. A critical role for Grp78/Bip in the tumor microenvironment for neovascularization during tumor growth and metastasis. Cancer Res. 2011, 71, 2848–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katanasaka, Y.; Ishii, T.; Asai, T.; Naitou, H.; Maeda, N.; Koizumi, F.; Miyagawa, S.; Ohashi, N.; Oku, N. Cancer antineovascular therapy with liposome drug delivery systems targeted to Bip/Grp78. Int. J. Cancer 2010, 127, 2685–2698. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Liu, H.; Zhang, X.; Zhang, L.; Li, X.; Wang, C.; Sun, S. Cell surface GRP78 accelerated breast cancer cell proliferation and migration by activating STAT3. PLoS ONE 2015, 10, e0125634. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-C.; Zhang, P.; Lee, A.S. The COOH-terminal proline-rich region of GRP78 is a key regulator of its cell surface expression and viability of tamoxifen-resistant breast cancer cells. Neoplasia 2019, 21, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Brauer, H.A.; D’Arcy, M.; Libby, T.E.; Thompson, H.J.; Yasui, Y.Y.; Hamajima, N.; Li, C.I.; Troester, M.A.; Lampe, P.D. Dermcidin expression is associated with disease progression and survival among breast cancer patients. Breast Cancer Res. Treat. 2014, 144, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bancovik, J.; Moreira, D.F.; Carrasco, D.; Yao, J.; Porter, D.; Moura, R.; Camargo, A.; Fontes-Oliveira, C.C.; Malpartida, M.G.; Carambula, S.; et al. Dermcidin exerts its oncogenic effects in breast cancer via modulation of ErbB signaling. BMC Cancer 2015, 15, 70. [Google Scholar] [CrossRef] [PubMed]
- Lager, T.W.; Conner, C.; Keating, C.R.; Warshaw, J.N.; Panopoulos, A.D. Cell surface Grp78 and dermcidin cooperate to regulate breast cancer cell migration through Wnt signaling. Oncogene 2021, 40, 4050–4059. [Google Scholar] [CrossRef]
- Zhang, Y.; Tseng, C.-C.; Tsai, Y.-L.; Fu, X.; Schiff, R.; Lee, A.S. Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production. PLoS ONE 2013, 8, e80071. [Google Scholar] [CrossRef]
- Cook, K.L.; Shajahan, A.N.; Wärri, A.; Jin, L.; Hilakivi-Clarke, L.A.; Clarke, R. Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer Res. 2012, 72, 3337–3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto-Pantoja, D.R.; Wilson, A.S.; Clear, K.Y.; Westwood, B.; Triozzi, P.L.; Cook, K.L. Unfolded protein response signaling impacts macrophage polarity to modulate breast cancer cell clearance and melanoma immune checkpoint therapy responsiveness. Oncotarget 2017, 8, 80545–80559. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zheng, H.; Yu, X.; Liu, L.; Li, H.; Zhu, H.; Zhang, Z.; Lei, P.; Shen, G. Tumor-secreted GRP78 promotes the establishment of a pre-metastatic niche in the liver microenvironment. Front. Immunol. 2020, 11, 584458. [Google Scholar] [CrossRef]
- Chou, C.W.; Yang, R.Y.; Chan, L.C.; Li, C.F.; Sun, L.; Lee, H.H.; Lee, P.C.; Sher, Y.P.; Ying, H.; Hung, M.C. The stabilization of PD-L1 by the endoplasmic reticulum stress protein GRP78 in triple-negative breast cancer. Am. J. Cancer Res. 2020, 10, 2621–2634. [Google Scholar]
- Zheng, Y.Z.; Cao, Z.G.; Hu, X.; Shao, Z.M. The endoplasmic reticulum stress markers GRP78 and CHOP predict disease-free survival and responsiveness to chemotherapy in breast cancer. Breast Cancer Res. Treat. 2014, 145, 349–358. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, Z.; Zou, Y.; Gao, G.; Liu, L.; Xu, H.; Liu, F. Expression of glucose-regulated protein 78 as prognostic biomarkers for triple-negative breast cancer. Histol. Histopathol. 2020, 35, 559–568. [Google Scholar]
- Bartkowiak, K.; Kwiatkowski, M.; Buck, F.; Gorges, T.M.; Nilse, L.; Assmann, V.; Andreas, A.; Müller, V.; Wikman, H.; Riethdorf, S.; et al. Disseminated tumor cells persist in the bone marrow of breast cancer patients through sustained activation of the unfolded protein response. Cancer Res. 2015, 75, 5367–5377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadhwa, R.; Takano, S.; Kaur, K.; Deocaris, C.C.; Pereira-Smith, O.M.; Reddel, R.R.; Kaul, S.C. Upregulation of mortalin/mtHsp70/Grp75 contributes to human carcinogenesis. Int. J. Cancer 2006, 118, 2973–2980. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.; Kaul, S.C.; Ryu, J.; Lee, J.S.; Ahn, H.M.; Kaul, Z.; Kalra, R.S.; Li, L.; Widodo, N.; Yun, C.O.; et al. Stress chaperone mortalin contributes to epithelial-mesenchymal transition and cancer metastasis. Cancer Res. 2016, 76, 2754–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, D.; Zhang, J.; Chen, P.; Guo, X.; Qiao, J.; Zhu, J.; Wang, L.; Lu, Z.; Liu, Z. HN1L promotes migration and invasion of breast cancer by up-regulating the expression of HMGB1. J. Cell. Mol. Med. 2021, 25, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Ji, M.; Chen, L.; Liu, Q.; Che, S.; Xu, M.; Lin, Z. The clinicopathological significance of mortalin overexpression in invasive ductal carcinoma of breast. J. Exp. Clin. Cancer Res. 2016, 35, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogeraki, A.; Giannikaki, E.; Tzardi, M.; Kafousi, M.; Ieromonachou, P.; Dariviannaki, K.; Askoxylakis, J.; Tsiftsis, D.; Stathopoulos, E.; Zoras, O. Correlation of heat shock protein (HSP70) expression with cell proliferation (MIB1), estrogen receptors (ER) and clinicopathological variables in invasive ductal breast carcinomas. J. Exp. Clin. Cancer Res. 2007, 26, 367–368. [Google Scholar]
- Somiari, R.I.; Sullivan, A.; Russell, S.; Somiari, S.; Hu, H.; Jordan, R.; George, A.; Katenhusen, R.; Buchowiecka, A.; Arciero, C.; et al. High-throughput proteomic analysis of human infiltrating ductal carcinoma of the breast. Proteomics 2003, 3, 1863–1873. [Google Scholar] [CrossRef]
- Davidson, B.; Valborg Reinertsen, K.; Trinh, D.; Reed, W.; Bøhler, P.J. BAG-1/SODD, HSP70, and HSP90 are potential prognostic markers of poor survival in node-negative breast carcinoma. Hum. Pathol. 2016, 54, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.Y.; Gabai, V.L. The role of Bag-3 in cell signaling. J. Cell. Biochem. 2021. online ahead of print. [Google Scholar] [CrossRef]
- Mena, S.; Rodríguez, M.L.; Ponsoda, X.; Estrela, J.M.; Jäättela, M.; Ortega, A.L. Pterostilbene-induced tumor cytotoxicity: A lysosomal membrane permeabilization-dependent mechanism. PLoS ONE 2012, 7, e44524. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, B.; Xiao, H.; Dong, J.; Li, Y.; Zhu, C.; Jin, Y.; Li, H.; Cui, M.; Fan, S. LncRNA HOTAIR enhances breast cancer radioresistance through facilitating HSPA1A expression via sequestering miR-449b-5p. Thorac. Cancer 2020, 11, 1801–1816. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Xu, Z.; Zhu, S.; Sun, W.; Wang, X.; Tan, C.; Zhang, Y.; Zhang, G.; Xu, Y.; Tang, J. Small extracellular vesicle-mediated Hsp70 intercellular delivery enhances breast cancer adriamycin resistance. Free Radic. Biol. Med. 2021, 164, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tian, Y.; Tian, W.; Sun, J.; Zhao, S.; Liu, Y.; Wang, C.; Tang, Y.; Ma, X.; Teng, Z.; et al. Selectively Sensitizing Malignant Cells to Photothermal Therapy Using a CD44-Targeting Heat Shock Protein 72 Depletion Nanosystem. ACS Nano 2016, 10, 8578–8590. [Google Scholar] [CrossRef]
- Zhou, A.; Du, J.; Jiao, M.; Xie, D.; Wang, Q.; Xue, L.; Ju, C.; Hua, Z.; Zhang, C. Co-delivery of TRAIL and siHSP70 using hierarchically modular assembly formulations achieves enhanced TRAIL-resistant cancer therapy. J. Control Release 2019, 304, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Fu, J.; Xiao, X.; Wu, J.; Wu, R.-C. MiR-34a regulates therapy resistance by targeting HDAC1 and HDAC7 in breast cancer. Cancer Lett. 2014, 354, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.; Cutress, R.I.; Johnson, P.W.M.; Packham, G.; Townsend, P.A. Short peptides derived from the BAG-1 C-terminus inhibit the interaction between BAG-1 and HSC70 and decrease breast cancer cell growth. FEBS Lett. 2009, 583, 3405–3411. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.L.C.H.B.; Kim, S.-A.; Choi, H.S.; Yoon, J.-H.; Ahn, S.-G. Epigallocatechin-3-gallate suppresses the expression of HSP70 and HSP90 and exhibits anti-tumor activity in vitro and in vivo. BMC Cancer 2010, 10, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massey, A.J.; Williamson, D.S.; Browne, H.; Murray, J.B.; Dokurno, P.; Shaw, T.; Macias, A.T.; Daniels, Z.; Geoffroy, S.; Dopson, M.; et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother. Pharmacol. 2010, 66, 535–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truman, A.W.; Kristjansdottir, K.; Wolfgeher, D.; Ricco, N.; Mayampurath, A.; Volchenboum, S.L.; Clotet, J.; Kron, C.J. Quantitative proteomics of the yeast Hsp70/Hsp90 interactomes during DNA damage reveal chaperone-dependent regulation of ribonucleotide reductase. J. Proteom. 2015, 112, 285–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, B.; Yang, H.; Zhang, X.; Li, H. Visualizing and Quantifying the Effect of the Inhibition of HSP70 on Breast Cancer Cells Based on Laser Scanning Microscopy. Technol. Cancer Res. Treat. 2018, 17, 1533033818785274. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.R.; Cesa, L.C.; Li, X.; Julien, O.; Zhuang, M.; Shao, H.; Chung, J.; Maillard, I.; Wells, J.A.; Duckett, C.S. Heat Shock Protein 70 (Hsp70) Suppresses RIP1-Dependent Apoptotic and Necroptotic Cascades. Mol. Cancer Res. 2018, 16, 58–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, R.; Mukherjee, S.; Biswas, J.; Roy, M. Sulphoraphane, a naturally occurring isothiocyanate induces apoptosis in breast cancer cells by targeting heat shock proteins. Biochem. Biophys. Res. Commun. 2012, 427, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Das, C.K.; Linder, B.; Bonn, F.; Rothweiler, F.; Dikic, I.; Michaelis, M.; Cinatl, J.; Mandal, M.; Kögel, D. BAG3 Overexpression and Cytoprotective Autophagy Mediate Apoptosis Resistance in Chemoresistant Breast Cancer Cells. Neoplasia 2018, 20, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Howe, M.K.; Bodoor, K.; Carlson, D.A.; Hughes, P.F.; Alwarawrah, Y.; Loiselle, D.R.; Jaeger, A.M.; Darr, D.B.; Jordan, J.L.; Hunter, L.M.; et al. Identification of an allosteric small-molecule inhibitor selective for the inducible form of heat shock protein 70. Chem. Biol. 2014, 21, 1648–1659. [Google Scholar] [CrossRef] [Green Version]
- Mawatari, T.; Ninomiya, I.; Inokuchi, M.; Harada, S.; Hayashi, H.; Oyama, K.; Makino, I.; Nakagawara, H.; Miyashita, T.; Tajima, H.; et al. Valproic acid inhibits proliferation of HER2-expressing breast cancer cells by inducing cell cycle arrest and apoptosis through Hsp70 acetylation. Int. J. Oncol. 2015, 47, 2073–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.; Cao, R.; Zhang, T.; Li, S.; Zhong, W. Design and synthesis of piperidine derivatives as novel human heat shock protein 70 inhibitors for the treatment of drug-resistant tumors. Eur. J. Med. Chem. 2015, 97, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fani, S.; Dehghan, F.; Karimian, H.; Lo, K.M.; Nigjeh, S.E.; Keong, Y.S.; Soori, R.; Chow, K.M.; Kamalidehghan, B.; Ali, H.M.; et al. Monobenzyltin Complex C1 Induces Apoptosis in MCF-7 Breast Cancer Cells through the Intrinsic Signaling Pathway and through the Targeting of MCF-7-Derived Breast Cancer Stem Cells via the Wnt/β-Catenin Signaling Pathway. PLoS ONE 2016, 11, e0160836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fani, S.; Kamalidehghan, B.; Lo, K.M.; Nigjeh, S.E.; Keong, Y.S.; Dehghan, F.; Soori, R.; Abdulla, M.A.; Chow, K.M.; Ali, H.M.; et al. Anticancer activity of a monobenzyltin complex C1 against MDA-MB-231 cells through induction of apoptosis and inhibition of breast cancer stem cells. Sci. Rep. 2016, 6, 38992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafavinia, S.E.; Khorashadizadeh, M.; Hoshyar, R. Antiproliferative and Proapoptotic Effects of Crocin Combined with Hyperthermia on Human Breast Cancer Cells. DNA Cell Biol. 2016, 35, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Kodiha, M.; Mahboubi, H.; Maysinger, D.; Stochaj, U. Gold Nanoparticles Impinge on Nucleoli and the Stress Response in MCF7 Breast Cancer Cells. Nanobiomedicine 2016, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, V.; Cascione, M.; Rizzello, L.; Manno, D.E.; Di Guglielmo, C.; Rinaldi, R. Synergistic Effect Induced by Gold Nanoparticles with Polyphenols Shell during Thermal Therapy: Macrophage Inflammatory Response and Cancer Cell Death Assessment. Cancers 2021, 13, 3610. [Google Scholar] [CrossRef]
- Zeng, Y.-Q.; Cao, R.-Y.; Yang, J.-L.; Li, X.-Z.; Li, S.; Zhong, W. Design, synthesis and biological evaluation of novel HSP70 inhibitors: N, N’-disubstituted thiourea derivatives. Eur. J. Med. Chem. 2016, 119, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Khan, G.N.; Kim, E.J.; Shin, T.S.; Lee, S.H. Azacytidine-induced Chemosensitivity to Doxorubicin in Human Breast Cancer MCF7 Cells. Anticancer Res. 2017, 37, 2355–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Houston, Z.H.; Simpson, J.D.; Chen, L.; Fletcher, N.L.; Fuchs, A.V.; Blakey, I.; Thurecht, K.J. Using Peptide Aptamer Targeted Polymers as a Model Nanomedicine for Investigating Drug Distribution in Cancer Nanotheranostics. Mol. Pharm. 2017, 14, 3539–3549. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Srinivasan, S.R.; Connarn, J.; Ahmad, A.; Young, Z.T.; Kabza, A.M.; Zuiderweg, E.R.P.; Sun, D.; Gestwicki, J.E. Analogs of the Allosteric Heat Shock Protein 70 (Hsp70) Inhibitor, MKT-077, as Anti-Cancer Agents. ACS Med. Chem. Lett. 2013, 4, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Yaglom, J.A.; Wang, Y.; Li, A.; Li, Z.; Monti, S.; Alexandrov, I.; Lu, X.; Sherman, M.Y. Cancer cell responses to Hsp70 inhibitor JG-98: Comparison with Hsp90 inhibitors and finding synergistic drug combinations. Sci. Rep. 2018, 8, 3010. [Google Scholar] [CrossRef] [Green Version]
- Shao, H.; Li, X.; Moses, M.A.; Gilbert, L.A.; Kalyanaraman, C.; Young, Z.T.; Chernova, M.; Journey, S.N.; Weissman, J.S.; Hann, B.; et al. Exploration of Benzothiazole Rhodacyanines as Allosteric Inhibitors of Protein-Protein Interactions with Heat Shock Protein 70 (Hsp70). J. Med. Chem. 2018, 61, 6163–6177. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, S.; Assimon, V.A.; Young, Z.T.; Morra, G.; Shao, H.; Taylor, I.R.; Gestwicki, J.E.; Colombo, G. A Local Allosteric Network in Heat Shock Protein 70 (Hsp70) Links Inhibitor Binding to Enzyme Activity and Distal Protein-Protein Interactions. ACS Chem. Biol. 2018, 13, 3142–3152. [Google Scholar] [CrossRef]
- You, C.; Li, Y.; Dong, Y.; Ning, L.; Zhang, Y.; Yao, L.; Wang, F. Low-Temperature Trigger Nitric Oxide Nanogenerators for Enhanced Mild Photothermal Therapy. ACS Biomater. Sci. Eng. 2020, 6, 1535–1542. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Gestwicki, J.E. Neutral analogs of the heat shock protein 70 (Hsp70) inhibitor, JG-98. Bioorg. Med. Chem. Lett. 2020, 30, 126954. [Google Scholar] [CrossRef] [PubMed]
- Pirali, M.; Taheri, M.; Zarei, S.; Majidi, M.; Ghafouri, H. Artesunate, as a HSP70 ATPase activity inhibitor, induces apoptosis in breast cancer cells. Int. J. Biol. Macromol. 2020, 164, 3369–3375. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Li, Y.; Qu, Y.-X.; Su, Y.; Peng, Y.; Zhao, Z.; Fu, T.; Wang, X.-Q.; Tan, W. Aptamer-Peptide Conjugates as Targeted Chemosensitizers for Breast Cancer Treatment. ACS Appl. Mater. Interfaces 2021, 13, 9436–9444. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-S.; Kumar, V.; Lee, D.-Y.; Chen, Y.; Wu, Y.-C.; Gao, J.-Y.; Chu, P.-C. Development of Novel Rhodacyanine-Based Heat Shock Protein 70 Inhibitors. Curr. Med. Chem. 2021, 28, 5431–5446. [Google Scholar] [CrossRef] [PubMed]
- Hyun, S.Y.; Le, H.T.; Min, H.-Y.; Pei, H.; Lim, Y.; Song, I.; Nguyen, Y.T.K.; Hong, S.; Han, B.W.; Lee, H.-Y. Evodiamine inhibits both stem cell and non-stem-cell populations in human cancer cells by targeting heat shock protein 70. Theranostics 2021, 11, 2932–2952. [Google Scholar] [CrossRef] [PubMed]
- Sannino, S.; Yates, M.E.; Schurdak, M.E.; Oesterreich, S.; Lee, A.V.; Wipf, P.; Brodsky, J.I. Unique integrated stress response sensors regulate cancer cell susceptibility when Hsp70 activity is compromised. eLife 2021, 10, e64977. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Liang, Z.; Lu, J.; Wang, X.; Jia, F.; Hu, Q.; Xiao, X.; Deng, X.; Wu, Y.; Sheng, W. A multifunctional nanodiamond-based nanoplatform for the enhanced mild-temperature photothermal/chemo combination therapy of triple negative breast cancer via an autophagy regulation strategy. Nanoscale 2021, 13, 13375–13389. [Google Scholar] [CrossRef]
- Wawruszak, A.; Halasa, M.; Okon, E.; Kukula-Koch, W.; Stepulak, A. Valproic Acid and Breast Cancer: State of the Art in 2021. Cancers 2021, 13, 3409. [Google Scholar] [CrossRef] [PubMed]
- Moses, M.A.; Kim, Y.S.; Rivera-Marquez, G.M.; Oshima, N.; Watson, M.J.; Beebe, K.E.; Wells, C.; Lee, S.; Zuehlke, A.D.; Shao, H.; et al. Targeting the Hsp40/Hsp70 Chaperone Axis as a Novel Strategy to Treat Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 4022–4035. [Google Scholar] [CrossRef] [Green Version]
- Nitika; Blackman, J.S.; Knighton, L.E.; Takakuwa, J.E.; Calderwood, S.K.; Truman, A.W. Chemogenomic screening identifies the Hsp70 co-chaperone DNAJA1 as a hub for anticancer drug resistance. Sci. Rep. 2020, 10, 13831. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Wang, S.; Li, C.; Qian, M.; Zheng, Y.; Yan, X.; Huang, R. TKD peptide as a ligand targeting drug delivery systems to memHsp70-positive breast cancer. Int. J. Pharm. 2016, 498, 40–48. [Google Scholar] [CrossRef]
- Shen, S.; Wei, C.; Fu, J. RNA-Sequencing Reveals Heat Shock 70-kDa Protein 6 (HSPA6) as a Novel Thymoquinone-Upregulated Gene That Inhibits Growth, Migration, and Invasion of Triple-Negative Breast Cancer Cells. Front. Oncol. 2021, 11, 667995. [Google Scholar] [CrossRef]
- Wang, J.; Yin, Y.; Hua, H.; Li, M.; Luo, T.; Xu, L.; Wang, R.; Liu, D.; Zhang, Y.; Jiang, Y. Blockade of GRP78 sensitizes breast cancer cells to microtubules-interfering agents that induce the unfolded protein response. J. Cell. Mol. Med. 2009, 13, 3888–3897. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.; Nalluri, S.; Kolhe, R.; Yang, Y.; Fiskus, W.; Chen, J.; Ha, K.; Buckley, K.M.; Balusu, R.; Coothankandaswamy, V.; et al. Treatment with panobinostat induces glucose-regulated protein 78 acetylation and endoplasmic reticulum stress in breast cancer cells. Mol. Cancer Ther. 2010, 9, 942–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Cheng, X.L.; Yang, Y.P.; Li, Z.Q. GRP78 mediates radiation resistance of a stem cell-like subpopulation within the MCF-7 breast cancer cell line. Oncol. Rep. 2013, 30, 2119–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, K.; Clarke, P.A.G.; Clarke, R. Targeting GRP78 and antiestrogen resistance in breast cancer. Future Med. Chem. 2013, 9, 1047–1057. [Google Scholar] [CrossRef]
- Parmar, J.H.; Cook, K.L.; Shajahan-Haq, A.N.; Clarke, P.A.G.; Tavassoly, I.; Clarke, R.; Tyson, J.J.; Baumann, W.T. Modelling the effect of GRP78 on anti-oestrogen sensitivity and resistance in breast cancer. Interface Focus 2013, 3, 20130012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, K.L.; Soto-Pantoja, D.R.; Clarke, P.A.G.; Cruz, M.I.; Zwart, A.; Wärri, A.; Hilakivi-Clarke, L.; Roberts, D.D.; Clarke, R. Endoplasmic Reticulum Stress Protein GRP78 Modulates Lipid Metabolism to Control Drug Sensitivity and Antitumor Immunity in Breast Cancer. Cancer Res. 2016, 76, 5657–5670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, X.; Tu, Y.; Xu, Y.; Guo, Y.; Yao, F.; Zhang, X. Endoplasmic reticulum stress confers 5-fluorouracil resistance in breast cancer cell via the GRP78/OCT4/lncRNA MIAT/AKT pathway. Am. J. Cancer Res. 2020, 10, 838–855. [Google Scholar]
- Patel, M.R.; Kozuch, S.D.; Cultrara, C.N.; Yadav, R.; Huang, S.; Samuni, U.; Koren, J.; Chiosis, G.; Sabatino, D. RNAi Screening of the Glucose-Regulated Chaperones in Cancer with Self-Assembled siRNA Nanostructures. Nano Lett. 2016, 16, 6099–6108. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.A.S.; Park, S.; Kim, S.-Y.; Min, D.-H.; Jeon, N.L.; Song, J.M. Liposomal co-delivery-based quantitative evaluation of chemosensitivity enhancement in breast cancer stem cells by knockdown of GRP78/CLU. J. Liposome Res. 2019, 29, 44–52. [Google Scholar] [CrossRef]
- Ermakova, S.P.; Kang, B.S.; Choi, B.Y.; Choi, H.S.; Schuster, T.F.; Ma, W.-Y.; Bode, A.M.; Dong, Z. (-)-Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res. 2006, 66, 9260–9269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Wang, J.; Jing, J.; Hua, H.; Luo, T.; Xu, L.; Wang, R.; Liu, D.; Jiang, Y. Synergistic promotion of breast cancer cells death by targeting molecular chaperone GRP78 and heat shock protein 70. J. Cell. Mol. Med. 2009, 13, 4540–4550. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhao, Y.; Zhang, Y.; Zhang, D. Fucoidan induces cancer cell apoptosis by modulating the endoplasmic reticulum stress cascades. PLoS ONE 2014, 9, e108157. [Google Scholar] [CrossRef]
- Wang, N.; Wang, Z.; Peng, C.; You, J.; Shen, J.; Han, S.; Chen, J. Dietary compound isoliquiritigenin targets GRP78 to chemosensitize breast cancer stem cells via β-catenin/ABCG2 signaling. Carcinogenesis 2014, 35, 2544–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Adhikary, A.; Chakraborty, S.; Bhattacharjee, P.; Mazumder, M.; Putatunda, S.; Gorain, M.; Chakraborty, A.; Kundu, G.C.; Das, T.; et al. Cross-talk between endoplasmic reticulum (ER) stress and the MEK/ERK pathway potentiates apoptosis in human triple negative breast carcinoma cells: Role of a dihydropyrimidone, nifetepimine. J. Biol. Chem. 2015, 290, 3936–3949. [Google Scholar] [CrossRef] [Green Version]
- Pujari, R.; Jose, J.; Bhavnani, V.; Kumar, N.; Shastry, P.; Pal, J.K. Tamoxifen-induced cytotoxicity in breast cancer cells is mediated by glucose-regulated protein 78 (GRP78) via AKT (Thr308) regulation. Int. J. Biochem. Cell Biol. 2016, 77, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Kawiak, A.; Domachowska, A.; Jaworska, A.; Lojkowska, E. Plumbagin sensitizes breast cancer cells to tamoxifen-induced cell death through GRP78 inhibition and Bik upregulation. Sci. Rep. 2017, 7, 43781. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-Q.; Aka, J.A.; Li, T.; Xu, D.; Doillon, C.J.; Lin, S.-X. Inhibition of 17beta-hydroxysteroid dehydrogenase type 7 modulates breast cancer protein profile and enhances apoptosis by down-regulating GRP78. J. Steroid Biochem. Mol. Biol. 2017, 172, 188–197. [Google Scholar] [CrossRef]
- Viswanath, A.N.I.; Lim, J.W.; Seo, S.H.; Lee, J.Y.; Lim, S.M.; Pae, A.N. GRP78-targeted in-silico virtual screening of novel anticancer agents. Chem. Biol. Drug Des. 2018, 92, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Peng, F.; Huang, X.; Xie, X.; Chen, B.; Shen, J.; Gao, F.; You, J.; Xie, X.; Chen, J. Neoisoliquiritigenin Inhibits Tumor Progression by Targeting GRP78-β-catenin Signaling in Breast Cancer. Curr. Cancer Drug Targets 2018, 18, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zheng, Y.; Gu, J.; Wang, S.; Wang, N.; Yang, B.; Zhang, F.; Wang, D.; Fu, W.; Wang, Z. Betulinic acid chemosensitizes breast cancer by triggering ER stress-mediated apoptosis by directly targeting GRP78. Cell Death Dis. 2018, 9, 636. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Li, S.; Zhang, X.; Lu, J.; Wang, W.; Zhou, S.; Zhang, J.; Wang, R.; Li, A. HHQ-4, a quinoline derivate, preferentially inhibits proliferation of glucose-deprived breast cancer cells as a GRP78 down-regulator. Toxicol. Appl. Pharmacol. 2019, 373, 10–25. [Google Scholar] [CrossRef]
- Liao, M.; Wang, C.; Yang, B.; Huang, D.; Zheng, Y.; Wang, S.; Wang, X.; Zhang, J.; Tang, C.; Xu, Z.; et al. Autophagy Blockade by Ai Du Qing Formula Promotes Chemosensitivity of Breast Cancer Stem Cells via GRP78/β-Catenin/ABCG2 Axis. Front. Pharmacol. 2021, 12, 659297. [Google Scholar] [CrossRef]
- Tan, W.W.; Allred, J.B.; Moreno-Aspitia, A.; Northfelt, D.W.; Ingle, J.N.; Goetz, M.P.; Perez, E.A. Phase I Study of Panobinostat (LBH589) and Letrozole in Postmenopausal Metastatic Breast Cancer Patients. Clin. Breast Cancer 2016, 16, 82–86. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.-C.; Stanciauskas, R.; Zhang, P.; Woo, D.; Wu, K.; Kelly, K.; Gill, P.S.; Yu, M.; Pinaud, F.; Lee, A.S. GRP78 regulates CD44v membrane homeostasis and cell spreading in tamoxifen-resistant breast cancer. Life Sci. Alliance 2019, 2, e201900377. [Google Scholar] [CrossRef] [Green Version]
- Arap, M.A.; Lahdenranta, J.; Mintz, P.J.; Hajitou, A.; Sarkis, A.S.; Arap, W.; Pasqualini, R. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell 2004, 6, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Staquicini, D.I.; D’Angelo, S.; Ferrara, F.; Karjalainen, K.; Sharma, G.; Smith, T.L.; Tarleton, C.A.; Jaalouk, D.E.; Kuniyasu, A.; Baze, W.B.; et al. Therapeutic targeting of membrane-associated GRP78 in leukemia and lymphoma: Preclinical efficacy in vitro and formal toxicity study of BMTP-78 in rodents and primates. Pharmacogenom. J. 2018, 18, 436–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passarella, R.J.; Spratt, D.E.; van der Ende, A.E.; Phillips, J.G.; Wu, H.; Sathiyakumar, V.; Zhou, L.; Hallahan, D.E.; Harth, E.; Diaz, R. Targeted nanoparticles that deliver a sustained, specific release of Paclitaxel to irradiated tumors. Cancer Res. 2010, 70, 4550–4559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobroff, A.S.; D’Angelo, S.; Eckhardt, B.L.; Ferrara, F.; Staquicini, D.I.; Cardó-Vila, M.; Staquicini, F.I.; Nunes, D.N.; Kim, K.; Driessen, W.H.P.; et al. Towards a transcriptome-based theranostic platform for unfavorable breast cancer phenotypes. Proc. Natl. Acad. Sci. USA 2016, 113, 12780–12785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Zhang, Y.; Yu, V.C.; Chong, Y.-S.; Yoshioka, T.; Ge, R. Isthmin targets cell-surface GRP78 and triggers apoptosis via induction of mitochondrial dysfunction. Cell Death Differ. 2014, 21, 797–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, C.; Chandna, R.; Ghode, A.; Dsouza, C.; Chen, M.; Larsson, A.; Lim, S.H.; Wang, M.; Cao, Z.; Zhu, Y.; et al. Proapoptotic Cyclic Peptide BC71 Targets Cell-Surface GRP78 and Functions as an Anticancer Therapeutic in Mice. EBioMedicine 2018, 33, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-H.; Chi-Lung Lee, A.C.-L.; Chen, I.-J.; Chang, N.-C.; Wu, H.-C.; Yu, H.-M.; Chang, Y.-J.; Lee, T.-W.; Yu, J.-C.; Yu, A.L.; et al. Structure-based optimization of GRP78-binding peptides that enhances efficacy in cancer imaging and therapy. Biomaterials 2016, 94, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Shan, M.; Gao, C.; Feng, X.; Yang, Y.; Zhang, R.; He, Y.; Zhang, G.; Zhang, L. PCDHGB7 Increases Chemosensitivity to Carboplatin by Inhibiting HSPA9 via Inducing Apoptosis in Breast Cancer. Dis. Markers 2019, 2019, 6131548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaul, S.C.; Aida, S.; Yaguchi, T.; Kaur, K.; Wadhwa, R. Activation of wild type p53 function by its mortalin-binding, cytoplasmically localizing carboxyl terminus peptides. J. Biol. Chem. 2005, 280, 39373–39379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grover, A.; Priyandoko, D.; Gao, R.; Shandilya, A.; Widodo, N.; Bisaria, V.S.; Kaul, S.C.; Wadhwa, R.; Sundar, D. Withanone binds to mortalin and abrogates mortalin-p53 complex: Computational and experimental evidence. Int. J. Biochem. Cell Biol. 2012, 44, 496–504. [Google Scholar] [CrossRef]
- Abunimer, A.N.; Mohammed, H.; Cook, K.L.; Soto-Pantoja, D.R.; Campos, M.M.; Abu-Asab, M.S. Mitochondrial autophagosomes as a mechanism of drug resistance in breast carcinoma. Ultrastruct. Pathol. 2018, 42, 170–180. [Google Scholar] [CrossRef]
- Nigam, N.; Grover, A.; Goyal, S.; Katiyar, S.P.; Bhargava, P.; Wang, P.-C.; Sundar, D.; Kaul, S.C.; Wadhwa, R. Targeting Mortalin by Embelin Causes Activation of Tumor Suppressor p53 and Deactivation of Metastatic Signaling in Human Breast Cancer Cells. PLoS ONE 2015, 10, e0138192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadhwa, R.; Nigam, N.; Bhargava, P.; Dhanjal, J.K.; Goyal, S.; Grover, A.; Sundar, D.; Ishida, Y.; Terao, K.; Kaul, S.C. Molecular Characterization and Enhancement of Anticancer Activity of Caffeic Acid Phenethyl Ester by γ Cyclodextrin. J. Cancer 2016, 7, 1755–1771. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.-B.; Gonzalez, R.R.; Lillard, J.; Bond, V.C. Secretion modification region-derived peptide blocks exosome release and mediates cell cycle arrest in breast cancer cells. Oncotarget 2017, 8, 11302–11315. [Google Scholar] [CrossRef] [Green Version]
- Elwakeel, A.; Sari, A.N.; Dhanjal, J.K.; Meidinna, H.N.; Sundar, D.; Kaul, S.C.; Wadhwa, R. Mutant p53L194F Harboring Luminal-A Breast Cancer Cells Are Refractory to Apoptosis and Cell Cycle Arrest in Response to MortaparibPlus, a Multimodal Small Molecule Inhibitor. Cancers 2021, 13, 3043. [Google Scholar] [CrossRef] [PubMed]
- Cable, J.; Greenbaum, B.; Pe’er, D.; Bollard, C.M.; Bruni, S.; Griffin, M.E.; Allison, J.P.; Wu, C.J.; Subudhi, S.K.; Mardis, E.R.; et al. Frontiers in cancer immunotherapy—A symposium report. Ann. N. Y. Acad. Sci. 2021, 1489, 30–47. [Google Scholar] [CrossRef] [PubMed]
- Bausero, M.A.; Gastpar, R.; Multhoff, G.; Asea, A. Alternative mechanism by which IFN-gamma enhances tumor recognition: Active release of heat shock protein 72. J. Immunol. 2005, 175, 2900–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, K.; Sheppe, A.E.F.; Singh, I.; Hui, W.W.; Edelmann, M.J.; Rinaldi, C. Exosomes released by breast cancer cells under mild hyperthermic stress possess immunogenic potential and modulate polarization in vitro in macrophages. Int. J. Hyperth. 2020, 37, 696–710. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Bao, D.; Chen, X.; Wu, Y.; Wei, Y.; Wu, Z.; Li, F.; Piao, J.-G. Microwave-triggered/HSP-targeted gold nano-system for triple-negative breast cancer photothermal therapy. Int. J. Pharm. 2021, 593, 120162. [Google Scholar] [CrossRef]
- Faure, O.; Graff-Dubois, S.; Bretaudeau, L.; Derré, L.; Gross, D.-A.; Alves, P.M.S.; Cornet, S.; Marie-Thérèse Duffour, M.-T.; Chouaib, S.; Miconnet, I.; et al. Inducible Hsp70 as target of anticancer immunotherapy: Identification of HLA-A*0201-restricted epitopes. Int. J. Cancer 2004, 108, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Hazama, S.; Tamada, K.; Udaka, K.; Irie, A.; Nishimura, Y.; Miyakawa, T.; Doi, S.; Nakajima, M.; Kanekiyo, S.; et al. Identification of a Promiscuous Epitope Peptide Derived from HSP70. J. Immunother. 2019, 42, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Majumder, N.; Lin, H.; Chen, J.; Falo, L.D.; You, Z. Enhanced immunity by NeuEDhsp70 DNA vaccine Is needed to combat an aggressive spontaneous metastatic breast cancer. Mol. Ther. 2005, 11, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, X.; Gao, W.; Yang, Y.; Ma, H.; Ren, X. A new purification method for enhancing the immunogenicity of heat shock protein 70-peptide complexes. Oncol. Rep. 2012, 28, 1977–1983. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Zhang, Y.; Durfee, J.; Weng, D.; Liu, C.; Koido, S.; Song, B.; Apostolopoulos, V.; Calderwood, S.K. A heat shock protein 70-based vaccine with enhanced immunogenicity for clinical use. J. Immunol. 2010, 184, 488–496. [Google Scholar] [CrossRef]
- Weng, D.; Calderwood, S.K.; Gong, J. A Novel Heat Shock Protein 70-based Vaccine Prepared from DC-Tumor Fusion Cells. Methods Mol. Biol. 2018, 1709, 359–369. [Google Scholar] [PubMed]
- Zhang, Y.; Luo, W.; Wang, Y.; Chen, J.; Liu, Y.; Zhang, Y. Enhanced antitumor immunity of nanoliposome-encapsulated heat shock protein 70 peptide complex derived from dendritic tumor fusion cells. Oncol. Rep. 2015, 33, 2695–2702. [Google Scholar] [CrossRef]
- Cook, K.L.; Soto-Pantoja, D.R. “UPRegulation” of CD47 by the endoplasmic reticulum stress pathway controls anti-tumor immune responses. Biomark. Res. 2017, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Bhowmik, A.; Bhandary, S.; Putatunda, S.; Laskar, A.; Biswas, A.; Dolui, S.; Banerjee, B.; Khan, R.; Das, N.; et al. Formulation and antitumorigenic activities of nanoencapsulated nifetepimine: A promising approach in treating triple negative breast carcinoma. Nanomedicine 2016, 7, 1973–1985. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Adhikary, A.; Chakraborty, S.; Nandi, P.; Mohanty, S.; Chakraborty, S.; Bhattacharjee, P.; Mukherjee, S.; Putatunda, S.; Chakraborty, S.; et al. Nifetepimine, a dihydropyrimidone, ensures CD4+ T cell survival in a tumor microenvironment by maneuvering sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA). J. Biol. Chem. 2012, 287, 32881–32896. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Liu, L.; Brown, N.J.; Christian, S.; Hornby, D. Quantum dot-conjugated anti-GRP78 scFv inhibits cancer growth in mice. Molecules 2012, 17, 796–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, S.; Staquicini, F.I.; Ferrara, F.; Staquicini, D.I.; Sharma, G.; Tarleton, C.A.; Nguyen, H.; Naranjo, L.A.; Sidman, R.L.; Arap, W.; et al. Selection of phage-displayed accessible recombinant targeted antibodies (SPARTA): Methodology and applications. JCI Insight 2018, 3, e98305. [Google Scholar] [CrossRef]
- Jubran, R.; Saar-Ray, M.; Wawruszak, A.; Ziporen, L.; Donin, N.; Bairey, O.; Fishelson, Z. Mortalin peptides exert antitumor activities and act as adjuvants to antibody-mediated complement-dependent cytotoxicity. Int. J. Oncol. 2020, 57, 1013–1026. [Google Scholar] [CrossRef]
- Wang, J.; Bhargava, P.; Yu, Y.; Sari, A.N.; Zhang, H.; Ishii, N.; Yan, K.; Zhang, Z.; Ishida, Y.; Terao, K.; et al. Novel Caffeic Acid Phenethyl Ester—Mortalin Antibody Nanoparticles Offer Enhanced Selective Cytotoxicity to Cancer Cells. Cancers 2020, 12, 2370. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HSP | Subtypes | ER+/PR+ | ER−/PR− | HER2+ | HER2− | Refs |
|---|---|---|---|---|---|---|
| HSP1A | − | + | − | − | − | [10] |
| HSPA2 | Luminal A, B | + | − | − | + | [10,11] |
| HSPA5 | Basal | − | − | − | − | [11] |
| HSPA6 | Basal | − | − | − | − | [11] |
| HSPA8 | − | − | − | + | − | [10] |
| SUPPRESSION | PROMOTION | PROMOTION | ||||
|---|---|---|---|---|---|---|
| Apoptosis | Senescence | Angiogenesis | EMT | Migration/Invasion | Metastases | |
| HSPA1 | [73,74] | [59,60] | [86,87,88,89,90,91] | [93] | [33,83,94,95,96,97] | [63,102,103] |
| HSPA2 | [110] | [108] | ND | ND | [110,111,112] * | ND |
| HSPA5 | [117] | ND | [117,129] | [126,127] | [27,123,124,125] | [27,28,123,124,127,128] |
| HSPA9 | ND | ND | ND | [33,34,145] | [33,34,145] | ND |
| Overexpression | Progression/Grade | Metastases | Mortality | |
|---|---|---|---|---|
| HSPA1 | [101,148,149] | [71,148,149] | [100,101,102] | [100,150] |
| HSPA2 | [107,108] | NC [110] | ND | [112] |
| HSPA5 | [113,114,115] | [101,123] | [123,131] | [141,142] |
| HSPA9 | [147] | [147] | [147] | [147] |
| Agent | Cells or Tumor | Molecular Target | Achieved Effects |
|---|---|---|---|
| BAG1-derived peptides [158] | MCF-7 and ZR-75-1 cells | HSC70–BAG-1 interactions | Inhibition of cell proliferation |
| Epigallocatechin-3-gallate [159] | MCF-7 cells, | HSP70 | Inhibited HSP70 expression, |
| xenografts | expression | reduced tumor size | |
| VER155008 [103,160,161,162,163] | BT474, MCF-7 and MDA-MB-231 cells | HSP70 ATPase | Induction of apoptosis, damage of mitochondria, sensitization to TNF, heating and gemcitabine |
| Sulphoraphane [164] | MDA-MB-231 | HSP70 expression | Downregulation of HSP70 and HSP90, apoptosis |
| and MCF-7 cells | |||
| YM-1 [86,165] | MCF-7 xenografts, MDA-MB-231 and BT-549 cells | HSP70–BAG-3 interactions | Inhibited xenograft growth, |
| sensitization to drug-induced apoptosis | |||
| HS-72 [166] | BT474 and MCF-7 cells, | HSP70–ATP | Antiproliferative activity, reduced tumor size |
| MMTV 1-neu model | affinity | ||
| Valproic acid [167] | SKBR3 cells | HDAC 2, | Increased HSP70 acetylation, cell cycle arrest, apoptosis |
| acetylated HSP70 | |||
| Piperidine derivatives [168] | BT474, BT/Lap(R)1.0, MDA-MB-231 and other cell lines | HSP70 ATPase | Inhibited cell proliferation, sensitization to lapatinib |
| Monobenzyltin complex C1 [169,170] | MCF-7, MDA-MB-231 cells and breast CSCs | HSP70 expression | Decreased HSP70 level, induction of apoptosis |
| Crocin [171] | MDA-MB-468 cells | HSP70 expression | Decreased HSP70 and HSP90 levels, induction of apoptosis |
| Gold NPs 3 [172,173] | MCF-7 cells | HSP70 expression | Downregulation of HSP70 and ribosome biogenesis, thermosensitization |
| Disubstituted thiourea [174] | BT474 cells | HSP70 ATPase | Sensitization to lapatinib |
| Azacytidine [175] | MCF-7 cells | HSP70 expression | Sensitization to doxorubicin |
| Peptide aptamers with high affinity to HSP70 (as components of NPs with doxorubicin) [176] | MDA-MB-468 cells, xenografts | Tumoral HSP70 | Tumor regression, sensitization to doxorubicin |
| JG-98 [22,163,177,178] | MDA-MB-231 and MCF-7 cells | HSP70 allosteric site in NBD 4, HSP70–BAG-3 interactions | Loss of c-IAP1 5 and XIAP 6, apoptosis and necroptosis, sensitization to drugs |
| JG-231 [179] | MCF-7, MDA-MB-231 | HSP70 allosteric site in NBD | Cell death, reduced tumor burden in xenografts |
| cells and xenografts | |||
| MKT-077 and its analog JG-237 [180] | MDA-MB-231 | “Loop 222” in | Antiproliferative |
| and MCF-7 cells | NBD of HSP70 | activities | |
| PES 7 (or pifithrin-μ) [22,181] | MDA-MB-231and MCF-7 cells, xenografts | HSP70 SBD 8 | Loss of c-IAP1 and XIAP, apoptosis, sensitization to photothermal therapy |
| Neutral analogs of JG-98 [182] | MCF-7 cells | HSP70 allosteric site | Antiproliferative activities |
| Artesunate [183] | 4T1 and MCF-7 cells | HSP70 ATPase, HSP70 expression | Inhibition of HSP70, induction of apoptosis |
| Aptamer peptide conjugates [184] | MCF-7 cells | Tumoral HSP70 | Sensitization to doxorubicin |
| Benzo-fused rhodacyanines [185] | Breast cancer cells | HSP70 chaperone Function | Antiproliferative activities, downregulation of client antiapoptotic proteins |
| Evodiamine [186] | MDA-MB-231 cells, CSCs, PDX 9 model | HSP70 NBD, HSP70 expression | Degradation of HSP70, inhibited cell proliferation, reduced tumor growth |
| MAL3-101 [187] | Cell lines from TNBC 10 and luminal subtypes | HSP70 ATPase | Induction of UPR and cell death via apoptosis |
| Apoptozole (as one of components of nano-diamond-based nano-platform) [188] | MDA-MB-231 cells and xenografts | HSP70 ATPase, HSP70 expression | Sensitization to photothermal chemo-combined therapy, inhibition of autophagy |
| Agent | Cells or Tumor | Molecular Target | Achieved Effects |
|---|---|---|---|
| Epigallocatechin gallate [203,204] | MDA-MB-231 and T47D cells | GRP78 ATPase | Sensitization to etoposide and quercetin, apoptosis |
| Panobinostat (LBH589) [195] | MDA-MB-231 | HDAC6 1, acetylated GRP78 | Induction of UPR and apoptosis |
| and MCF-7 cells | |||
| Fukoidan [205] | MDA-MB-231 cells | GRP78 expression | Downregulation of GRP78 and apoptosis |
| Isoliquiritigenin [206] | MCF-7 and MDA-MB-231 | GRP78 ATPase, GRP78/β-catenin/ABCG2 2 signaling | Sensitization to epirubicin in vitro and in vivo |
| cells, sorted CSCs, xenografts from CSCs | |||
| Nifetepimine [207] | MDA-MB-468 and MDA-MB-231 cells, xenografts | GRP78 expression | Attenuated GRP78 induction, apoptosis, reduced tumor growth |
| VER155008 [208] | MDA-MB-231 and MCF-7 cells | GRP78 ATPase | Sensitization to tamoxifen, apoptosis |
| Plumbagin [209] | MCF-7 and T47D cells | GRP78 expression | Downregulation of GRP78, apoptosis, sensitization to tamoxifen |
| INH7 [210] | MCF-7 cells | 17β-HSD7 3, GRP78 expression | Downregulation of GRP78, apoptosis |
| VH1019, VH1011 [211] | MCF-7 cells | GRP78 structure- based docking | Antiproliferative and cytotoxic effects |
| Neoisoliquiritigenin [212] | Breast cancer cells and xenografts | GRP78 ATPase, GRP78/β-catenin signaling | Inhibition of proliferation, apoptosis |
| Betulinic acid [213] | MDA-MB-231 and MCF-7 cells | GRP78 ATPase | Sensitization to taxol, apoptosis |
| HHQ-4 [214] | Glucose-deprived breast cancer cells | GRP78 expression | Downregulation of GRP78, inhibition of proliferation |
| Indolylkojil methane analog (IKM5) [127] | MDA-MB-231, MDA-MB-468, MCF-7 and 4T1 cells (in mice) | GRP78 SBD 4, GRP78–TIMP-1 5 interactions | Inhibited expression of EMT markers, suppression of invasion, tumor growth and lung metastases |
| Ai Du Qing formula [215] | MDA-MB-231, MCF-7 cells, breast CSCs and xenografts | GRP78 expression, GRP78/β-catenin/ /ABCG2 axis | Downregulation of GRP78, β-catenin degradation, repressing and chemo-sensitizing effects on cancer cells, CSCs and xenografts |
| HA15 [121] | HCC1954 and SKBR3 cells | GRP78 ATPase | Apoptosis, suppressed cell proliferation |
| Agent | Cells or Tumor | Molecular Target | Achieved Effects |
|---|---|---|---|
| p53 carboxyl-terminus peptides [226] | MCF-7 cells | Mortalin–p53 interaction | Disruption of the p53–mortalin |
| complex, activation of p53, growth arrest | |||
| Withanone [227] | MCF-7 cells | Mortalin–p53 interaction | Abrogation of the p53–mortalin |
| complex, activation of p53, growth arrest or apoptosis | |||
| MKT-077 [227,228] | MCF-7 cells | p53-binding region of mortalin | Mitochondrial toxicity, selective killing of tumor cells |
| Embelin [229] | MDA-MB-231 and MCF-7 cells | Mortalin–p53 interaction, mortalin expression | Abrogation of the p53–mortalin |
| complex, activation of p53, growth arrest and inactivation of metastatic signaling | |||
| CAPE 1 and its complex with γ-cyclodextrin [230] | MDA-MB-231 and MCF-7 cells | Mortalin–p53 interaction | Disruption of the p53–mortalincomplex, activation of p53, growth arrest, suppression of metastases |
| PEG-SMRwt-CLU 2 [231] | MDA-MB-231 and MCF-7 cells | Exosome secretion, mortalin (suggested) | Blockade of exosome release, growth arrest |
| Mortaparib Plus 3 [232] | MCF-7 cells | Mortalin–p53 interaction | Abrogation of the p53–mortalincomplex, activation of p53, growth arrest, apoptosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kabakov, A.E.; Gabai, V.L. HSP70s in Breast Cancer: Promoters of Tumorigenesis and Potential Targets/Tools for Therapy. Cells 2021, 10, 3446. https://doi.org/10.3390/cells10123446
Kabakov AE, Gabai VL. HSP70s in Breast Cancer: Promoters of Tumorigenesis and Potential Targets/Tools for Therapy. Cells. 2021; 10(12):3446. https://doi.org/10.3390/cells10123446
Chicago/Turabian StyleKabakov, Alexander E., and Vladimir L. Gabai. 2021. "HSP70s in Breast Cancer: Promoters of Tumorigenesis and Potential Targets/Tools for Therapy" Cells 10, no. 12: 3446. https://doi.org/10.3390/cells10123446
APA StyleKabakov, A. E., & Gabai, V. L. (2021). HSP70s in Breast Cancer: Promoters of Tumorigenesis and Potential Targets/Tools for Therapy. Cells, 10(12), 3446. https://doi.org/10.3390/cells10123446